
Abstract
Genetic variants such as single nucleotide polymorphisms (SNPs) have been suggested as potential molecular biomarkers to predict the functional outcome of psychiatric disorders. To assess the schizophrenia’ functional outcomes such as Quality of Life Scale (QLS) and the Global Assessment of Functioning (GAF), we leveraged a bagging ensemble machine learning method with a feature selection algorithm resulting from the analysis of 11 SNPs (AKT1 rs1130233, COMT rs4680, DISC1 rs821616, DRD3 rs6280, G72 rs1421292, G72 rs2391191, 5-HT2A rs6311, MET rs2237717, MET rs41735, MET rs42336, and TPH2 rs4570625) of 302 schizophrenia patients in the Taiwanese population. We compared our bagging ensemble machine learning algorithm with other state-of-the-art models such as linear regression, support vector machine, multilayer feedforward neural networks, and random forests. The analysis reported that the bagging ensemble algorithm with feature selection outperformed other predictive algorithms to forecast the QLS functional outcome of schizophrenia by using the G72 rs2391191 and MET rs2237717 SNPs. Furthermore, the bagging ensemble algorithm with feature selection surpassed other predictive algorithms to forecast the GAF functional outcome of schizophrenia by using the AKT1 rs1130233 SNP. The study suggests that the bagging ensemble machine learning algorithm with feature selection might present an applicable approach to provide software tools for forecasting the functional outcomes of schizophrenia using molecular biomarkers.
Similar content being viewed by others

Introduction
Precision psychiatry is a newly-developed interdisciplinary study of precision medicine and psychiatry1,2, where state-of-the-art artificial intelligence and machine learning methods are integrated with molecular biomarkers such as genetic variants to provide personalized arrangements during all phases of medical intervention3,4,5,6. For instance, studies in precision psychiatry using machine learning algorithms involve the prediction of diagnosis of schizophrenia7,8 and the prediction of treatment response in patients with major depressive disorder9,10. On another note, functional outcomes in schizophrenia, which are normally determined by the tools such as Quality of Life Scale (QLS)11 and the Global Assessment of Functioning (GAF) Scale12, may affect the diagnosis and treatment of schizophrenia patients. As a result, it is vital to establish potential biomarkers that affect functional outcomes in schizophrenia13. Accordingly, we hypothesized that machine learning algorithms may be capable of forecasting probable biomarkers that influence functional outcomes in schizophrenia by using molecular biomarkers such as genetic variants.
Genetic variants such as single nucleotide polymorphisms (SNPs) have been a focus of attention in schizophrenia research. Various SNPs have been indicated as potential molecular biomarkers with respect to the developmental etiology of schizophrenia (Supplementary Table S1), including AKT1 rs1130233, COMT rs4680, DISC1 rs821616, DRD3 rs6280, G72 rs1421292, G72 rs2391191, 5-HT2A rs6311, MET rs2237717, MET rs41735, MET rs42336, and TPH2 rs4570625. For example, a previous association study by Emamian et al.14 indicated that there was a significant association of schizophrenia with the rs1130233 variant in the AKT1 gene. Another study by Chen et al.15 also reported that COMT rs4680 contributed to schizophrenia in Irish patents. In addition, a study by Callicott et al.16 showed that DISC1 rs821616 significantly influenced hippocampal structure and increased the risk for schizophrenia. Moreover, Talkowski et al.17 implicated that DRD3 rs6280 was markedly associated with schizophrenia in the U.S. samples. In order to differentiate schizophrenia patients from healthy individuals, Lin et al.8 employed machine learning algorithms (such as logistic regression, naive Bayes, and C4.5 decision tree) to construct classification models by using G72 rs1421292, G72 rs2391191, and G72 protein. Furthermore, a link between 5-HT2A rs6311 and a sensorimotor gating deficit in schizophrenia was observed in schizophrenia patients18. Additionally, Burdick et al.19 detected the association of MET rs2237717, MET rs41735, and MET rs42336 with schizophrenia risk and general cognitive ability in schizophrenia patients. The association of TPH2 rs4570625 with schizophrenia was not statistically significant in Korean schizophrenia patients20; however, it was related with social cognition21.
In a previous study, Lin et al.13 reported that clinical symptoms contribute to the link between cognitive behaviors and functional outcomes in schizophrenia by applying the structural equation modeling method. Additionally, it has been suggested that machine learning methods incorporating with feature selection techniques possess the advantages of improved prediction in precision psychiatry studies10,22,23. Here, we employed the same cohort of 302 schizophrenia patients and performed the first study on the QLS and GAF functional outcome prediction in schizophrenia with 11 aforementioned molecular biomarkers (namely 11 SNPs) by using a bagging ensemble machine learning method24. Moreover, in order to predict functional outcomes with improved performance, we utilized the M5 Prime feature selection algorithm25 to identify a small subset of suitable biomarkers from the 11 SNPs. We inferred that our bagging ensemble machine learning method would be capable of forecasting the QLS and GAF functional outcomes of schizophrenia by utilizing a small subset of chosen genetic variants. To the best of our knowledge, no preceding studies have been conducted to assess predictive algorithms for functional outcomes in schizophrenia with molecular biomarkers by utilizing the bagging ensemble machine learning method with the M5 Prime feature selection algorithm. We chose the bagging ensemble machine learning method due to its merits in lower variance and less overfitting; and thereby this method is widely leveraged to deal with complicated prediction and classification studies24,25. This study precisely scrutinized the performance of the bagging ensemble machine learning method to other broadly-used machine learning models, including support vector machine (SVM), multi-layer feedforward neural networks (MFNNs), linear regression, and random forests. The analysis showed that the bagging ensemble machine learning method with the M5 Prime feature selection algorithm led to improved performance.
Results
The functional outcomes of the study cohort
The participants encompassed 302 schizophrenia patients in the Taiwanese population. Study measures in regard to demographic characteristics and the QLS and GAF of schizophrenia were detailed before13.
Genetic variants
There were 11 genetic variants including AKT1 rs1130233, COMT rs4680, DISC1 rs821616, DRD3 rs6280, G72 rs1421292, G72 rs2391191, 5-HT2A rs6311, MET rs2237717, MET rs41735, MET rs42336, and TPH2 rs4570625. Their genotype frequencies are shown in Table 1. All of them, except G72 rs1421292, did not deviate from the Hardy–Weinberg equilibrium.
Feature selection using genetic variants
We completed a series of various biomarker combinations using the 11 genetic variants (Table 2; the Feature-A–Feature-C sets) to forecast the QLS and GAF of schizophrenia. Note that the Feature-A set encompasses the 11 genetic variants.
First, for forecasting the QLS, we utilized the M5 Prime feature selection algorithm (see Methods) to find two biomarkers (such as G72 rs2391191 and MET rs2237717) from the 11 genetic variants, where the Feature-B dataset comprises these two selected biomarkers (Supplementary Figure S1).
Second, for forecasting the GAF, we utilized the M5 Prime feature selection algorithm to identify one biomarker (such as AKT1 rs1130233) from the 11 genetic variants, where the Feature-C dataset comprises this selected biomarker (Supplementary Figure S2).
Prediction of the QLS and GAF of schizophrenia using genetic variants
We utilized genetic variants (namely the Feature-A–Feature-C datasets) to create the predictive algorithms for the QLS and GAF of schizophrenia, respectively. Table 2 shows the results of repeated tenfold cross-validation experiments for the predictive algorithms using genetic variants by the bagging ensemble algorithm with feature selection (Supplementary Figures S1 and S2), the bagging ensemble algorithm (Supplementary Figure S3), SVM (Supplementary Figure S4), MFNNs (Supplementary Figure S5), linear regression (Supplementary Figure S6), and random forests (Supplementary Figure S7). Furthermore, we utilized the RMSE values to assess the performance of the predictive algorithms.
As shown in Table 2, to forecast the QLS, the bagging ensemble algorithm with feature selection (Supplementary Figure S1) obtained the RMSE value of 8.6766 ± 1.0421 using the Feature-B dataset (namely G72 rs2391191 and MET rs2237717).
Moreover, to forecast the GAF, the bagging ensemble algorithm with feature selection (Supplementary Figure S2) obtained the RMSE value of 9.6982 ± 1.3354 using the Feature-C dataset (namely AKT1 rs1130233) (Table 2).
Benchmarking
We scrutinized the results (Table 2) for forecasting the QLS of schizophrenia among machine learning predictive models including the bagging ensemble algorithm with feature selection (Supplementary Figure S1), the bagging ensemble algorithm (Supplementary Figure S3), SVM (Supplementary Figure S4), MFNNs (Supplementary Figure S5), linear regression (Supplementary Figure S6), and random forests (Supplementary Figure S7) using two biomarker datasets (namely Feature-A and Feature-B). We found that the bagging ensemble algorithm with feature selection (using Feature-B; Supplementary Figure S1) performed best to forecast the QLS. The best RMSE value for forecasting the QLS was 8.6766 ± 1.0421 (Table 2).
In addition, we scrutinized the results (Table 2) for forecasting the GAF of schizophrenia among machine learning predictive models including the bagging ensemble algorithm with feature selection (Supplementary Figure S2), the bagging ensemble algorithm (Supplementary Figure S3), SVM (Supplementary Figure S4), MFNNs (Supplementary Figure S5), linear regression (Supplementary Figure S6), and random forests (Supplementary Figure S7) using two biomarker datasets (namely Feature-A and Feature-C). We found that the bagging ensemble algorithm with feature selection (using Feature-C; Supplementary Figure S2) performed best to forecast the GAF. The best RMSE value for forecasting the GAF was 9.6982 ± 1.3354 (Table 2).
Here, we observed that the bagging ensemble algorithm with feature selection using the chosen biomarkers from SNPs achieved best outcome forecasting in terms of both QLS and GAF when compared to other state-of-the-art models, including SVM, MFNNs, linear regression, and random forests. Our analysis suggested that the bagging ensemble algorithm with feature selection was well-adapted for predictive algorithms in the functional outcomes of schizophrenia.
Discussion
To our knowledge, this is the first study to date to explore a bagging ensemble machine learning method with the M5 Prime feature selection algorithm using molecular biomarkers for constructing predictive algorithms of functional outcomes in schizophrenia among Taiwanese patients. In addition, we conducted the first study to search probable biomarkers for functional outcomes of schizophrenia by using genetic biomarkers. The findings indicated that the bagging ensemble machine learning method with feature selection using two genetic biomarkers (G72 rs2391191 and MET rs2237717 SNPs) surpassed other state-of-the-art predictive models in terms of RMSE for forecasting the QLS outcome. Moreover, for forecasting the GAF outcome, we observed that the bagging ensemble machine learning method with feature selection using one genetic biomarker (AKT1 rs1130233) surpassed other state-of-the-art predictive algorithms in terms of RMSE.
By taking advantage of the genetic biomarkers, we created the predictive algorithms of functional outcomes in schizophrenia patients using the bagging ensemble machine learning method with the M5 Prime feature selection algorithm. This study is a proof of concept of a machine learning predictive framework for forecasting functional outcomes of schizophrenia. The results suggest that the bagging ensemble machine learning method may provide a clinically feasible tool for predicting functional outcomes of schizophrenia.
In addition, it is worthwhile to discuss the M5 Prime feature selection algorithm for discovering probable biomarkers in this study. We found that the bagging ensemble machine learning method with the selected biomarkers of the M5 Prime feature selection algorithm consistently surpassed the bagging ensemble machine learning method without using feature selection. For example, the bagging ensemble machine learning method with the Feature-B dataset excelled the bagging ensemble machine learning method with the Feature-A in forecasting the QLS outcome. Likewise, the bagging ensemble model with the Feature-C dataset surpassed the bagging ensemble machine learning method with the Feature-A dataset in forecasting the GAF outcome. In other words, the bagging ensemble machine learning method with feature selection inclined to obtain lower RMSE values (the better the performance). The findings suggest that the M5 Prime feature selection algorithm may have a better potential to single out biomarkers affecting functional outcomes of schizophrenia. In accordance, it has been reported that machine learning methods with feature selection outperformed the ones without feature selection in predicting the diagnosis and treatment outcome of psychiatric disorders10,22,23.
Remarkably, we further speculated the synergistic effects of chosen biomarkers (namely the Feature-B dataset), which were pinpointed by the M5 Prime feature selection algorithm when a biomarker dataset of 11 genetic variants was utilized to forecast the QLS outcome. As indicated in “Results” section the Feature-B dataset comprised 2 SNPs (namely G72 rs2391191 and MET rs2237717) for the QLS outcome. Subsequently, the bagging ensemble machine learning method with feature selection using the Feature-B dataset performed best in predicting the QLS outcome among the predictive algorithms. To our knowledge, scanty studies have been investigated to assess causal links between genetic variants. The biological mechanisms of these causal links in the functional outcomes of schizophrenia remain to be elucidated. It has been demonstrated that MET rs2237717 was linked to schizophrenia19 and G72 rs2391191 was also associated with schizophrenia8. Based on the previous findings8,19, it is hypothesized that synergistic interactions between genetic variants may provide a hallmark of molecular effects on the functional outcomes of schizophrenia.
In conclusion, we built a bagging ensemble machine learning method with feature selection for predicting functional outcomes of schizophrenia in Taiwanese patients by using genetic biomarkers. The analysis reveals that the bagging ensemble machine learning method with feature selection may present a plausible tool to construct predictive models for functional outcomes of schizophrenia in terms of favorable performance. Nonetheless, it is fundamental to further investigate the role of the bagging ensemble machine learning method by more replication studies. Ultimately, we would expect that the findings of the present study may be generalized in precision psychiatry to predict the diagnosis and treatment outcomes for various psychiatric disorders. Furthermore, the findings may be presumably leveraged to develop molecular diagnostic and prognostic tools in the near future.
Materials and methods
Study population
The study cohort composed of 302 schizophrenia patients, who were recruited from the China Medical University Hospital and affiliated Taichung Chin-Ho Hospital in Taiwan13. In this study, schizophrenia patients were aged 18–65 years and were healthy in the physical conditions. After presenting a complete description of this study to the subjects, we obtained written informed consents from a parent and/or legal guardian in line with the institutional review board guidelines. Details of the diagnosis of schizophrenia were published previously13. This study was approved by the institutional review board of the China Medical University Hospital in Taiwan and was performed in accordance with the Declaration of Helsinki.
Functional outcomes
We assessed functional outcomes by employing the QLS11 and the GAF Scale of the DSM-IV12. The QLS is a clinical tool for assessing the functional outcomes in patients with schizophrenia, including anhedonia, aimless inactivity, capacity for empathy, curiosity, emotional interaction, motivation, sense of purpose, social activity, social initiatives, and social withdrawal11. The GAF is a clinical tool for evaluating the global psychological, social, and occupational functioning in patients with schizophrenia12.
Laboratory assessments: genotyping
DNA was extracted from venous blood. In this study, the panel of genetic variants consisted of the aforementioned 11 SNPs. Their genotyping methods were detailed previously: AKT1 rs113023326, COMT rs468021, DISC1 rs82161627, DRD3 rs628028, G72 rs14212928, G72 rs23911918, 5-HT2A rs631129, MET rs223771726, MET rs4173526, MET rs4233626, and TPH2 rs457062521. These 11 genetic variants were used to create the predictive algorithms for the QLS and GAF of schizophrenia.
Statistical analysis
For genetic variants, we assessed the genotype frequencies for Hardy–Weinberg equilibrium by using a chi-squared goodness-of-fit test with 1 degree of freedom30. The criterion for failure to achieve Hardy–Weinberg equilibrium was set at P < 0.05. Data are presented as the mean ± standard deviation.
Bagging ensemble machine learning method
We applied a key ensemble machine learning method called bagging predictors24 and employed the Waikato Environment for Knowledge Analysis (WEKA) software (which is available from https://www.cs.waikato.ac.nz/ml/weka/)25 to conduct the bagging ensemble machine learning method. All the experiments were carried out on a computer with Intel (R) Core (TM) i5-4210U, 4 GB RAM, and Windows 77.
In principle, the bagging ensemble machine learning method (Supplementary Figure S3) takes advantage of averaging the predictive performance of multiple versions of a base model to obtain a combined model with better performance24. The multiple versions of the base model are generated by bootstrap reproductions, where the bootstrap technique is one of the most suitable data resampling approaches employed in statistical analysis. In other words, the bootstrap technique produces the multiple versions of the base model, that is, the Model-version #1 to the Model-version #n (Supplementary Figure S3). Subsequently, the combined model summarizes the predictive performance of these base models from 1 to n. The technique of bagging models inclines to lower variance and prevent overfitting. The base model we used was linear regression. Here, we utilized the default tuning parameters of WEKA, such as 100 for the batch size, 100 for the percentage of the bag size, and 10 for the number of iterations7,10.
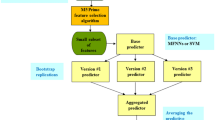
Figure 1 demonstrates the illustrative diagram of the bagging ensemble machine learning method with feature selection. For the feature selection task, we utilized the M5 Prime algorithm (as described below).

The schematic illustration of the bagging ensemble machine learning method with feature selection. First, the M5 Prime feature selection algorithm is conducted to find a small subset of biomarkers, which serves as the input to the bagging ensemble machine learning method. The concept of the bagging ensemble machine learning method is to create the multiple versions of a base model by bootstrap reproductions. Then, the ultimate prediction is generated by averaging the predictive performance of the multiple versions. The base model was chosen as linear regression in this study.
M5 Prime feature selection algorithm
In the present study, we used an Akaike information criterion (AIC)-based method called the M5 Prime algorithm25,31 for the feature selection function. The M5 Prime algorithm builds a decision tree with multivariate linear models at the terminal nodes and iteratively eliminates the biomarker with the smallest normalized coefficient until no further improvement in the evaluated error specified by the AIC32,33. We chose the M5 Prime algorithm due to its merits in dealing with the large number of biomarkers, performing fast during training, and being a straightforward approach25,31. In addition, the relevant features of the M5 Prime algorithm include robustness in handling missing values and enumerated attributes25,31.
To forecast the QLS and GAF, we utilized the M5 Prime algorithm to choose biomarkers from a biomarker dataset, which includes 11 genetic variants (Fig. 1). By using 11 genetic variants, the M5 Prime algorithm generated the first feature dataset including two genetic variants (Supplementary Figure S1). In addition, by using 11 genetic variants, the M5 Prime algorithm generated the second feature dataset including one genetic variant (Supplementary Figure S2).
Machine learning algorithms for benchmarking
For the benchmarking task in the present study, we employed four state-of-the-art machine learning models including SVM, MFNNs, linear regression, and random forests (Supplementary Figures S4–S7). We performed the analyses for these four machine learning models using the WEKA software25 and a computer with Intel (R) Core (TM) i5-4210U, 4 GB RAM, and Windows 77.
First, the SVM model34 (Supplementary Figure S4) is a popular approach for pattern recognition and classification7,35,36,37. Given a training set, the SVM model applies a kernel function to find a linear relationship between input variables and the predicted output34,38. The SVM model then determines the best predicted output by minimizing both the coefficients of the cost function and the predictive errors34,38. In this study, we utilized the WEKA’s tuning parameter for the polynomial kernel with the exponent value of 1.07,10.
Second, an MFNN model (Supplementary Figure S5) comprises one input layer, one or multiple hidden layers, and one output layer, where links among neuron nodes actually have no directed cycles7,39. In the learning stage of the MFNN model, the back-propagation algorithm40 is achieved for the learning task. In the retrieval stage, the MFNN model reiterates by way of all the neuron nodes to accomplish the retrieval task at the output layer based on the inputs of test data7,41. In this study, we utilized the architecture incorporating one hidden layer. For instance, we utilized the following WEKA’s tuning parameters for training the MFNN model with one hidden layer: the momentum = 0.01, the learning rate = 0.01, and the batch size = 1007,42.
Next, the linear regression model (Supplementary Figure S6), the conventional approach for prediction issues in clinical studies, was utilized as a basis for the benchmarking task7,25.
Finally, the random forests model (Supplementary Figure S7) is an ensemble learning approach which consists of a group of decision trees throughout training and produces a better prediction by aggregating the predictive results among the individual decision trees7,35,36,37,43. Here, we utilized the default tuning parameters of WEKA for the random forests model; for instance, 100 for the batch size and 100 for the number of iterations7.
Evaluation of the predictive performance
In this study, we employed one of the most popular standards, the root mean square error (RMSE), to examine the performance of predictive algorithms22,38,44. The RMSE estimates the difference between the measured values and the predicted values by a predictive algorithm. The better the prediction algorithm, the lower the RMSE22,44. In addition, we applied the repeated tenfold cross-validation method to assess the generalization of predictive models45. Firstly, the whole dataset was randomly fragmented into ten individual partitions. Secondly, the predictive model was trained using nine-tenths of the partitions and was tested using the remaining tenth of the partitions to estimate the predictive performance. Next, the previous step was repeated nine more times by choosing different nine-tenths of the partitions for training and a different tenth of the partitions for testing. Lastly, the final estimation was evaluated by averaging the aforementioned ten runs. In the present study, we reported the performance of all predictive models using the repeated tenfold cross-validation method.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Bzdok, D. & Meyer-Lindenberg, A. Machine learning for precision psychiatry: opportunities and challenges. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 3, 223–230 (2018).
Lin, E., Lin, C.-H. & Lane, H.-Y. Precision psychiatry applications with pharmacogenomics: Artificial intelligence and machine learning approaches. Int. J. Mol. Sci. 21, 969 (2020).
Lane, H. Y., Tsai, G. E. & Lin, E. Assessing gene-gene interactions in pharmacogenomics. Mol. Diagn. Ther. 16, 15–27. https://doi.org/10.2165/11597270-000000000-00000 (2012).
Lin, E. & Chen, P. S. Pharmacogenomics with antidepressants in the STAR*D study. Pharmacogenomics 9, 935–946. https://doi.org/10.2217/14622416.9.7.935 (2008).
Lin, E. & Lane, H. Y. Genome-wide association studies in pharmacogenomics of antidepressants. Pharmacogenomics 16, 555–566. https://doi.org/10.2217/pgs.15.5 (2015).
Lin, E. & Lane, H.-Y. Machine learning and systems genomics approaches for multi-omics data. Biomark. Res. 5, 2 (2017).
Lin, E., Lin, C.-H., Hung, C.-C. & Lane, H.-Y. An ensemble approach to predict schizophrenia using protein data in the N-methyl-D-aspartate receptor (NMDAR) and tryptophan catabolic pathways. Front. Bioeng. Biotechnol. 8, 569 (2020).
Lin, E. et al. Combination of G72 genetic variation and G72 protein level to detect schizophrenia: machine learning approaches. Front. Psychiatry 9, 566 (2018).
Lin, E. et al. A deep learning approach for predicting antidepressant response in major depression using clinical and genetic biomarkers. Front. Psychiatry 9, 290 (2018).
Lin, E. et al. Prediction of antidepressant treatment response and remission using an ensemble machine learning framework. Pharmaceuticals 13, 305 (2020).
Heinrichs, D. W., Hanlon, T. E. & Carpenter, W. T. Jr. The quality of life scale: an instrument for rating the schizophrenic deficit syndrome. Schizophr. Bull. 10, 388–398 (1984).
Startup, M., Jackson, M. C. & Bendix, S. The concurrent validity of the Global Assessment of Functioning (GAF). Br. J. Clin. Psychol. 41, 417–422 (2002).
Lin, C.-H. et al. Clinical symptoms, mainly negative symptoms, mediate the influence of neurocognition and social cognition on functional outcome of schizophrenia. Schizophr. Res. 146, 231–237 (2013).
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M. & Gogos, J. A. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 36, 131–137 (2004).
Chen, X., Wang, X., O’neill, A., Walsh, D. & Kendler, K. Variants in the catechol-o-methyltransferase (COMT) gene are associated with schizophrenia in Irish high-density families. Mol. Psychiatry 9, 962–967 (2004).
Callicott, J. H. et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. 102, 8627–8632 (2005).
Talkowski, M. E. et al. Novel, replicated associations between dopamine D3 receptor gene polymorphisms and schizophrenia in two independent samples. Biol. Psychiatiatry 60, 570–577 (2006).
Quednow, B. B. et al. Sensorimotor gating of schizophrenia patients is influenced by 5-HT2A receptor polymorphisms. Biol. Psychiatiatry 64, 434–437 (2008).
Burdick, K. E., DeRosse, P., Kane, J. M., Lencz, T. & Malhotra, A. K. Association of genetic variation in the MET proto-oncogene with schizophrenia and general cognitive ability. Am. J. Psychiatry 167, 436–443 (2010).
Serretti, A. et al. Influence of TPH2 variants on diagnosis and response to treatment in patients with major depression, bipolar disorder and schizophrenia. Psychiatry Res. 189, 26–32 (2011).
Lin, C.-H. et al. Synergistic effects of COMT and TPH2 on social cognition. Psychiatry Interpers. Biol. Process. 76, 273–294 (2013).
Huang, L. C., Hsu, S. Y. & Lin, E. A comparison of classification methods for predicting Chronic Fatigue Syndrome based on genetic data. J. Transl. Med. 7, 81. https://doi.org/10.1186/1479-5876-7-81 (2009).
Shahamat, H. & Pouyan, A. A. Feature selection using genetic algorithm for classification of schizophrenia using fMRI data. J. AI Data Min. 3, 30–37 (2015).
Breiman, L. Bagging predictors. Mach. Learn. 24, 123–140 (1996).
Witten, I. H. & Frank, E. Data Mining: Practical Machine Learning Tools and Techniques (Morgan Kaufmann Publishers, 2005).
Lin, M.-T. et al. MET and AKT genetic influence on facial emotion perception. PLoS ONE 7, e36143 (2012).
Chang, J.P.-C., Huang, K.-H., Lin, C.-H. & Lane, H.-Y. Genetic effects of DISC1 and G72 (DAOA) on visual learning of patients with schizophrenia. Neuropsychiatr. Dis. Treat. 16, 771 (2020).
Lane, H.-Y. et al. Dopamine D3 receptor Ser9Gly polymorphism and risperidone response. J. Clin. Psychopharmacol. 25, 6–11 (2005).
Tsuang, H.-C. et al. Impaired impulse control is associated with a 5-HT2A receptor polymorphism in schizophrenia. Psychiatry Res. 208, 105–110 (2013).
Lin, E., Kuo, P.-H., Liu, Y.-L., Yang, A. & Tsai, S.-J. Association and interaction effects of interleukin-12 related genes and physical activity on cognitive aging in old adults in the Taiwanese population. Front. Neurol. 10, 1065 (2019).
Wang, Y. & Witten, I. H. Induction of model trees for predicting continuous classes. in Proceedings of the European Conference on Machine Learning, Prague 128–137 (1996).
Bozdogan, H. Model selection and Akaike’s information criterion (AIC): the general theory and its analytical extensions. Psychometrika 52, 345–370 (1987).
Quinlan, J. R. Learning with continuous classes. in 5th Australian Joint Conference on Artificial Intelligence 343–348 (World Scientific, 1992)..
Vapnik, V. The Nature of Statistical Learning Theory (Springer, 2013).
Hasan, M. M. et al. Meta-i6mA: an interspecies predictor for identifying DNA N6-methyladenine sites of plant genomes by exploiting informative features in an integrative machine-learning framework. Brief. Bioinf. Online ahead of print (2020).
Hasan, M. M., Khatun, M. S. & Kurata, H. iLBE for Computational identification of linear B-cell epitopes by integrating sequence and evolutionary features. Genomics Proteomics Bioinform. S1672-0229, 30274-2 (2020).
Hasan, M. M. et al. HLPpred-Fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 36, 3350–3356 (2020).
Lin, E. & Hwang, Y. A support vector machine approach to assess drug efficacy of interferon-alpha and ribavirin combination therapy. Mol. Diagn. Ther. 12, 219–223 (2008).
Bishop, C. M. Neural Networks for Pattern Recognition (Clarendon Press, 1995).
Rumelhart, D. E. H., Hinton G.E., William, R.J. Learning internal representation by error propagation. in The Micro-structure of Cognition Vol. 1 (MIT Press, 1996).
Kung, S. Y. & Hwang, J. N. Neural networks for intelligent multimedia processing. Proc. IEEE 86, 1244–1272 (1998).
Lin, E., Mukherjee, S. & Kannan, S. A deep adversarial variational autoencoder model for dimensionality reduction in single-cell RNA sequencing analysis. BMC Bioinform. 21, 1–11 (2020).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Linden, A. Measuring diagnostic and predictive accuracy in disease management: an introduction to receiver operating characteristic (ROC) analysis. J. Eval. Clin. Pract. 12, 132–139. https://doi.org/10.1111/j.1365-2753.2005.00598.x (2006).
Lin, E., Lin, C.-H. & Lane, H.-Y. Relevant applications of generative adversarial networks in drug design and discovery: molecular de novo design, dimensionality reduction, and de novo peptide and protein design. Molecules 25, 3250 (2020).
Acknowledgements
This work was supported by National Health Research Institutes, Taiwan (NHRI-EX109-10731NI), Ministry of Science and Technology in Taiwan (MOST 109-2622-B-039-001-CC2; 109-2314-B-039-001; 109-2314-B-039-039-MY3), China Medical University Hospital (DMR-108-097).
Author information
Authors and Affiliations
Contributions
E.L., C.H.L. and H.Y.L. designed the study. C.H.L. and H.Y.L. conducted the study. E.L. analyzed the data. E.L. drafted the manuscript. E.L., C.H.L. and H.Y.L. revised the manuscript. All authors provided the final approval of the version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, E., Lin, CH. & Lane, HY. Prediction of functional outcomes of schizophrenia with genetic biomarkers using a bagging ensemble machine learning method with feature selection. Sci Rep 11, 10179 (2021). https://doi.org/10.1038/s41598-021-89540-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89540-6
