
Abstract
The Rift Valley fever virus (RVFV) is a zoonotic arbovirus and pathogenic to both humans and animals. Currently, no proven effective RVFV drugs or licensed vaccine are available for human or animal use. Hence, there is an urgent need to develop effective treatment options to control this viral infection. RVFV glycoprotein N (GN), glycoprotein C (GC), and nucleocapsid (N) proteins are attractive antiviral drug targets due to their critical roles in RVFV replication. In present study, an integrated docking-based virtual screening of more than 6000 phytochemicals with known antiviral activities against these conserved RVFV proteins was conducted. The top five hit compounds, calyxin C, calyxin D, calyxin J, gericudranins A, and blepharocalyxin C displayed optimal binding against all three target proteins. Moreover, multiple parameters from the molecular dynamics (MD) simulations and MM/GBSA analysis confirmed the stability of protein–ligand complexes and revealed that these compounds may act as potential pan-inhibitors of RVFV replication. Our computational analyses may contribute toward the development of promising effective drugs against RVFV infection.
Similar content being viewed by others

Introduction
The Rift Valley fever is an emerging mosquito and aerosol borne disease that caused by Rift Valley fever virus (RVFV) and associated with endemic to sub-Saharan Africa and Arabian Peninsula1. The RVFV (genus Phlebovirus, family Bunyaviridae) is an infectious pathogen that can cause disease ranging from a mild illness to hemorrhagic fever and encephalitis in humans1. Livestock such as cattle, goats and sheep are also susceptible to RVFV infection2. Currently, the options to treat RVFV infected individuals and livestock are limited. The antiviral agent, Ribavirin, was used during past outbreaks, however, its use was limited due to undesirable side effects and the high potential to cause birth defects3. Additionally, favipiravir has been also proposed as a broad-spectrum inhibitor of viral hemorrhagic fever4. However, the efficacy of drugs in RVFV infected humans or livestock have not yet been verified. The paucity of licensed drugs needed to treat RVFV infection as well as the ability of RNA viruses to mutate and develop resistance to drugs emphasize the continued need for identification of anti-viral agents.
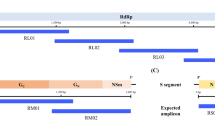
Like other bunyavirus family members, the RVFV genome consists of three negative-sense RNA segments labelled as Large (L), Medium (M) and Small (S) segment5. The L segment contains the viral RNA-dependent-RNA polymerase (RdRp) that is essential for viral replication cycle6. The M segment encodes the two major structural glycoproteins, referred to as glycoprotein N (GN), glycoprotein C (GC) as well as two nonstructural (NS) proteins, NSm1 and NSm27. The structural glycoproteins, GN and GC, of RVFV, assemble around the outer lipid envelope of RVFV and are required for host cell entry8,9. The viral entry into the host cell depends mainly on the ability of these glycoproteins to bind host cellular proteins and to efficiently prompt fusion of the virus envelope with the host cell membrane. They are usually the prime targets of neutralizing antibodies10. The S segment, on the other hand, encodes the nonstructural NSs protein, a major RVFV’s virulence factor, and the nucleocapsid (N) protein11,12. The RVFV N protein is a 27 kDa protein and encapsulates the RVFV genome by coating the viral RNA9 (Fig. 1).

Virion and genome structure of RVFV.
The encapsulation process acts as a shield to protect the viral RNA and prevents the activation of the host anti-viral response by the formation of double-stranded RNA during replication. Therefore, the N protein is essential for several steps in viral replication and transcription cycle9,12,13. The N protein is also involved in the virus assembly via the interactions with the glycoproteins (GN and GC)9. The blocking of viral fusion activity by targeting viral glycoproteins (GN and GC) as well as the inhibition of viral nucleocapsid N protein function represent an attractive antiviral therapeutic strategy due to their essential role in the viral life cycle.
The use of computational approaches to discover small molecules has become increasingly important in early drug development in recent years14,15,16,17,18. Moreover, molecular docking is a widely used tool for prediction of the interaction mechanism between ligands and the target protein17,19,20,21,22,23,24. Putative antiviral compounds have already been found using hierarchical virtual screening approaches against a wide spectrum of viruses including influenza25, Ebola18,26, Zika27,28, human immunodeficiency virus (HIV)29, hepatitis C virus HCV16 and Dengue Fever30,31. MD simulations, which are relied on a general model of the physics governing interatomic interactions, predict how each atom in a protein or other molecular system will move over time32. MM/PBSA and MM/GBSA have already been extensively used in biomolecular studies including protein–ligand binding, protein folding, protein–protein interaction, and so on33. The prediction of ADMET properties is critical in the drug design process because these properties are responsible for the failure of approximately 60% of all drugs in the clinical phases34 This research revolves around the sequential computational screening methods including structure-based virtual screening, MD simulations, MMGBSA calculation and ADME-T analysis of ~ 6000 phytochemicals against three key RVFV structural proteins (GN, GC, and N).
Material and methods
Proteins preparation
The x-ray structures of Glycoprotein (C) (PDB ID: 4HJC), Glycoprotein (N) (PDB ID: 6F8P) and Nucleocapsid (N) (PDB ID: 3OV9) were retrieved from RCSB Protein Data Bank35. The X-ray diffraction analysis demonstrated that all these three structures have resolution up to 4.15 Å, and 1.60 Å, and 1.60 Å, respectively. The Molecular Operating Environment (MOE) was used to prepare the protein structures for docking. Mainly, refinement of structures such as removal of H2O molecules, 3D protonation and energy minimization were performed through MOE using default parameters36. Minimized structures were further used for molecular docking.
Ligand database preparation
A library of ~ 6000 phytochemicals was prepared by collecting phytochemicals with known antiviral activity from several databases such as PubChem, MAPS, MPD3 and ZINC in .sdf format37,38,39. Energy minimization for each ligand was performed using the following parameters, Gradient: 0.05, Force Field: MMFF94X and Chiral Constraint: Current Geometry. The minimized ligands were then saved into the MOE database in .mol format.
Molecular docking
The phytochemicals were docked against GC, GN, and N proteins using MOE Dock tool while setting the specific docking sites36. Site Finder tool was used to predict the active sites of GC, GN, and N proteins40,41,42. MOE possesses multiple docking algorithms to get the best poses of docked complexes. In this analysis, triangular matcher algorithm was utilized with ten iterations to get the best poses for further analysis43. Docking binding scores were utilized as a key evaluation criterion to filter out promising compounds. For each docked complexes, the model with the maximum absolute value of binding energy were considered accurate.
Receptor ligand interaction analysis
Two dimensional (2D) diagram of protein–ligand complexes were obtained using LigX tool in MOE to clearly visualize the ligand-receptor interaction of best-docked complexes44. LigX tool in MOE creates a 2D graph showing interacting forces (such as covalent and non-covalent interaction) for compounds within the active sites of RVFV proteins. Later, 3D diagram of best-docked complexes were displayed using PyMOL and Discovery Studio36,45.
Drug scan/ADME toxicity
The analysis of the drug-likeness as well as ADME-T (absorption, distribution, metabolism, and excretion—toxicity) properties of drug molecules is a crucial phase in the drug discovery pipeline46. Theses parameters were determined using the canonical simplified molecular input line-entry system (SMILES) of each molecule as input file. The drug likeliness of docked molecules was calculated using the drug scan tools at Molinspiration web-server following "Lipinski's Rule of Five" criteria47,48. Furthermore, the ADME properties were predicted using ADMETlab 2.0 and Swiss ADME webservers. The Protox II webserver was used to predict the acute oral toxicity of molecules49. This server categorized compounds into six toxicity classes (1–6), with class 1 being the most dangerous and poisonous, with an estimated fatal dose (LD50) of less than 5, and class 6 denoting non-toxicity with an LD50 > 5000.
Molecular dynamics simulation
Molecular Dynamic (MD) simulation is a critical computational approach for investigating the structural stability and dynamics of docked complexes. The AMBER1850 software was used to perform MD simulations of the antiviral compounds in complex with target proteins. The top docked complexes were deliberately solvated with H2O molecules, and then counter ions were added in order to create a neutral system. The TIP3P solvent model was then used to generate a water box with a thickness of 12 Å to encircle the complexes51. The docked complexes were simulated by employing periodic boundary conditions32. Further, for non-bounded interactions, a boundary value of 8 Å was set. After minimizing water molecules for 500 cycles, the entire system was minimized for 1000 rounds. The temperature of each system was then steadily increased to 300 K. The systems were equilibrated for 100 ps using the NPT ensemble. During the equilibration of counter ions and water molecules, solutes in the first phase were restricted for 50 ps, and protein side chains were then relaxed. A 100 ns MD simulation was run for two fs at 300 K and 1 atm using the NPT ensemble. The SHAKE algorithm52 was employed to restrain the hydrogen and covalent bonding, while Langevin dynamics53 were used to regulate system temperature. The initial structure was employed as a baseline, and AMBER's CPPTRAJ54,55 was used to generate a RMSD plot to ensure that the system MD simulation was converging56. The structural flexibilities of ligands were determined using the ligand RMSD method57. RoG was studied for the compactness and three-dimensional packaging of the complex. The RMSF reflects the average root mean square distance between an atom and its average geometric position in a certain dynamics58.
Binding free energy calculations
The MM-GBSA method, implemented in AMBER 18, was used to calculate the binding free energies (ΔGtol) of RVFV proteins complexed with the most potential hit compounds59,60. Briefly, 10,000 snapshots were generated from the last 20 ns stable trajectories with a 2 ps interval for each system. The total binding free energy is calculated as the solvation free energy (ΔGsol) and the sum of the molecular mechanics binding energy (ΔEMM), as shown below.
where, ΔEMM is further divided into electrostatic energy (ΔEele), internal energy (ΔEint) and van der Waals energy (ΔEvdw). The sum of non-polar (ΔGNp) and polar (ΔGp) components contribute to the total solvation free energy (Gsol). The MM-GBSA method is well demonstrated in binding free estimation for antiviral inhibitors29,61,62,63.
Results and discussion
Molecular docking
The developed phytochemicals database was docked against RVFV proteins (GN, GC, and N). Molecular docking is a technique for predicting how ligands will bind to their protein targets. As a result, molecular docking become an important tool in virtual screening and the development of novel antiviral medicines to combat severe disorders30,64. Docked compounds were selected by applying a strict filtering criterion that took into account following conditions: strength of H-bond interaction, binding pocket maximum occupancy with the lowest Gibbs free energy and docking score/strength compared to reported native ligand. Reported native ligands 1,2-ethanediol; 3-Aminophthalylhydrazido-N-acetyl-beta-glucosaminide; and Nitrite Ion of RVFV GN, GC and N proteins, respectively, were used as control and only those compounds which showed stronger binding affinity were chosen for further analyses. Calyxin C, calyxin D, calyxin J, gericudranins A, and blepharocalyxin C (Fig. 2) were discovered, binding with the interacting residues of all 3 target proteins at high binding affinity (Table 1).

Two-dimensional presentation of top compounds binding to RVFV proteins.
Calyxin C was bound to GN protein with a score of -12.32 kJ/mol, forming hydrogen bonds with the side chains of Arg-461, Lys-199, Leu-299 and Lys-247 and calyxin D was bound with a binding score of −11.30 kcal/mol, forming hydrogen bonds with side chains of His-249, Asp-301, Lys-247 and Arg-461. Calyxin C and calyxin D showed strong binding with GN active residues followed by calyxin J, gericudranins A, and blepharocalyxin C with binding scores of −14.15 kcal/mol, −11.74 kcal/mol, and −10.96 kcal/mol, respectively (Table 1). Except for gericudranins A, all ligands showed strong hydrogen bonding with the conserved Lys-247 (Fig. 3).

Binding modes and interaction mechanisms of novel GN protein inhibitors. (A) Inhibitory binding modes of all ligands. A 3D close view into the binding mode of calyxin C (B), calyxin D (C), Calyxin J (D), Gericudranins A (E) and Blepharocalyxin C (F). 2D interaction analysis of calyxin C (G), Calyxin D (H), Calyxin J (I), Gericudranins A (J) and Blepharocalyxin C (K).
Likewise, in GC protein calyxin C, calyxin D, calyxin J, gericudranins A, and blepharocalyxin C have been found to bind through significant hydrogen bonds having binding scores of −14.95 kcal/mol, −10.68 kcal/mol, −11.45 kcal/mol, −13.39 kcal/mol, and −12.10 kcal/mol respectively. All the essential residues (Arg-810, Arg-949, and Lys-813) that comprise the active site was found to serve as electron donors in the formation of a H-bond network. Other active site residues (Leu-789, Asn-592, Asp-793) showed strong non-covalent and hydrophobic interactions, as detailed in Table 1 and illustrated in Fig. 4.

Binding modes and interaction mechanisms of novel GC protein inhibitors. (A) Inhibitory binding modes of all ligands. A 3D close view into the binding mode of calyxin C (B), calyxin D (C), calyxin J (D), Gericudranins A (E) and Blepharocalyxin C (F). 2D interaction analysis of calyxin C (G), Calyxin D (H), Calyxin J (I), Gericudranins A (J) and blepharocalyxin C (K).
Similarly, top five inhibitors (calyxin C, calyxin D, calyxin J, gericudranins A, and blepharocalyxin C) which were found to inhibit glycoproteins (GN and GC) were also observed as inhibiting N protein. The binding energies of the five active compounds were in the range of −18.02 kcal/mol to −16.72 kcal/mol (Table 1). Most compounds established hydrogen bonds with Arg B64, Lys B74, and Arg B70, which indicates that these compounds have potential role to play in disease management. Hydrogen interactions between the side chains and backbone atoms of these N protein residues stabilized the inhibitors spatially within the pocket. All ligands showed strong hydrogen bonding with the conserved Arg B70 (Fig. 5).

Binding modes and interaction mechanisms of novel N protein inhibitors. (A) Inhibitory binding modes of all ligands. A 3D close view into the binding mode of calyxin C (B), Calyxin D (C), Calyxin J (D), Gericudranins A (E) and Blepharocalyxin C (F). 2D Interaction analysis of calyxin C (G), Calyxin D (H), Calyxin J (I), Gericudranins A (J) and blepharocalyxin C (K).
All top five inhibitors were making strong bonds with functionally and structurally key interacting sites of the RVFV proteins. However, calyxin C was ranked first because it had the highest binding score and affinity. The compounds discovered in present study may have synergistic or additive effects against RVFV. This is an important aspect in case of viruses, which are constantly evolving due to a higher mutation rate. In case of HCV and HIV infections, the advantages of synergistic treatment techniques have already been documented65,66.
Drug scan/ADME toxicity
The drug likeness (DL) properties of final active compounds were identified using Molinspiration server which offers ADMET tool to scan the pharmacokinetic properties. The rule states that a compound must contain less than five H-bond donors, less than ten H-bond acceptors, a molecular mass of less than five hundred daltons, and log P should be greater than five. Drug-likeness properties of potential compounds are enlisted in Table 2. According to the results, all compounds showed only one violation of Lipinski rule of five i.e., MW > 500 daltons.
Additional analyses were performed on the ADMET properties of selected compounds. The assessment of compounds' ADMET properties is a key step in the drug discovery toolbox. A major portion of proposed drug candidates failed to reach the final step because of toxicity and poor pharmacokinetic properties66,67. ADMET lab 2.0 and Swiss ADMET were used to predict ADMET properties of screened compounds, and their results are presented in Table 3.
Drug distribution and absorption of drug molecules are indicated by gastrointestinal absorption (GI) and blood–brain barrier (BBB) permeation68,69,70. Table 3 shows that all compounds have low gastro-intestinal absorption and no BBB permeation. The compounds' absorption was further demonstrated by caco-2 permeability values ranging from −5.35 to −6.57 log unit. In ADMETlab 2.0 server, permeability greater than −5.15 log unit indicates optimal caco-2 absorption. Furthermore, several cytochromes (CYPs) regulate drug metabolism, with CYP2C19, CYP2C9, CYP3A4, CYP1A2, and CYP2D6 being essential for drug molecules biotransformation71,72. Furthermore, p-glycoprotein inhibitors decrease the bioavailability of drugs known to be transported73,74,75. Calyxin D, and blepharocalyxin C are inhibitors of p-glycoprotein while others are non-inhibitors. Similarly, calyxin D and calyxin J are negative substrates of p-glycoprotein, while others are substrates, which explain their good absorption profiles. Following that, toxicity prediction research was carried out to assess the compounds' safety profile. All selected compounds were found to be non-toxic and non-carcinogenic. These findings suggested that no toxicophore associated with these compounds and could be developed into safer drugs.
Molecular dynamics simulation
MD simulation is a powerful approach in biophysical research that offers important dynamic values of protein–ligand interactions53,76,77. A number of studies showed that some systems require MD simulations to discover the accurate binding conformations21,29,77,78,79,80,81 and therefore, it has profound importance in computer-aided drug discovery18,82. For the present study, MD simulation were carried out on the top models obtained through docking with calyxin C and calyxin D inhibitors. To explicate the dynamic stability and ensure the rationality of the ligand sampling, the RMSD values of GN, GC and N protein, and heavy atoms of calyxin C and calyxin D inhibitors relative to the respective initial structures were calculated, and RMSD trajectories were analyzed over a period of throughout 100 ns. The RMSD plots of protein–ligand complexes are displayed in Fig. 6. RMSD values of complexes were predicted as: GN-calyxin C (maximum, 1.8 Å; mean, 1.2 Å), GN-calyxin D (maximum, 2.5 Å; mean, 1.9 Å), GC-calyxin C (maximum, 2.6 Å; mean, 1.8 Å) , GC-calyxin D (maximum, 2.8 Å; mean, 2.25 Å) , N-calyxin C (maximum, 3.5 Å; mean 2.45 Å) and N-calyxin D (maximum, 2.6 Å; mean, 2.15 Å) (Fig. 6). In terms of 3D structure, all the receptors are relatively stable, and no secondary structure flexibility was observed. Hence, calyxin C and calyxin D binding poses remained unchanged, indicating stable and strong complexes formation. Although the conformations of the complex were expanded, the RMSD remained converge under 3 Å. The RMSD of protein backbone atoms of the order of 1–3 Å with no high conformational change certainly favours that the system is well equilibrated and calyxin C and calyxin D inhibitors binds more stably with the binding pocket of GN, GC and N proteins, which is an acceptable measure in protein–ligand simulation systems76,83.

(A) Root mean square deviation (RMSD) analysis of RVFV GN in complex with two candidate compounds (calyxin C and calyxin D) during 100 ns MD simulation. (B) RMSD analysis of RVFV GC in complex with calyxin C and calyxin D. (C) RMSD analysis of RVFV N in complex with calyxin C and calyxin D.
The stability and residual flexibility of proteins in presence of calyxin C and calyxin D was further computed through RMSF analysis. Mean RMSF for GN- calyxin C is 3.4 Å GN-calyxin D is 3.8 Å, GC-calyxin C is 0.6 Å, GC-calyxin D is 1.7 Å, N-calyxin C is 1.2 Å and N-calyxin D is 2.2 Å (Fig. 7). These values indicate a high level of agreement on intermolecular stability. Generally, the GN-calyxin D shows high rates of fluctuation starting from residue 150 to 250 exhibiting a high tendency to fluctuate.

(A) Root mean square fluctuation (RMSF) analysis of RVFV GN in complex with two candidate compounds (calyxin C and calyxin D) during 100 ns MD simulation. (B) RMSF analysis of RVFV GC in complex with calyxin C and calyxin D. (C) RMSF analysis of RVFV N in complex with calyxin C and calyxin D.
Furthermore, Rg analysis was conducted to assess structural equilibrium and protein compactness over the simulation time. An optimum Rg value should be low in case of globular proteins, however, the Rg value for protein form with a greater number of turns and loops could be significantly larger83. The Rg values of the complexes are follows; GN-calyxin C (maximum, 44.8 Å; mean, 42.5 Å), GN-calyxin D (maximum, 45 Å; mean, 43 Å), GC-calyxin C (maximum, 90 Å; mean, 84 Å), GC-calyxin D (maximum, 94.5 Å; mean, 86 Å), N-calyxin C (maximum, 80 Å; mean, 70 Å) and N-calyxin D (maximum, 77 Å; mean, 67 Å) (Fig. 8). For RVFV GN-calyxin C and GN-calyxin D, both complexes showed compactness, while RVFV GC/N-calyxin C and GC/N-calyxin D complexes indicated slight loss of compactness at the end. Overall, no significant loss in compactness was observed during the simulation period in all complexes.

(A) Radius of gyration (Rg) analysis of RVFV GN in complex with two candidate compounds (calyxin C and calyxin D) during 100 ns MD simulation. (B) Rg analysis of RVFV GC in complex with calyxin C and calyxin D. (C) Rg analysis of RVFV N in complex with calyxin C and calyxin D.
Binding free energy calculations
Binding free energies of best-docked complexes with calyxin C and calyxin D inhibitors were calculated by employing MMGBSA methods to better understand the complexes binding ability with RVFV proteins. The binding energies of the complexes are given in detail in the Table 4. The results from MM/GBSA analysis demonstrated the favorable affinity of calyxin C and calyxin D inhibitors within the binding pocket RVFV GN/GC and N proteins. From the calculated results, RVFV GN/GC and N proteins showed more favorable total binding free energy (ΔGtol) in complex with calyxin C (−27, −18.64, and −33.54 kcal/mol) as compared to calyxin D (−21.58, −17.92, and −23.54 kcal/mol), respectively. Whereas RVFV GN/GC and N proteins with bound inhibitors showed a considerable increase in negative values for vdW interaction energies (ΔGvdW range from −27.11 to −36.99 kcal/mol) as compared to electrostatic interactions (ΔGelec range from 3.08 to − 45.35 kcal/mol). In protein/ligand systems, vdW interactions (ΔGvdW) are considered important in overall strength of nonpolar interactions84,85. Among all complexes, vdW highly contributed towards stabilising the complexes with comparatively higher negative values than electrostatic interactions (ΔGelec). Besides, polar solvation energy (ΔGsol) displayed the energy associated with dissolving calyxin C and calyxin D inhibitors within the solvent, and highly positive polar solvation energies (ΔGsol ranges from 13 to 60.17 kcal/mol) were obtained, which was demonstrated to be unfavorable. The more favorable total binding free energy in complex with calyxin C determined a more stable protein–ligand interaction profile within the binding site GN/GC and N proteins, which was evident from the less residual flexibility compared to calyxin D (Fig. 7).
Conclusions
RVFV is a pathogenic agent and associated with hemorrhagic fever and liver damage. Previous studies reported that few antiviral drugs such as; benzavir-2, favipiravir T-705, and 2′-fluoro-2′-deoxycytidine (2′-FdC), have shown anti-RVFV activities and currently are under development process86,87,88,89. However, no proven RVFV drug or licensed vaccine are available to date in market. Natural molecules-based drug discovery through a pipeline of modern computational tools could be an essential framework towards identifying potential hits against RVFV infection. As an initial step, we designed this study and utilized an integrated computational approach, that identified five novel hit compounds from a focused library of 6000 natural compounds, bearing specific scaffolds which can inhibit the crucial proteins (GN, GC and N) of RVFV. Our discovered drug-like molecules, including calyxin C, calyxin D displayed stable interactions and favorable binding energies. Experimental evaluation of drug targets and subsequent drug molecules designing against any target is time consuming and costly work. Therefore, the results of our study will greatly facilitate drug development process against RVFV. We acknowledge that computational analyses have certain limitations, thus further in-vitro and in-vivo studies are warranted to validate the inhibitory potential of selected promising candidates against RVFV.
References
Linthicum, K. J., Britch, S. C. & Anyamba, A. Rift Valley fever: An emerging mosquito-borne disease. Annu. Rev. Entomol. 61, 395–415 (2016).
Ikegami, T. & Makino, S. The pathogenesis of Rift Valley fever. Viruses 3, 493–519 (2011).
Sidwell, R. W., Huffman, J. H., Barnett, B. B. & Pifat, D. Y. In vitro and in vivo Phlebovirus inhibition by ribavirin. Antimicrob. Agents Chemother. 32, 331–336 (1988).
Furuta, Y., Komeno, T. & Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 93, 449–463 (2017).
Schreur, P. J. W., Kormelink, R. & Kortekaas, J. Genome packaging of the Bunyavirales. Curr. Opin. Virol. 33, 151–155 (2018).
Muller, R., Argentini, C., Bouloy, M., Prehaud, C. & Bishop, D. Completion of the genome sequence of Rift Valley fever phlebovirus indicates that the L RNA is negative sense and codes for a putative transcriptase-replicase [corrected]. Nucleic Acids Res. 19, 5433 (1991).
Schmaljohn, C. S. et al. Baculovirus expression of the M genome segment of Rift Valley fever virus and examination of antigenic and immunogenic properties of the expressed proteins. Virology 170, 184–192 (1989).
Terasaki, K., Murakami, S., Lokugamage, K. G. & Makino, S. Mechanism of tripartite RNA genome packaging in Rift Valley fever virus. Proc. Natl. Acad. Sci. 108, 804–809 (2011).
Liu, L., Celma, C. C. & Roy, P. Rift Valley fever virus structural proteins: Expression, characterization and assembly of recombinant proteins. Virol. J. 5, 1–13 (2008).
Huiskonen, J. T., Overby, A. K., Weber, F. & Grünewald, K. Electron cryo-microscopy and single-particle averaging of Rift Valley fever virus: evidence for GN-GC glycoprotein heterodimers. J. Virol. 83, 3762–3769 (2009).
Brennan, B., Welch, S. R. & Elliott, R. M. The consequences of reconfiguring the ambisense S genome segment of Rift Valley fever virus on viral replication in mammalian and mosquito cells and for genome packaging. PLoS Pathog. 10, e1003922 (2014).
Ferron, F. et al. The hexamer structure of the Rift Valley fever virus nucleoprotein suggests a mechanism for its assembly into ribonucleoprotein complexes. PLoS Pathog. 7, e1002030 (2011).
Raymond, D. D., Piper, M. E., Gerrard, S. R. & Smith, J. L. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. 107, 11769–11774 (2010).
Mirza, M. U. & Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharmaceut. Anal. 10, 320–328 (2020).
Muneer, I., Tusleem, K., Abdul Rauf, S., Hussain, H. M. & Siddiqi, A. R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anticancer Drugs 30, 363–373 (2019).
Khalid, H. et al. Discovery of novel Hepatitis C virus inhibitor targeting multiple allosteric sites of NS5B polymerase. Infect. Genet. Evol. 84, 104371 (2020).
Durdagi, S. et al. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 85, 122–129. https://doi.org/10.1016/j.jmgm.2018.07.010 (2018).
Mirza, M. U. et al. Perspectives towards antiviral drug discovery against Ebola virus. J. Med. Virol. 91, 2029–2048 (2019).
Saleh, N. A. The QSAR and docking calculations of fullerene derivatives as HIV-1 protease inhibitors. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 136, 1523–1529 (2015).
Ahmad, S. et al. Fragment‐based in silico design of SARS CoV‐2 main protease inhibitors. Chem. Biol. Drug Des. 98, 604–619 (2021).
Durrani, F. G. et al. Mutagenesis of DsbAss is crucial for the signal recognition particle mechanism in Escherichia coli: Insights from molecular dynamics simulations. Biomolecules 9, 133 (2019).
Ikram, N. et al. Inhibition of oncogenic kinases: An in vitro validated computational approach identified potential multi-target anticancer compounds. Biomolecules 9, 124 (2019).
Khalid, H. et al. Discovery of novel hepatitis C virus inhibitor targeting multiple allosteric sites of NS5B polymerase. Infect. Genet. Evolut. 84, 104371 (2020).
Mirza, M. U. et al. In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo hemorrhagic fever virus inhibitors. Sci. Rep. 9, 1–18 (2019).
Du, J., Cross, T. A. & Zhou, H.-X. Recent progress in structure-based anti-influenza drug design. Drug Discov. Today 17, 1111–1120 (2012).
Shurtleff, A. C., Nguyen, T. L., Kingery, D. A. & Bavari, S. Therapeutics for filovirus infection: Traditional approaches and progress towards in silico drug design. Expert Opin. Drug Discov. 7, 935–954 (2012).
Nitsche, C. Strategies towards protease inhibitors for emerging flaviviruses. in Dengue and Zika: Control and Antiviral Treatment Strategies. 175–186. (2018).
Rehman, A., Ashfaq, U. A., Shahid, F., Noor, F. & Aslam, S. The screening of phytochemicals against NS5 polymerase to treat Zika virus infection: Integrated computational based approach. in Combinatorial Chemistry & High Throughput Screening. (2021).
Mirza, M. U. et al. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach. Eur. J. Pharm. Sci. 155, 105537 (2020).
ul Qamar, M. T. et al. Discovery of novel dengue NS2B/NS3 protease inhibitors using pharmacophore modeling and molecular docking based virtual screening of the zinc database. Int. J. Pharmacol. 12, 621–632 (2016).
ul Qamar, M. T., Mumtaz, A., Ashfaq, U. A., Adeel, M. M. & Fatima, T. Potential of plant alkaloids as dengue ns3 protease inhibitors: Molecular docking and simulation approach. Bangladesh J. Pharmacol. 9, 262–267 (2014).
Karplus, M. & McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9, 646–652 (2002).
Wang, E. et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 119, 9478–9508 (2019).
Norinder, U. & Bergström, C. A. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 1, 920–937 (2006).
Sussman, J. L. et al. Protein data bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D. Biol. Crystallogr. 54, 1078–1084 (1998).
Vilar, S., Cozza, G. & Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 8, 1555–1572 (2008).
Kim, S. et al. PubChem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213 (2016).
Mumtaz, A. et al. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 31, 1228–1236 (2017).
Irwin, J. J. & Shoichet, B. K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182 (2005).
Mottram, T. J. et al. Mutational analysis of Rift Valley fever phlebovirus nucleocapsid protein indicates novel conserved, functional amino acids. PLoS Negl. Trop. Dis. 11, e0006155 (2017).
Guardado-Calvo, P. et al. Mechanistic insight into bunyavirus-induced membrane fusion from structure-function analyses of the hantavirus envelope glycoprotein Gc. PLoS Pathol. 12, e1005813 (2016).
Wang, Q. et al. Neutralization mechanism of human monoclonal antibodies against Rift Valley fever virus. Nat. Microbiol. 4, 1231–1241 (2019).
Podvinec, M. et al. Novel inhibitors of dengue virus methyltransferase: Discovery by in vitro-driven virtual screening on a desktop computer grid. J. Med. Chem. 53, 1483–1495 (2010).
Khalifa, I., Zhu, W., Mohammed, H. H. H., Dutta, K. & Li, C. Tannins inhibit SARS-CoV-2 through binding with catalytic dyad residues of 3CLpro: An in silico approach with 19 structural different hydrolysable tannins. J. Food Biochem. 44, e13432 (2020).
DeLano, W. L. The PyMOL Molecular Graphics System. (Delano Scientific, San Carlos, 2002).
Wang, Y. et al. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 48, 488–515 (2015).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 (1997).
Jarrahpour, A. et al. Petra, Osiris and Molinspiration (POM) together as a successful support in drug design: antibacterial activity and biopharmaceutical characterization of some azo Schiff bases. Med. Chem. Res. 21, 1984–1990 (2012).
Banerjee, P., Eckert, A. O., Schrey, A. K. & Preissner, R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263 (2018).
Weiner, P. K. & Kollman, P. A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 2, 287–303 (1981).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210 (2013).
Kräutler, V., Van Gunsteren, W. F. & Hünenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001).
Bussi, G. & Parrinello, M. Accurate sampling using Langevin dynamics. Phys. Rev. E 75, 056707 (2007).
Bhowmik, D. et al. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 84, 104451 (2020).
Roe, D. R. & Cheatham, T. E. III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Kouznetsova, V. L., Zhang, A., Tatineni, M., Miller, M. A. & Tsigelny, I. F. Potential COVID-19 papain-like protease PLpro inhibitors: Repurposing FDA-approved drugs. PeerJ 8, e9965 (2020).
Turner, P., McLennan, A., Bates, A. & White, M. BIOS Instant Notes in Molecular Biology (Garland Science, 2007).
Kuzmanic, A. & Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J . 98, 861–871 (2010).
Homeyer, N. & Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inf. 31, 114–122 (2012).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Srivastava, H. K. & Sastry, G. N. Molecular dynamics investigation on a series of HIV protease inhibitors: Assessing the performance of MM-PBSA and MM-GBSA approaches. J. Chem. Inf. Model. 52, 3088–3098 (2012).
Sun, H. et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. PCCP 20, 14450–14460 (2018).
Sun, H., Li, Y., Tian, S., Xu, L. & Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. PCCP 16, 16719–16729 (2014).
Lengauer, T. & Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 6, 402–406 (1996).
Nakata, H. et al. Potent synergistic anti-human immunodeficiency virus (HIV) effects using combinations of the CCR5 inhibitor aplaviroc with other anti-HIV drugs. Antimicrob. Agents Chemother. 52, 2111–2119 (2008).
Lin, B., He, S., Yim, H. J., Liang, T. J. & Hu, Z. Evaluation of antiviral drug synergy in an infectious HCV system. Antivir. Ther. 21, 595 (2016).
Tsaioun, K., Bottlaender, M. & Mabondzo, A. ADDME–Avoiding drug development mistakes early: Central nervous system drug discovery perspective. BMC Neurol. 9, 1–11 (2009).
Benet, L. Z., Kroetz, D., Sheiner, L., Hardman, J. & Limbird, L. Pharmacokinetics: the dynamics of drug absorption, distribution, metabolism, and elimination. Goodman Gilman’s Pharmacol. Basis Ther. 3, e27 (1996).
Chillistone, S. & Hardman, J. G. Factors affecting drug absorption and distribution. Anaesth. Intensive Care Med. 18, 335–339 (2017).
Schanker, L. S. Mechanisms of drug absorption and distribution. Annu. Rev. Pharmacol. 1, 29–45 (1961).
Donato, M. T. & Castell, J. V. Strategies and molecular probes to investigate the role of cytochrome P450 in drug metabolism. Clin. Pharmacokinet. 42, 153–178 (2003).
Zanger, U. M. & Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141 (2013).
Balayssac, D., Authier, N., Cayre, A. & Coudore, F. Does inhibition of P-glycoprotein lead to drug–drug interactions?. Toxicol. Lett. 156, 319–329 (2005).
Srivalli, K. M. R. & Lakshmi, P. K. Overview of P-glycoprotein inhibitors: A rational outlook. Braz. J. Pharm. Sci. 48, 353–367 (2012).
Varma, M. V. S., Ashokraj, Y., Dey, C. S. & Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 48, 347–359 (2003).
Guterres, H. & Im, W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J. Chem. Inf. Model. 60, 2189–2198 (2020).
Sakano, T., Mahamood, M. I., Yamashita, T. & Fujitani, H. Molecular dynamics analysis to evaluate docking pose prediction. Biophys. Physicobiol. 13, 181–194 (2016).
Makeneni, S., Thieker, D. F. & Woods, R. J. Applying pose clustering and MD simulations to eliminate false positives in molecular docking. J. Chem. Inf. Model. 58, 605–614 (2018).
Anwar, F. et al. New naphthalene derivative for cost-effective AChE inhibitors for Alzheimer’s treatment: In silico identification, in vitro and in vivo validation. Comput. Biol. Chem. 89, 107378 (2020).
Iman, K. et al. In silico structure-based identification of novel acetylcholinesterase inhibitors against Alzheimer’s disease. CNS Neurol. Disord. Drug Targets (Formerly Curr. Drug Targets-CNS Neurol. Disord.) 17, 54–68 (2018).
Vanmeert, M. et al. Rational design of an XNA ligase through docking of unbound nucleic acids to toroidal proteins. Nucleic Acids Res. 47, 7130–7142 (2019).
Salo-Ahen, O. M. H. et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9, 71 (2021).
Shukla, R. & Tripathi, T. Computer-Aided Drug Design 133–161 (Springer, 2020).
Bulusu, G. & Desiraju, G. R. Strong and weak hydrogen bonds in protein–ligand recognition. J. Indian Inst. Sci. 100, 31–41 (2020).
Cramer, D. L. et al. Some thermodynamic effects of varying nonpolar surfaces in protein-ligand interactions. Eur. J. Med. Chem. 208, 112771 (2020).
Islam, M. K. et al. Anti-Rift Valley fever virus activity in vitro, pre-clinical pharmacokinetics and oral bioavailability of benzavir-2, a broad-acting antiviral compound. Sci. Rep. 8, 1–12 (2018).
Scharton, D. et al. Favipiravir (T-705) protects against peracute Rift Valley fever virus infection and reduces delayed-onset neurologic disease observed with ribavirin treatment. Antiviral Res. 104, 84–92 (2014).
Borrego, B., de Ávila, A. I., Domingo, E. & Brun, A. Lethal mutagenesis of Rift Valley fever virus induced by favipiravir. Antimicrob. Agents Chemother. 63, e00669-e1619 (2019).
Atkins, C. & Freiberg, A. N. Recent advances in the development of antiviral therapeutics for Rift Valley fever virus infection. Futur. Virol. 12, 651–665 (2017).
Acknowledgements
The researchers would like to thank the Deanship of Scientific Research, Qassim University for funding the publication of this project.
Author information
Authors and Affiliations
Contributions
Conceptualization, S.A., M.T.u.Q. and A.A.; Data curation, I.F., S.A., F.S., E.A.A., F.F.A.A. and W.A.-M.; Formal analysis, I.F., S.A., M.U.M., M.T.u.Q. and A.R.; Funding acquisition, A.A.; Investigation, I.F., S.A., M.A. and A.R.; Methodology, M.U.M., M.T.u.Q., F.S., E.A.A., F. F.A.A. and W.A.-M.; Project administration, M.T.u.Q. and A.A.; Resources, W.A.-M. and A.A.; Software, S.A., M.A. and M.U.M.; Supervision, M.T.u.Q. and A.Al.; Validation, M.A., M.T.u.Q., A.R., F.S., E.A A., F.F.A.A., W.A.-M. and A.A.; Visualization, I.F., S.A. and M.U.M.; Writing – original draft, I.F., S.A., M.U.M., M.T.u.Q. and F.S.; Writing – review & editing, M.A., A.R., E.A.A., F.F.A.A., W.A.-M. and A.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fatima, I., Ahmad, S., Alamri, M.A. et al. Discovery of Rift Valley fever virus natural pan-inhibitors by targeting its multiple key proteins through computational approaches. Sci Rep 12, 9260 (2022). https://doi.org/10.1038/s41598-022-13267-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-022-13267-1
This article is cited by
-
Microbial pigments as potential anti-rift valley fever virus drugs
BMC Chemistry (2026)
-
Computational prediction of phytochemical inhibitors against the cap-binding domain of Rift Valley fever virus
Molecular Diversity (2024)
