
Abstract
Alport syndrome is the commonest inherited kidney disease and nearly half the pathogenic variants in the COL4A3–COL4A5 genes that cause Alport syndrome result in Gly substitutions. This study examined the molecular characteristics of Gly substitutions that determine the severity of clinical features. Pathogenic COL4A5 variants affecting Gly in the Leiden Open Variation Database in males with X-linked Alport syndrome were correlated with age at kidney failure (n = 157) and hearing loss diagnosis (n = 80). Heterozygous pathogenic COL4A3 and COL4A4 variants affecting Gly (n = 304) in autosomal dominant Alport syndrome were correlated with the risk of haematuria in the UK 100,000 Genomes Project. Gly substitutions were stratified by exon location (1 to 20 or 21 to carboxyl terminus), being adjacent to a non-collagenous region (interruption or terminus), and the degree of instability caused by the replacement residue. Pathogenic COL4A5 variants that resulted in a Gly substitution with a highly destabilising residue reduced the median age at kidney failure by 7 years (p = 0.002), and age at hearing loss diagnosis by 21 years (p = 0.004). Substitutions adjacent to a non-collagenous region delayed kidney failure by 19 years (p = 0.014). Heterozygous pathogenic COL4A3 and COL4A4 variants that resulted in a Gly substitution with a highly destabilising residue (Arg, Val, Glu, Asp, Trp) were associated with an increased risk of haematuria (p = 0.018), and those adjacent to a non-collagenous region were associated with a reduced risk (p = 0.046). Exon location had no effect. In addition, COL4A5 variants adjacent to non-collagenous regions were over-represented in the normal population in gnomAD (p < 0.001). The nature of the substitution and of nearby residues determine the risk of haematuria, early onset kidney failure and hearing loss for Gly substitutions in X-linked and autosomal dominant Alport syndrome.
Similar content being viewed by others


Introduction
Alport syndrome (AS) is an inherited basement membrane disease characterised by progressive kidney failure, sensorineural hearing loss and ocular abnormalities1. Estimates of its disease frequency range from a prevalence of one in 5000 people in Utah2 to one in 53,000 live births in Finland3, but the number of people with a predicted genetic risk of disease is even higher4.
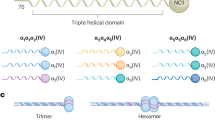
AS results from pathogenic variants in COL4A55, COL4A3 or COL4A46. These genes encode the collagen IV α5, α3 and α4 chains respectively, that trimerise to form a triple helical structure and chickenwire network typical of basement membranes7,8. X-Linked AS is the commonest form that causes kidney failure9. Males are more severely affected than females and 70% have kidney failure by the age of 3010. Females are affected twice as often as males but normally have a milder and more variable phenotype11 due in part to non-random X- chromosome inactivation12.
Autosomal recessive AS is less common, and results from two pathogenic variants affecting the COL4A3 or COL4A4 genes6. These may be homozygous6, or compound heterozygous variants in trans13. Digenic variants result most often from one pathogenic variant in COL4A3 and one in COL4A414. Pathogenic heterozygous variants in COL4A3 or COL4A4 result in autosomal dominant AS (also known as ‘thin basement membrane nephropathy’), with haematuria15, and late-onset kidney failure in up to 25% of affected individuals in hospital-based series16,17,18.
Each collagen IV α chain comprises an intermediate collagenous domain of Gly Xaa Yaa triplet repeats19,20,21. However, the collagen IV α chains differ from most other collagen types in that the collagenous domain includes 21–26 short non-collagenous interruptions21. These provide flexibility to an otherwise rigid molecule, and may include important ligand binding sites22. Collagen IV chains also differ in that the mature chains retain their non-collagenous amino and carboxyl termini, which interact with the termini of neighbouring trimers to create the network8. The carboxyl terminus is also the site of chain recognition where trimerisation begins, proceeding in a zipper-like manner towards the amino terminus23.
Previous genotype–phenotype correlations in males with pathogenic COL4A5 variants have demonstrated that truncating variants and large deletions lead to the most severe phenotype, with the youngest age at kidney failure10,24. The corresponding mRNA is degraded by the podocytes, the and there is no collagen IV α5 chain incorporated into the trimer or staining in a kidney biopsy25. Missense variants usually result in milder disease10,24 and the abnormal chain may be incorporated into mature trimers that are secreted into the basement membrane25 in reduced amounts26. The abnormal chains are also often retained within the podocytes, activating the unfolded protein response and increasing endoplasmic reticulum stress27,28. Similar studies in females with heterozygous COL4A5 variants have not found such a clear genotype–phenotype correlation with age at kidney failure11,12, but a recent study suggested that females with missense variants were less likely to develop proteinuria and had better kidney function than those with other variant types29.
In autosomal recessive AS, individuals with at least one truncating variant in COL4A3 or COL4A4 are more likely to progress to kidney failure before the age of 30 years than those with non-truncating variants30. Disease progression also correlates with the number of missense variants, where individuals with at least one missense variant have a delayed onset of kidney failure and hearing loss compared with those with none31. In individuals with autosomal dominant AS, heterozygous truncating variants in COL4A3 or COL4A4 are associated with an earlier age at kidney failure than those with missense variants16.
Missense variants affecting Gly residues in the collagenous Gly Xaa Yaa repeats are the commonest pathogenic type32. These residues are critical to the structure since Gly is the only amino acid small enough to fit within the core of triple helix and allow close packing of the chains32,33. Substitution with any other amino acid may destabilise the trimer, interfere with triple helix propagation, and cause disease34.
The clinical phenotype associated with pathogenic Gly missense variants is highly variable. Gly substitution with a bulky or charged amino acid usually leads to more severe clinical features35, but contrary examples also exist36. In other collagen types, Gly substitutions in the amino exons result in a milder phenotype37, but evidence for this in collagen IV is conflicting24,38. Location adjacent to a non-collagenous interruption has also been associated with a milder phenotype39, but again this is variable35.
The aim of this study was to better understand the molecular features of Gly substitutions in the COL4A3–COL4A5 genes that affect disease severity.
Methods
Variant databases
Three variant databases were examined. The Leiden Open Variation Database (LOVD) is an open source database of genomic variants with associated phenotypes (https://www.lovd.nl)40. It includes variants published in the literature in addition to those submitted directly by laboratories, and has recently been updated to include a total of 3869 (including 2988 pathogenic) COL4A5 variants. Pathogenicity was assessed by the submitting laboratory, or where none was provided, using the VarSome scores (https://varsome.com) based on the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG/AMP) criteria. Varsome scores automatically include previous ClinVar or other published assessments (PP5). Many variants also included clinical data such as gender, age at kidney failure, hearing loss and ocular abnormalities. This database was used to determine whether molecular features of pathogenic COL4A5 variants that resulted in Gly substitutions affected age at kidney failure or hearing loss diagnosis using survival analysis.
The Genomics England 100,000 Genomes Project (100kGP) is a database comprising genomic and clinical data from individuals and families with various diseases, including familial haematuria and other inherited kidney disease (https://www.genomicsengland.co.uk; version 10 data release)41. This was used to determine whether molecular features of heterozygous pathogenic COL4A3 and COL4A4 variants that resulted in Gly substitutions were associated with haematuria.
The Genome Aggregation Database (gnomAD) comprises exomes and genomes from individuals recruited as part of various disease-specific and population genetic studies (gnomAD version 2.1.1; https://gnomad.broadinstitute.org; accessed 11 September 2021)42. These primarily include participants and controls from studies of cardiovascular disease, diabetes or psychiatric disorders, who have not been selected for kidney disease but rather to represent a cross-section of the population. This database was used to determine whether milder molecular characteristics of COL4A5 Gly substitutions were increased the general population.
The individuals whose variants and other deidentified information were included in these databases had provided informed consent at the time of recruitment under the supervision of the corresponding institutional review boards.
Reference sequences
COL4A5 variants were described using the collagen IV α5 chain isoform 2 reference sequence comprising 53 exons (NM_033380.3). COL4A3 and COL4A4 variants were described using the reference sequences for the collagen IV α3 (NM_000091.5) and α4 (NM_000092.5) chains respectively.
Predicted splicing changes
Previous studies have demonstrated that exonic nucleotide substitutions affecting Gly codons in COL4A5 sometimes result in abnormal splicing43,44, and recent evidence suggests that this is common when the affected base is the final nucleotide of an exon45. To ensure that unknown splicing variants were not unintentionally included in this study, all variants occurring within 3 bases of a splice site were analysed using MaxEntScan to determine whether they were likely to affect normal splicing46. MaxEntScan was chosen since it is freely available and was able to correctly identify 6 known exonic splice changes previously reported for COL4A5 (data not shown). Both mutant and wild type sequences were scored using the maximum entropy model. Variants were considered to affect splicing where the mutant score was more than 15% lower than the wild type score47. All variants predicted to affect normal splicing were excluded to ensure that any phenotypic effect attributed to a variant was due solely to a missense change, rather than an unreported splicing change (Supplemental Table 1).
Molecular characteristics
Three molecular features were examined for each Gly substitution. These were the molecular location of the variant (exons 1 to 20, or exons 21 to carboxyl terminus), whether the variant was adjacent to a non-collagenous interruption or terminus (also called ‘non-collagenous boundary’ variants) (Supplemental Table 2), and the degree of instability caused by the residue replacing Gly (Ala, Ser, Cys were considered mildly destabilising; Arg, Val, Glu, Asp, Trp were considered highly destabilising)34.
In addition a subgroup of variants was examined separately to determine the effect of a variant’s relative location with respect to its local collagenous region (a single uninterrupted stretch of Gly XY repeats flanked by two non-collagenous interruptions/termini). This subgroup excluded all non-collagenous boundary variants to ensure that the significantly different phenotypes usually observed for these variants did not obscure any small effect size. The remaining variants were considered in three groups: variants affecting the 2 Gly residues at the amino end of a local collagenous region (not counting the boundary Gly), variants affecting the 2 Gly residues at the carboxyl end of a local collagenous regions, and all other variants falling between these two ends (Fig. 1).

Terminology used in this study to describe the position of Gly residues with respect to their local collagenous region. One single ‘local’ collagenous region is shown, bounded by non-collagenous interruptions at either end. The boxes could also represent either of the two non-collagenous termini. The non-collagenous boundary Gly residues are a special case, so were not included in this subgroup. The larger arrow indicates the direction of trimerisation, which is initiated at the carboxyl terminus and then proceeds in the amino direction. NC, non-collagenous.
COL4A5
Kidney failure
COL4A5 variants reported in LOVD were filtered to include those with entries including any of the following keywords: ‘renal’, ‘failure’, ‘ESRD’, ‘ESRF’, ‘ESKD’, ‘ESKF’, ‘transplant’, ‘dialysis’ (Supplemental Fig. 1a). Only variants affecting a Gly residue in a collagenous sequence (GlyXaaYaa) and reported as ‘Pathogenic’ or ‘Likely Pathogenic’ were included. Predicted splicing variants were excluded as described. Each entry was then examined manually to determine the age and kidney failure status of male participants. Age at kidney failure was defined as the age at diagnosis of kidney failure, or where this was not reported, the age at commencement of dialysis, or at first kidney transplant. Unclear or ambiguous entries in LOVD were resolved by referring to the original manuscripts.
Each family was included once only. Where multiple affected males were reported in the same family, the mean age at kidney failure was used (or median age at kidney failure, if this was the only value available). Where only a range of ages for kidney failure was reported, the midpoint of this range was used. Where a male had not yet progressed to kidney failure, the age of the male at the most recent report was used as a censored data point. Families with only affected females, or participants who had multiple variants in the COL4A3-COL4A5 genes were excluded.
Hearing loss
COL4A5 variants reported in LOVD were filtered to include those which had entries with the following keywords: ‘hearing’, ‘hypoacusia’, ‘deaf’, ‘sensorineural’, or ‘audio’ (Supplemental Fig. 1b). Onset of hearing loss is often poorly recognised and probably occurs much earlier than reported, so here the age at diagnosis of hearing loss was used as a measure of hearing loss severity. Ages were extracted as for kidney failure, using similar inclusion and exclusion criteria.
COL4A3/COL4A4
Haematuria
Individuals with and without haematuria were identified from the 100kGP database. The haematuria cohort included unrelated individuals with any haematuria-related terms (HP:0000790, hematuria; HP:0002907, microscopic hematuria; HP:0012587, macroscopic hematuria), excluding those with a diagnosis of kidney or bladder cancer, or other less common causes (n = 2221). The ancestry-matched control cohort included all individuals who did not have any documented haematuria in their medical records (n = 37,200). Individuals were then filtered to include only those reported to have a heterozygous Gly missense variant in COL4A3 or COL4A4. Variants near splice sites were assessed for predicted splice changes as described.
All variants included were assessed for the same molecular characteristics as described above, and a logistic regression model used to identify which features were associated with a difference in risk for haematuria. All variants not adjacent to a non-collagenous region were also examined separately to determine any associations with local collagenous location.
Prevalence of COL4A5 non-collagenous boundary variants in normals
To determine whether non-collagenous boundary variants were increased in the general population, the collagenous location and prevalence of all Gly substitutions in gnomAD were investigated. Predicted splicing variants were excluded.
The expected proportion of variants affecting non-collagenous boundary Gly variants was calculated using a neighbour-dependent substitution rate model48. This model was chosen since it takes into account the higher rates of substitution seen for transitions than transversions, as well as the effect of the neighbouring nucleotides. This model has also been used previously in the context of Gly substitutions in collagen molecules34. The expected proportion of variants affecting non-collagenous boundary Gly residues was compared with the proportion observed in gnomAD. The population prevalence of individuals with these variants was then calculated based on the allele frequencies reported in gnomAD, correcting for numbers reported in homozygous females.
Statistical analysis
All statistical analyses were performed using R (version 3.6.2). Survival analysis was performed using the survival package49,50. Separate survival curves were produced for each molecular feature using the Kaplan–Meier method, and compared using the log-rank test. The individual contribution of each covariate was then analysed using a Cox proportional hazards model. The overall significance of this model was assessed using the likelihood ratio test.
Logistic regression models were used to examine the associations between the molecular features and haematuria. The proportion of variance in haematuria explained by the model was assessed using McFadden’s pseudo-R2 and calculated using the blorr package51.
Expected and observed proportions of variants in gnomAD were compared with the exact binomial test. For all analyses, a p-value less than 0.05 was considered significant. Figures were produced using the survminer52 and forestplot53 packages.
Results
COL4A5
Kidney failure
One hundred and fifty-seven families were studied, including 129 with at least one male with kidney failure (Table 1a, Fig. 2a–c). Overall, the median age at kidney failure was 26 years (95% CI 25–28). Age at kidney failure did not differ for variants located within the first 20 exons compared with those in exons 21 to 53 (p = 0.41). Substitution of a non-collagenous boundary Gly residue delayed median time to kidney failure by 19 years compared with those not adjacent to a non-collagenous region (p = 0.014), while substitution with a highly destabilising residue shortened median time to kidney failure by 7 years compared with mildly destabilising residues (p = 0.002). Substitution of a non-collagenous boundary Gly residue was independently associated with a decreased risk of kidney failure (p = 0.025), while substitution with a highly destabilising residue was independently associated with an increased risk (p = 0.003) (Fig. 3a).

Proportion of cases without kidney failure for COL4A5 Gly missense variants reported in LOVD. Variants were stratified by (a) molecular location (p = 0.41), (b) collagenous location (p = 0.014) and (c) substituting residue (p = 0.002). (d) Excluding variants affecting NC boundary residues, variants were further stratified by relative location within their local collagenous region (p = 0.14). Censored data points are not shown. NC, non-collagenous.

Cox proportional hazards model of kidney failure risk for COL4A5 Gly missense variants reported in LOVD. Analysis was performed for (a) all Gly missense variants (overall significance of model, p < 0.001) and (b) excluding NC boundary Gly variants (overall significance of model, p = 0.014). HR (95% CI), Hazard ratio (95% confidence interval); NC, non-collagenous; ref, Reference group.
Considering only the subgroup excluding the non-collagenous boundary variants, location within a local collagenous region did not affect the median time to kidney failure (p = 0.14) (Table 1b, Fig. 2d). However, substitution of a Gly residue at the carboxyl end of a local collagenous region was independently associated with an increased risk of kidney failure compared with substitutions in the central region (p = 0.031) (Fig. 3b). Substitution with a highly destabilising residue remained significantly associated with an increased risk of kidney failure for this subgroup (p = 0.004).
Hearing loss
Eighty families were studied, including 42 with at least one report of hearing loss in a male (Table 2a, Fig. 4a–c). Median age at hearing loss diagnosis did not differ for variants located within the first 20 exons compared with those located in exons 21 to 53 (p = 0.85). Unlike with kidney failure, median age at hearing loss diagnosis did not differ between variants affecting non-collagenous boundary Gly residues and variants not adjacent to a non-collagenous region (p = 0.38). However, the sample size for the non-collagenous boundary variants was small (n = 8), and only 2 of these eight families had a report of hearing loss at the time of the study. Substitution with a highly destabilising residue shortened median time to diagnosis of hearing loss by 21 years compared with mildly destabilising residues (p = 0.004), and this was the only molecular feature independently associated with an increased risk of hearing loss (p < 0.001) (Fig. 5a).

Proportion of cases without a hearing loss diagnosis for COL4A5 Gly missense variants reported in LOVD. Variants were stratified by (a) molecular location (p = 0.85), (b) collagenous location (p = 0.38) and (c) substituting residue (p = 0.004). (d) Excluding variants affecting NC boundary residues, variants were further stratified by relative location within their local collagenous region (p = 0.77). Censored data points are not shown. NC, non-collagenous.

Cox proportional hazards model of hearing loss diagnosis risk for COL4A5 Gly missense variants reported in LOVD. Analysis was performed for (a) all Gly missense variants (overall significance of model, p = 0.002) and (b) excluding NC boundary Gly variants (overall significance of model, p = 0.004). HR (95% CI), Hazard ratio (95% confidence interval); NC, non-collagenous; ref, reference group.
Excluding all non-collagenous boundary variants, location within a local collagenous region did not affect the median time to hearing loss diagnosis (p = 0.77) (Table 2b, Fig. 4d). Risk of hearing loss also did not differ for substitutions at the amino (p = 0.18) or carboxyl ends (p = 0.96) (Fig. 5b). Substitution with a highly destabilising residue remained significantly associated with an increased risk of hearing loss for this subgroup (p < 0.001).
COL4A3/COL4A4
Haematuria
This cohort comprised 304 individuals from the 100kGP, including 48 with documented haematuria (Table 3). In total, 153 unique heterozygous COL4A3 and COL4A4 Gly missense variants were studied, and most (n = 105) were found once only.
Location in exons 1–20 was not associated with a difference in risk for haematuria compared with exons 21–53 (p = 0.51). Substitution of a non-collagenous boundary Gly residue was associated with a lower risk for haematuria (p = 0.046), while substitution with a highly destabilising residue was associated with a higher risk (p = 0.018) (Table 4a). However, these features only explained a small proportion of the total variance in haematuria risk (pseudo-R2McFadden = 0.040). Excluding all variants affecting a non-collagenous boundary residue, those affecting the amino or carboxyl ends of a local collagenous region were not associated with a difference in risk for haematuria compared with centrally located variants (p = 0.23, p = 0.20 respectively) (Table 4b). Substitution with a highly destabilising residue did not remain significantly associated with a higher risk for haematuria in this subgroup (p = 0.09).
Two COL4A3 variants were reported significantly more often in this cohort than any other variant in either gene (Supplemental Fig. 2). These were Gly695Arg (n = 21) and Gly1277Ser (n = 30). Together these accounted for 51/304 (16.8%) of all variants. To ensure that these two variants did not overly influence the results obtained, the logistic regression model was re-evaluated excluding both variants (Supplemental Table 3). Substitution of a non-collagenous boundary residue remained significantly associated with a lower risk for haematuria (p = 0.031), but substitution with a highly destabilising residue now fell just outside the nominal significance level (p = 0.064). The total variance in haematuria explained by the predictor variables remained low (pseudo-R2McFadden = 0.043). Interestingly, in the subgroup excluding the non-collagenous boundary variants, location at the carboxyl end of a local collagenous region was now associated with a higher risk for haematuria (p = 0.041).
Prevalence of COL4A5 non-collagenous boundary variants in normals
Forty-five unique COL4A5 Gly missense variants were reported in gnomAD. The proportion of these variants affecting a non-collagenous boundary residue (15/45 = 33.3%) was higher than the expected frequency of 10.1% (p < 0.001). Of interest, the 5 most commonly reported Gly missense variants all affected a non-collagenous boundary residue (Table 5). These 5 variants predominated in single ancestral groups.
Assuming equal numbers of males and females in gnomAD, missense variants affecting non-collagenous boundary residues were present in 0.6% of the general population (1 in 179 individuals). The majority of these cases were due to a single variant, Gly953Val, which is highly prevalent in East Asian and South Asian populations and currently considered benign54. Excluding this variant, missense variants affecting non-collagenous boundary Gly residues were present in 0.05% of the population (1 in 2078 individuals). In contrast, missense variants affecting all other collagenous Gly residues were only present in 0.03% of the general population (1 in 3000 individuals) despite there being nine times as many of these Gly residues in the α5 chain.
Discussion
Previous studies have demonstrated that the clinical severity of X-linked AS in males is closely associated with truncating, large deletion, splice site, and missense variant types in COL4A510,24. This study found that missense variants affecting collagenous Gly residues can be further classified by molecular features that also correlate with severity.
Gly substitutions with highly destabilising residues were associated with an earlier median age at kidney failure. This is consistent with previous studies in other collagen chain genes such as COL1A1 and COL1A2, where Gly substitutions with Arg, Val, Glu or Asp were more likely to result in a lethal phenotype of osteogenesis imperfecta37. Similar observations have been seen for COL4A5 where substitutions with bulkier amino acids lead to an earlier age at kidney failure35. Our results are also consistent with the underrepresentation of substitutions with Ala and Ser in pathogenic databases34,55, which suggests that they are associated with milder and possibly undiagnosed disease.
Substitutions affecting non-collagenous boundary Gly residues resulted in a delayed age at kidney failure. In general, the non-collagenous interruptions within the collagen type IV chains contribute flexibility and non-pathogenic variants are common in these regions56. Gly residues at the boundary of these interruptions may inherit some of this flexibility, which could account for variants’ milder phenotypes. Interestingly, almost all (13/15 = 87%) non-collagenous boundary variants occurred on the amino side of an interruption. Trimerisation occurs in the carboxyl to amino direction, so the side of an interruption that a variant occurs on may affect severity.
Conclusions of the effect of a variant’s molecular location on phenotype severity have been conflicting. Some studies have found that Gly missense variants at the amino end of the collagen IV α5 chain give rise to a milder disease phenotype38, while others have found no such relationship24. Our study has demonstrated that molecular location was not associated with a difference in age at kidney failure. These results also contradict studies in other collagen genes such as COL1A1, that have demonstrated that Gly missense variants occurring at the amino end are more likely to be non-lethal37. However, these other collagen types differ from collagen type IV in a number of structural features such as the presence of interruptions and retention of non-collagenous termini, so that a direct comparison may not be appropriate.
In order to deal with the uncertainty associated with the non-collagenous interruptions and the effect of molecular location, each of the 23 local collagenous Gly XY regions was considered as an individual domain with its own amino and carboxyl ends. Surprisingly, analogous to observations in COL1A1, variants at the carboxyl end of their local collagenous region were associated with an increased risk of kidney failure. Considering that variants affecting non-collagenous boundary residues have a milder effect than most other Gly variants, the variants affecting the next Gly along were also expected to be milder. This difference may be due to the trimer assembly of the three collagen IV α-chains beginning at the carboxyl end of each chain. Boundary variants on the amino side of an interruption may not be as destructive since they only expand the flexible interruption by one or two residues. However, since the trimers are assembled in the carboxyl to amino direction, a substitution of one or two Gly residues further along may affect the next nucleation-zippering event for trimerization, and result in a more severe phenotype. To our knowledge, this is the first report of such an observation. A previous study of the effect of the distance of a variant to its nearest interruption on age at kidney failure did not demonstrate any relationship35.
Substitution with a highly destabilising residue was the only molecular feature associated with an earlier age at diagnosis of hearing loss. The median ages reported here do not predict the age at hearing loss onset, but rather the age at diagnosis. In severe disease, hearing loss onset generally occurs in the first decade, but is often unrecognised and underreported. Affected boys often only undergo audiometry after kidney disease is detected, and usually only when the hearing loss is obvious rather than as a screening test. Nonetheless, these results provide a proof of concept that substitution with a highly destabilising residue has a negative effect on hearing loss phenotype. Variants affecting non-collagenous boundary residues would be expected to also result in a milder hearing loss, but the sample size of this study precluded the demonstration of any differences.
In COL4A3 and COL4A4, substitution of a non-collagenous boundary Gly residue was associated with a lower risk of haematuria, while substitution with a more destabilising residue was associated with a higher risk. This is consistent with our findings in COL4A5, where these features were associated with later and earlier ages at kidney failure respectively. However, the proportion of variance in haematuria explained by these features alone was low, and it is likely that other genetic and environmental factors also contributed to haematuria risk. In addition, the control group used here were individuals where haematuria was not formally noted in their medical records. They were not necessarily individuals with a negative urinalysis, and undiagnosed haematuria in this group was probably higher than reported.
A higher proportion of variants affecting non-collagenous boundary Gly residues was observed in gnomAD than expected. The higher frequency of these variants in the general population supports our conclusion that missense variants involving boundary residues are likely to be much milder. Some may even be benign54. Additionally, all of the 5 most frequently reported variants affected a boundary residue. One of these (Gly624Asp) has been demonstrated to have originated in Central/East Europe due to a founder effect 750–900 years ago57, and other variants have probably also arisen in similar circumstances. We have demonstrated previously that the number of people with a genetic risk for AS in the general population is likely to be higher than currently recognised4, and this study’s results suggest that variants affecting non-collagenous boundary residues are major contributors to this largely undiagnosed population.
In this study we have stratified pathogenic COL4A5 Gly missense variants causing AS into clinically relevant subgroups. We identified molecular features of these variants which are likely to contribute to the clinical severity and disease progression in affected individuals, and provide estimates for the age at kidney failure for each subgroup. However, even within these groups much phenotypic variation still exists, and other genetic factors such as whether a variant affects a ligand binding site58 or is located near a proline-rich sequence59 may also be important. Environmental factors such as blood pressure control and obesity may also affect the clinical course60. Accurate predictions of disease severity and rate of progression are helpful for patients, their clinicians and genetic counsellors, and genetic and clinical data should be considered together when managing patients with AS.
References
Gubler, M. C. et al. Alport’s syndrome: A report of 58 cases and a review of the literature. Am. J. Med. 70, 493–505 (1981).
Hasstedt, S. J. & Atkin, C. L. X-linked inheritance of Alport syndrome: Family P revisited. Am. J. Hum. Genet. 35, 1241 (1983).
Pajari, H., Kääriäinen, H., Muhonen, T. & Koskimies, O. Alport’s syndrome in 78 patients: Epidemiological and clinical study. Acta Paediatr. 85, 1300–1306 (1996).
Gibson, J. et al. Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J. Am. Soc. Nephrol. 32, 2273–2290 (2021).
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227 (1990).
Mochizuki, T. et al. Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat. Genet. 8, 77–82 (1994).
Yurchenco, P. D. & Ruben, G. C. Basement membrane structure in situ: Evidence for lateral associations in the type IV collagen network. J. Cell Biol. 105, 2559–2568 (1987).
Sundaramoorthy, M., Meiyappan, M., Todd, P. & Hudson, B. G. Crystal structure of NC1 domains: Structural basis for type IV collagen assembly in basement membranes. J. Biol. Chem. 277, 31142–31153 (2002).
Feingold, J. et al. Genetic heterogeneity of Alport syndrome. Kidney Int. 27, 672–677 (1985).
Jais, J. P. et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 11, 649–657 (2000).
Jais, J. P. et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 14, 2603–2610 (2003).
Rheault, M. N. Women and Alport syndrome. Pediatr. Nephrol. 27, 41–46 (2012).
Lemmink, H. H. et al. Mutations in the type IV collagen α3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum. Mol. Genet. 3, 1269–1273 (1994).
Mencarelli, M. A. et al. Evidence of digenic inheritance in Alport syndrome. J. Med. Genet. 52, 163–174 (2015).
Savige, J. et al. Thin basement membrane nephropathy. Kidney Int. 64, 1169–1178 (2003).
Matthaiou, A., Poulli, T. & Deltas, C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clin. Kidney J. 13, 1025–1036 (2020).
Kamiyoshi, N. et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin. J. Am. Soc. Nephrol. 11, 1441–1449 (2016).
Marcocci, E. et al. Autosomal dominant Alport syndrome: Molecular analysis of the COL4A4 gene and clinical outcome. Nephrol. Dial. Transplant. 24, 1464–1471 (2009).
Zhou, J., Hertz, J. M., Leinonen, A. & Tryggvason, K. Complete amino acid sequence of the human α5(IV) collagen chain and identification of a single-base mutation in exon 23 converting glycine 521 in the collagenous domain to cysteine in an Alport syndrome patient. J. Biol. Chem. 267, 12475–12481 (1992).
Mariyama, M., Leinonen, A., Mochizuki, T., Tryggvason, K. & Reeders, S. T. Complete primary structure of the human α3(IV) collagen chain: Coexpression of the α3(IV) and α4(IV) collagen chains in human tissues. J. Biol. Chem. 269, 23013–23017 (1994).
Leinonen, A., Mariyama, M., Mochizuki, T., Tryggvason, K. & Reeders, S. T. Complete primary structure of the human type IV collagen α4(IV) chain: Comparison with structure and expression of the other α(IV) chains. J. Biol. Chem. 269, 26172–26177 (1994).
Khoshnoodi, J., Pedchenko, V. & Hudson, B. G. Mammalian collagen IV. Microsc. Res. Tech. 71, 357–370 (2008).
Khoshnoodi, J. et al. Mechanism of chain selection in the assembly of collagen IV: A prominent role for the α2 chain. J. Biol. Chem. 281, 6058–6069 (2006).
Bekheirnia, M. R. et al. Genotype–phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 21, 876–883 (2010).
Hashimura, Y. et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int. 85, 1208–1213 (2014).
Kashtan, C. E. Alport syndrome and thin basement membrane disease. Curr. Diagn. Pathol. 8, 349–360 (2002).
Wang, D. et al. The chemical chaperone, PBA, reduces ER stress and autophagy and increases collagen IV α5 expression in cultured fibroblasts from men with X-linked Alport syndrome and missense mutations. Kidney Int. Rep. 2, 739–748 (2017).
Pieri, M. et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J. Am. Soc. Nephrol. 25, 260–275 (2014).
Mastrangelo, A. et al. X-Linked Alport syndrome in women: Genotype and clinical course in 24 cases. Front. Med. 7, 807 (2020).
Storey, H., Savige, J., Sivakumar, V., Abbs, S. & Flinter, F. A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 24, 1945–1954 (2013).
Lee, J. M. et al. Features of autosomal recessive Alport syndrome: A systematic review. J. Clin. Med. 8, 178 (2019).
Savige, J. et al. Consensus Statement on Standards and Guidelines for the Molecular Diagnostics of Alport Syndrome: Refining the ACMG Criteria. Eur. J. Hum. Genet. 29,1186–1197 (2021).
Bella, J., Eaton, M., Brodsky, B. & Berman, H. M. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science 266, 75–81 (1994).
Persikov, A. V. et al. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 24, 330–337 (2004).
Tsiakkis, D. et al. Genotype–phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin. Genet. 82, 297–299 (2012).
Kaneko, K. et al. A family with X-linked benign familial hematuria. Pediatr. Nephrol. 25, 545–548 (2010).
Marini, J. C. et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–221 (2007).
Gross, O., Netzer, K. O., Lambrecht, R., Seibold, S. & Weber, M. Meta-analysis of genotype–phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol. Dial. Transplant. 17, 1218–1227 (2002).
Demosthenous, P. et al. X-linked Alport syndrome in Hellenic families: Phenotypic heterogeneity and mutations near interruptions of the collagen domain in COL4A5. Clin. Genet. 81, 240–248 (2012).
Fokkema, I. F. A. C. et al. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 32, 557–563 (2011).
Genomics England: The National Genomics Research and Healthcare Knowledgebase v5 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Nozu, K. et al. X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin. J. Am. Soc. Nephrol. 9, 1958–1964 (2014).
Horinouchi, T. et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 29, 2244–2254 (2018).
Aoto, Y. et al. Last nucleotide substitutions of COL4A5 exons cause aberrant splicing. Kidney Int. Rep. 7, 108–116 (2022).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Houdayer, C. et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 33, 1228–1238 (2012).
Hess, S. T., Blake, J. D. & Blake, R. D. Wide variations in neighbor-dependent substitution rates. J. Mol. Biol. 236, 1022–1033 (1994).
Therneau, T: A Package for survival analysis in R. R package version 3.2-11. https://CRAN.R-project.org/package=survival (2021).
Therneau, T. M. & Grambsch, P. M. Modeling Survival Data: Extending the Cox Model (Springer, 2000).
Hebbali, A. blorr: Tools for developing binary logistic regression models. R package version 0.3.0. https://CRAN.R-project.org/package=blorr (2020).
Kassambara, A., Kosinski, M. & Biecek, P. survminer: Drawing survival curves using 'ggplot2'. R package version 0.4.9. https://CRAN.R-project.org/package=survminer (2021).
Gordon, M. & Lumley, T. forestplot: Advanced forest plot using 'grid' graphics. R package version 1.9. https://CRAN.R-project.org/package=forestplot (2019).
Zhang, Y. et al. Reassessing the pathogenicity of c.2858G>T(p.(G953V)) in COL4A5 gene: Report of 19 Chinese families. Eur. J. Hum. Genet. 28, 244–252 (2020).
Savige, J. et al. X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS ONE 11, e0161802 (2016).
Knebelmann, B. et al. Spectrum of mutations in the COL4A5 collagen gene in X-linked Alport syndrome. Am. J. Hum. Genet. 59, 1221 (1996).
Żurowska, A. M. et al. Mild X-linked Alport syndrome due to the COL4A5 G624D variant originating in the Middle Ages is predominant in Central/East Europe and causes kidney failure in midlife. Kidney Int. 99, 1451–1458 (2021).
Parkin, J. D. et al. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum. Mutat. 32, 127–143 (2011).
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009).
Yamamura, T. et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 98, 1605–1614 (2020).
Acknowledgements
The authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provide exome and genome variants data to this resource. A full list of contributing groups can be found at https://gnomad.broadinstitute.org/about. The authors would also like to thank the Leiden Open Variation Database (LOVD) and its contributors and administrators. We also acknowledge Olga Bielska (Department of Pediatrics, Nephrology and Hypertension, Medical University of Gdańsk) for their work in establishing the Polish National Registry.
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. Prof Daniel P Gale is the contact person for the work that involved the Genomics England Consortium at d.gale@ucl.ac.uk. Authors of this manuscript who were also Consortium members included MMY Chan, O Sadeghi-Alavijeh, Daniel P Gale and Judy Savige.
Funding
BSLZ is supported by the Polish National Science Center grant 2017/25/N/NZ5/00466.
Author information
Authors and Affiliations
Consortia
Contributions
J.T.G. designed the study, undertook the analysis and interpretation of data, produced all visualisations, and drafted and revised the manuscript. J.S. designed the study, contributed to the interpretation of data, and drafted and revised the manuscript. M.H., M.S.C.D. and K.S. updated the LOVD database. M.M.Y.C., O.S.A. and D.P.G. helped with the acquisition of the 100kGP data. H.R., P.H., C.D., H.S., B.S.L.Z. and A.C. provided new variants for the LOVD update. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gibson, J.T., Huang, M., Shenelli Croos Dabrera, M. et al. Genotype–phenotype correlations for COL4A3–COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci Rep 12, 2722 (2022). https://doi.org/10.1038/s41598-022-06525-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06525-9
This article is cited by
-
Pathogenic variants in the Alport genes are prevalent in the Singapore multiethnic population with highest frequency in the Chinese
Scientific Reports (2025)
-
A guide to gene–disease relationships in nephrology
Nature Reviews Nephrology (2025)
-
Detection of Alport gene variants in children and young people with persistent haematuria
Pediatric Nephrology (2025)
-
Identification of a novel intronic variant in COL4A2 gene associated with fetal severe cerebral encephalomalacia and subdural hemorrhage
BMC Medical Genomics (2024)
-
Clinical and diagnostic utility of genomic sequencing for children referred to a Kidney Genomics Clinic with microscopic haematuria
Pediatric Nephrology (2023)
