
Abstract
Breast cancer is one of the most common cancer types which is described as the leading cause of cancer death in women. After competitive endogenous RNA (ceRNA) hypothesis was proposed, this triple regulatory network has been observed in various cancers, and increasing evidences reveal that ceRNA network plays a significant role in the migration, invasion, proliferation of cancer cells. In the current study, our target is to construct a CD24-associated ceRNA network, and to further identify key prognostic biomarkers in breast cancer. Using the transcriptom profiles from TCGA database, we performed a comprehensive analysis between CD24high tumor samples and CD24low tumor samples, and identified 132 DElncRNAs, 602 DEmRNAs and 26 DEmiRNAs. Through comprehensive analysis, RP1-228H13.5/miR-135a-5p/BEND3 and SIM2 were identified as key CD24-associated biomarkers, which exhibited highly significance with overall survival, immune microenvironment as well as clinical features. To sum up the above, the current study constructed a CD24-associated ceRNA network, and RP1-228H13.5/miR-135a-5p/BEND3 and SIM2 axis worked as a potential therapeutic target and a predictor for BRCA diagnosis and prognosis.
Similar content being viewed by others


Introduction
Breast cancer is one of the most common cancers worldwide, and makes up the largest proportion of cancer associated death in women. Over 2 million new female breast cancer cases were diagnosed and approximately 685,000 women died of breast cancer among 185 countries in 20201. In addition, in 2022, the estimated number of new female breast cancer cases in the United States is up to 2878502. As a complex disease with genetic and clinical heterogeneity, breast cancer is generally categorized into 4 molecular subtypes, including Luminal A (ER+/PR+, HER2-, low proliferation), Luminal B (ER+/PR+, HER2-, high proliferation), HER2 enriched (HER2+, ER−, PR−; HER2+, ER+/PR+), Basal-like (HER2−, ER−, PR−) subtype3,4,5. Compared with other subtypes, luminal A presents a better clinical prognosis4. Luminal B subtype exhibits a lower expression of hormone receptors (ER and PR) but a higher expression of proliferation marker (Ki67) than Luminal A subtype6. HER2 enriched subtype has high sensitivity for anti-HER2 therapy7. Basal-like subtype, also known as triple-negative breast cancer (TNBC), is a highly aggressive tumor with poor prognosis, a high tendency to metastasis and recurrence, and less sensitivity to chemotherapy and radiotherapy8,9. What’s more, it was reported that multiple subtypes co-existed within a breast cancer10. The mortality of female breast cancer has decreased since 1989 due to early diagnosis and improvements in treatment methods1. The therapy strategy should be decided by molecular subtypes and locoregional tumor load. Generally, surgery, endocrine treatment, radiotherapy, neoadjuvant chemotherapy, and chemotherapy are basic therapy methods for breast cancer. Nowadays, breast conservation is technically feasible in many early breast cancers without metastasis, and the application of neoadjuvant chemotherapy extend the indication of breast conservation surgery3,11,12. In addition to the treatments mentioned above, some novel immunotherapy treatments are gradually adopted in clinical13,14, such as immune checkpoint blockades15, CAR-T cell treatment16, and mRNA vaccine technology17. Immunotherapy is a promising method for people with resistance to endocrine therapy or chemotherapy. For example, anti-PD-L1 therapy exhibited good efficacy in TNBC that lacks specific targets of endocrine therapy or chemotherapy but is rich in PD-L118,19. Nonetheless, immunotherapy in breast cancer remains copious challenges. Some patients exhibited less insensitive to immunotherapy or resistant to immunotherapy after initial improvement19. Hence, it is crucial to explore the mechanism of immune regulation in breast cance. CD24 is a novel anti-cancer target which contributes to the immune escape and chemotherapy resistance in multiple tumors20. However, anti-CD24 therapy is controversial. CD24 is expressed not only in tumor cells but also in immune cells. CD24 is required for homeostatic proliferation of T cell, the blockage of which may contribute to the over-activation of proinflammatory reaction20,21,22. The role of CD24 in breast cancer is gaining increasing attention. It was reported that the subcellular translocation of CD24 was correlated with drug resistance in breast cancer23. In addition, CD24 is downregulated by ZBTB28, which enhances phagocytosis of macrophages and consequently inhibits breast cancer24.
Non-coding RNAs (ncRNAs), constitute the majority of the transcriptional products, and play significant roles in the regulatory of biological processes25. NcRNAs comprise circular RNA (circRNA), long non-coding NRA (lncNRA), microNRA (miRNA) and PIWI interacting RNA (piRNA), and their functions are variable26. Some ncRNAs can encode peptides or proteins27, whereas some can regulate the development of diseases via competing endogenous RNA (ceRNA) network28. CeRNA hypothesis proposes a post-transcriptional modulation that transcripts compete for translation via binding shared microRNAs29. In the past decades of years, ceRNA network has been identified involved in the initiation, development and drug resistance of various cancers30,31,32,33. In colorectal cancer, lncRNA CRART16 was found to facilitate expression of ERBB3 and induce cetuximab resistance via binding miR-371a-5p34. In human hepatocellular carcinoma, CD24 is regulated by MALAT1/miR-3064-5p/FOXA1 ceRNA axis, which consequently suppresses angiogenesis35.In breast cancer, ceRNA networks also play key roles. It was reported that tumor-derived exosome-mediated lncRNA SNHG16 induced CD73 + γδT cells in breast cancer samples from Chinese patients36. Regrettably, the ceRNA regulatory network associated with CD24 in breast cancer remains unclear. The studies for ceRNA network can provide us a novel insight into the pathological process of cancer, as well as help to dig out promising drug target or prognosis biomarkers. Therefore, we performed this study in order to identify significant ceRNA network in breast cancer.
Materials and methods
Data download and processing
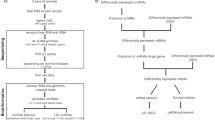
Our study processing was shown in Fig. 1. We downloaded transcriptional data and clinical information of breast invasive carcinoma (BRCA) from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Totally, gene expression raw data, miRNA expression raw data and clinical information for 1098 BRCA patients were downloaded. Finally, 1072 primary tumor sample were included in the study. In addition, 112 pairs of tumor samples and adjacent normal samples were extracted.

The flowchart of CD24 associated ceRNA network construction in breast invasive cancer (BRCA).
Significant role of CD24 in breast cancer
First, our team obtained expression levels of CD24 in normal tissues through GTEx (https://www.gtexportal.org/). Then, we compared the expression level of CD24 between breast cancer samples and adjacent normal samples, and estimated its prognostic value through GEPIA web tool (http://gepia.cancer-pku.cn/). GTEx database contains gene expression data of 54 non-diseased tissues from nearly 1000 individuals37. GEPIA is a web tool, which provides various customizable functions to dig out significant genes, including differential expression analysis, pan-cancer analysis, survival analysis, correlation analysis and so on38.
Screening of differentially expressed genes (DEGs)
To explore the role of CD24 underlying the pathogenesis of breast cancer, our study divided tumor samples into CD24high tumor and CD24low tumor using median expression level (read count = 42,946.5) as a criterion. Then, the current study performed a differential expression analysis between the two groups, and collected differentially expressed lncRNAs (DElncRNAs) with standard of p < 0.05 and |log2 fold change|> 0.3, differentially expressed miRNAs (DEmiRNAs) with standard of p < 0.05 and |log2 fold change|> 0.5 and differentially expressed mRNAs (DEmRNAs) with standard of p < 0.05 and |log2 fold change|> 0.7.
Functional enrichment analysis of DEmRNAs
The R package “clusterprofile” was used to conduct the functional enrichment analysis of DEmRNAs through GO and KEGG database, with the restrictions of p < 0.05 and minimum count of 3. The GO enrichment result was visualized through “GOplot” package, whereas the KEGG pathway enrichment result was visualized by ggplot2 package.
Construction of ceRNA network in BRCA and identification of hub RNAs
According to the hypothesis of ceRNA triple network, lncRNA is the first class of network and regulates the translation level of mRNAs via interacting with miRNA. Hence, the current study firstly explored lncRNA-miRNA pairs with high confidence via miRcode database (http://www.mircode.org). We then predicted mRNA targets of selected miRNAs using TargetScan (http://www.targetscan.org/) and miRDB (http://mirdb.org/), and extracted the same portion of their prediction results. The visualization of ceRNA network was created using Cytoscape 3.8.2, a visualized software used widely. The miRcode database predicts the interactions between lncRNAs and miRNAs based on seed complementarity and evolutionary conservation39. The targetScan database gets putative targets of miRNAs via matching the seed region of miRNAs and conserved 8mer, 7mer and 6mer sites of targets40. The miRDB database provides miRNAs targets that predicted by MirTarget tool, which was developed based on the interaction data of miRNA-target from high-throughput sequencing experiments41.
The filtration of hub genes integrated the results of survival analysis and lncRNA subcellular localization. LncRNA is a classical epigenetic regulative method and have a huge impact on the transcription of mRNA via interacting with miRNA. Hence, we explored hub lncRNAs first. We estimated prognostic value of DElncRNAs in ceRNA using logRank test and univariate Cox regression analyses. According to ceRNA hypothesis, the cellular location of lncRNA is highly correlated with its function. The lncRNA, localized in cytoplasm, can function as a sponge to regulate the expression of downstream genes. We downloaded the sequences of the prognosis associated lncRNAs from the LNCipedia database (https://lncipedia.org/)42, then we predicted the subcellular localization of lncRNAs via lncLocator web tool (http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/)43. This web provided scores for subcellular location, including cytoplasm, cytosol, exosome, nucleus and ribosome, which reflected the possibility of localization of lncRNAs. In addition, we also analyzed the differences of tumor expression levels of key RNAs between tumor samples, normal samples, as well as paired adjacent normal samples in R software. Finally, we evaluated the correlations between key RNAs.
Clinical correlation analysis
In order to further explore the significance of selected key genes in clinical application, the current study estimated the correlation between these genes and the following clinical features: age (< 35, 35–60, ≥ 60), clinical stage and TMN pathological stage.
Immune infiltrate analysis of BEND3 and SIM2
The current study estimated the correlations between immune cell infiltration and hub mRNAs through TIMER database (https://cistrome.shinyapps.io/timer/) and TISIDB database (http://cis.hku.hk/TISIDB/index.php). Additionally, we also applied TISIDB to explore the relationships between immune checkpoint genes and hub mRNAs. TIMER is an online web application that provides researchers with correlation analysis between various immune cells and any gene of interest44. TISIDB provides a web resource for exploring and visualizing the interactions between immune system and genes45.
Methylation analysis of BEND3 and SIM2
The current study applied three online web tools to estimate the methylation levels of BEND3 and SIM2, including UALCAN database (http://ualcan.path.uab.edu/), MEXPRESS database (https://www.mexpress.be/) and MethSurv database (https://biit.cs.ut.ee/methsurv/). UALCAN database provides access to public cancer data, and allows researchers to explore and visualize the correlation of gene expression and promoter methylation46. This database applies beta value to indicate promoter methylation level. Beta value 0.7–0.5 means hypermethylation whereas 0.3–0.25 means hypomethylation47. MEXPRESS is an online tool to analysis and visualize correlation between DNA methylation and clinical information based on TCGA public data48,49. MethSurv is a web tool providing information of single CpG50.
Statistical analysis
The current study performed total statistical analysis via R software version 4.0.3. We performed difference analysis through “DEseq” R package, and performed survival analysis via “survival” R package. The expression levels of hub genes between tumor samples, normal samples and paired adjacent normal samples were estimated by Wilcoxon test. The correlations between hub ceRNA network members were identified by Pearson test. Wilcoxon test was used to compare the expression levels of hub genes between non-metastasis and metastasis groups, and Kruskal–Wallis test was used to estimate the expression differences of hub genes in clinical stage, degree of spread to regional lymph nodes, tumor size and age. *P < 0.05, **P < 0.01, ***P < 0.001.
Result
The tumor promoter role and clinical value of CD24 overexpression in BRCA
In order to investigate the potential role of CD24 in BRCA, the current study explore the RNA expression of CD24 in normal breast tissue and BRCA tissue. We found that CD24 was highly expressed in breast mammary tissues based on GTEx database (Fig. 2A). Furthermore, based on GEPIA database, compared with normal breast tissues, CD24 was found upregulated in BRCA tissues (Fig. 2B).

The function analysis of CD24 in BRCA. (A) Expression distribution of CD24 in various normal tissues using GTEx database. (B) Expression levels of CD24 in BRCA and normal tissues using GEPIA database. (C). Survival analysis of CD24 using GEPIA database.
Given that CD24 was abnormally overexpressed in BRCA tissues, the current study then explored the clinical values of CD24 expression in BRCA patients. The Kaplan–Meier survival curve (Fig. 2C) from GEPIA database showed that higher RNA expression of CD24 was correlated with poor prognostic result (Logrank test, p = 0.00016), and its HR score (HR = 1.9, p = 0.00021) revealed that CD24 served as a risk factor in BRCA.
The identification of DEGs in BRCA
Our above analyses shed a light that CD24 was a key regulatory in BRCA, and the ceRNA network associated with CD24 had a potential to work as prognostic model for BRCA patients. Hence, the current study downloaded RNA sequence data and miRNA data of BRCA from TCGA database and performed further research.
According to the median of CD24 RNA expression, BRCA samples were divided into CD24high and CD24low groups, and we then performed difference analysis between the transcriptomic data of the two groups to identify the DEmRNAs, DElncRNAs and DEmiRNAs. We set p < 0.05 and |log2 fold change|> 0.7 as the DEmRNAs selection criteria, p < 0.05 and |log2 fold change|> 0.3 as the DElncRNAs selection criteria, and p < 0.05 and |log2 fold change|> 0.5 as DEmiRNAs selection criteria. In total, we selected 602 DEmRNAs (290 downregulated and 312 upregulated), 132 DElncRNAs (113 downregulated and 19 upregulated), and 26 DEmiRNAs (7 downregulated and 19 upregulated) from BRCA samples. The volcano plots exhibited the distribution of DElncRNAs, DEmiRNAs and DEmRNAs (Fig. 3A/B/C), and the heatmaps showed the RNA expression of 15 significant DE RNAs (Fig. 3D/E/F).

Differential expression analysis between tumor samples with high expression of CD24 and low expression of CD24. (A–C) Volcano maps of 132 DElncRNAs (p < 0.05 and |log2 fold change| > 0.3), 26 DEmiRNAs (p < 0.05 and |log2 fold change| > 0.5), 602 DEmRNAs (p < 0.05 and |log2 fold change| > 0.7). (D–F) Heatmaps of top 15 DElncRNAs, DEmiRNAs and DEmRNAs (sorted by p value).
Functional enrichment analysis of DEmRNAs
To dig out the potential biological mechanisms related with CD24 involved in BRCA, the current study explored the function of CD24-associated DEmRNAs via Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using ClusterProfiler R package. GO dot plot showed GO terms with gene counts > 3 and p value < 0.05 (Fig. 4A). The most enriched GO term in biological process (BP) was “positive regulation of ion transport digestion”, one in cellular component (CC) was “postsynaptic membrane”, and one in molecular function (MF) were “channel activity”. The lollipop chart exhibited total KEGG pathways with p value < 0.05 (Fig. 4B). Neuroactive ligand-receptor interaction was dominant pathway with highest gene counts (n = 34) and lowest p value (p = 2.2e7).

Functional enrichment analysis of DEmRNAs. (A) Annotation of MF, BP and CC for DEmRNAs. (B) KEGG pathway enrichment analysis of DEmRNAs.
Construction of the ceRNA network and identification of hub RNAs
The current study took use of two databases to identify lncRNA-miRNA-mRNA ceRNA pairs. Finally, 24 lncRNA-miRNA interaction pairs and 29 miRNA-mRNA interaction pairs were identified. The ceRNA network (Fig. 5) which was constructed via Cytoscape 3.8, which included 21 DElncRNAs (6 upregulated and 15 downregulated), 2 DEmiRNAs (2 downregulated) and 29 DEmRNAs (12 upregulated and 17 downregulated).

Construction of CD24-associated ceRNA network.
Through survival analysis, three lncRNAs were identified with P < 0.05 (logRank test) and P < 0.05 (univariate Cox regression analysis) (Fig. 6A). According to the result of survival analysis with logRank test (Fig. 6B), patients with lower expression of RP1-228H13.5 (P = 0.003) and RP11-384L8.1 (P = 0.001) had better prognostic results, whereas those with lower expression of CTD-3247F14.2 had poor prognostic results. Univariate Cox regression analyses showed the similar result (Fig. 6C). RP1-228H13.5 (hazard ratio = 1.247, P = 0.028) and RP11-384L8.1 (hazard ratio = 1.217, P = 0.001) were identified as risk features, whereas CTD-3247F14.2 (hazard ratio = 0.913, P = 0.010) was identified as a favorable factor.

Comprehensive analysis of DElncRNAs. (A) Veen diagram of prognosis-related DElncRNAs in CD24-based ceRNA network. (B) Kaplan-Meier curve analysis of key DElncRNAs. (C) Univariate Cox analysis of key DElncRNAs. (D) Predicted results of subcellular localization of DElncRNAs through lncLocator web tool. (E) The expression levels of key DElncRNAs in tumor samples and paired adjacent normal samples. (F) The expression levels of key DElncRNAs in two groups: CD24high samples and CD24low samples.
The current study predicted the location of three lncRNAs via lncLocator tool. The prediction results of LncLocator web (Fig. 6D) showed that RP1-228H13.5 was mainly located in cytoplasm, CTD-3247F14.2 mainly located in nucleus, and RP11-384L8.1 mainly located in exosome. Additionally, we verified different expression level of lncRNAs in 112 paired BRCA samples from TCGA database. RP1-228H13.5 was upregulated in tumor samples, whereas CTD-3247F14.2 and RP11-384L8.1 were downregulated in tumor samples (Fig. 6E). Figure 6F exhibited expression levels of 3 lncRNAs between CD24high and CD24low groups and there were obvious differences between two groups.
Comprehensive analysis of the above indicated that RP1-228H13.5 was the hub lncRNA in the ceRNA network. Hsa-miR-135a-5p, the target of lncRNA RP1-228H13.5, was comprehensively analyzed. According to survival analysis, hsa-miR-135a-5p was identified as a protective factor (Fig. 7B) and the patients with higher expression level of hsa-miR-135a-5p had better results (Fig. 7A). Moreover, hsa-miR-135a-5p was downregulated in the tumor tissues compared with paired normal tissues (P = 0.0132) (Fig. 7C), and there was significant difference of expression level between CD24high and CD24low groups (Fig. 7D). In contrast, another miRNA, hsa-miR-10a-5p, seemed less important. There was no significant relationship between hsa-miR-10a-5p and prognostic results of patients with BRCA, although there was significant difference in differential analysis of hsa-miR-10a-5p between paired normal and tumor samples (Fig. 7C) as well as between the groups with high CD24 expression and low CD24 expression (Fig. 7D).

Comprehensive analysis of DEmiRNAs. (A) Kaplan-Meier curve analysis of DEmiRNAs. (B) Univariate Cox analysis of DEmiRNAs. (C) The expression levels of DEmiRNAs in tumor samples and paired adjacent normal samples. (D) The expression levels of DEmiRNAs in two groups: CD24high samples and CD24low samples.
Among the target mRNAs of hsa-miR-135a-5p, the current study identified that BEND3 and SIM2 were hub mRNAs. We estimated the prognostic value of target genes via Kaplan–Meier curves (logRank test) and univariate Cox regression analysis. As shown in Fig. 8A, BEND3 and SIM2 were the mRNAs with P < 0.05 in both logRank test and univariate Cox regression analysis. Kaplan–Meier curve showed that the patients of BRCA with lower expression level of BEND3 (LogRank test, P = 0.015) and SIM2 (LogRank test, P = 0,009) had better prognostic results (Fig. 8B). And forest plot exhibited that BEND3 and SIM2 served as risk factors in the development of BRCA (P = 0.003 and P = 0.006 respectively) (Fig. 8C). The expression level of BEND3 in paired tumor and tissue samples had no significant difference (Fig. 8D), whereas the difference of expression level of BEND3 between groups of CD24high and CD24low was significant (Fig. 8E). SIM2 was upregulated in the paired tumor tissues compared with paired normal tissues (P < 0.001) (Fig. 8D), and was downregulated in the CD24low group compared with CD24high group (P = 0.0014) (Fig. 8E).

Comprehensive analysis of DEmRNAs. (A) Veen diagram of prognosis-related DEmRNAs in CD24-based ceRNA network. (B) Kaplan-Meier curve analysis of key DEmRNAs. (C) Univariate COX analysis of key DEmRNAs. (D) The expression levels of key DEmRNAs in tumor samples and paired adjacent normal samples. (E) The expression levels of key DEmRNAs in two groups: CD24high samples and CD24low samples.
To sum up the above analysis, we identified that RP1-228H13.5, hsa-miR-135a-5p, BEND3 and SIM2 composed hub ceRNA pair which was highly associated with CD24 (Fig. 9A). The binding sites of RP1-228H13.5, BEND3, SIM2 and hsa-miR-135a-5p were shown in Fig. 9B. Then, we performed expression correlation analysis for ceRNA network (Fig. 9C). It was confirmed that RP1-228H13.5 was positively correlated with BEND3 (R = 0.613, P < 0.001) and SIM2 (R = 0.204, P < 0.001). What’s more, CD24 was found highly correlated with genes of ceRNA network as well.

The identification significant ceRNA axis. (A) Schematic diagram of significant ceRNA axis. (B) The sequence of significant genes in ceRNA network. (C) Co-expression analysis of significant genes in ceRNA network.
Clinical relevance of ceRNA network in BRCA patients
To further explore clinical relevance of ceRNA network, the current study analyzed the relationships between hub RNAs (RP1-228H13.5, hsa-miR135a-5p, BEND3 and SIM2) and various clinical features, such as age, clinical stage, tumor status, distant metastasis and lymph code metastasis. The ceRNA network was found highly related with lymph code metastasis (Fig. 10C) and patients’ age (Fig. 10E), but was rarely related with other clinical information (clinical stage, M/T stage) (Fig. 10A/B/D).

Clinical correlation analysis of key genes in ceRNA network. (A–E) Correlation analysis between key genes and clinical stage, pathological metastasis stage, pathological N stage, pathological T stage and age at initial pathologic diagnosis.
Relationship between immune infiltration and expression levels of BEND3/SIM2
Immune infiltration is a mature factor involved in the initiation, development and prognostic of tumors. Hence, we further explored the relationships between immune infiltration and expression of BEND3/SIM2 via TIMER database. As shown in Fig. 11A/C, several immune cells are correlated with the altered copy number of BEND3/SIM2. Figure 11B/D exhibited the relationship between BEND3/SIM2 and tumor purity, B cells, CD8 + T cell, CD4 + T cell, Macrophage, Neutrophil and Dendritic cell. BEND3 was correlated with CD8 + T cell to a certain extent (Partial.cor = 0.17, p = 9.78e−08), whereas SIM2 had no notable correlation with these immune cells.

Comprehensive analysis of BEND3 (A, B) and SIM2 (C, D) and tumor immune microenvironment based on the TIMER database.
Then, based on TISIDB database, we explored the relationship between BEND3/SIM2 expression and abundance of 28 tumor-infiltrating lymphocytes (TILs) (Figs. 12, 13). We found that BEND3 had notable correlation with Act CD4 + T cell (r = 0.338, P < 2.2e−16), Th2 cell (r = 0.24, P = 7.8e−16), NK cell (r = − 0.259, P = 3.08e−18), pDC, Eosinophil cell (− 0.254, P = 1.6e−17) and Neutrophil cell (r = − 0.252, P = 2.47e−17). On the contrast, SIM2 had no notable correlation with TILs.

Comprehensive analysis of BEND3 and tumor immune microenvironment based on the TISIDB database. (A) The correlations of BEND3 with immune cells across cancers. (B–U) The significant correlations between BEND3 and immune cells in BRCA.

Comprehensive analysis of SIM2 and tumor immune microenvironment based on the TISIDB database. (A) The correlations of SIM2 with immune cells across cancers. (B–K) The significant correlations between SIM2 and immune cells in BRCA.
An increasing number of evidences has proved that immune checkpoint inhibitor (ICI) is a significant immunotherapy strategy with great prospects. Through searching in TISIDB database, we got the correlations between BEND3/SIM2 and over 40 common immune checkpoint genes in BRCA (Fig. 14). We found that BEND3 had highly negative correlation with TGFβ1 (r = − 0.363, P < 2.2e−16), whereas showed significantly positive correlation with PVR (r = 0.329, P < 2.2e−16) and ULBP1 (r = 0.377, P < 2.2e−16). SIM2 exhibited significantly positive correlation with PVR (r = 0.252, P = 2.66e−17) and ULBP1 (r = 0.279, P = 5.82e−21).

Comprehensive analysis of BEND3 and SIM2 and various immune checkpoints based on the TISIDB database. (A–B) The correlations of BEND3 with immune inhibitor genes and immune stimulator genes. (C–D) The correlations of SIM2 with immune inhibitor genes and immune stimulator genes.
Relationship between methylation and expression of BEND3/SIM2
In order to further explore the dysregulated mechanisms of BEND3/SIM2, the current study performed an analysis for their methylation status. We found that there was no significant difference between the promoter methylation level of BEND3 in normal tissues and primary tumor tissues (Fig. 15A). However, the promoter methylation level of SIM2 in BRCA tissues was significantly higher than normal tissues (Fig. 16A). The methylation of BEND3 occurred on some CpG islands, including cg23339693, cg23796329 (r = − 0.209, − 0.314, respectively) (Fig. 15C). In the meanwhile, the methylation of SIM2 occurred on a great number of sites (Fig. 16C). Figures 15B and 16B, respectively, showed the correlations of methylation sites of BEND3 and SIM2 with BRCA clinical factors.

Analysis of BEND3 and methylation. (A) The promoter methylation level of BEND3 in BRCA. (B) The correlations between methylation sites of BEND3 and clinical features in BRCA. (C) The methylation information of CpG islands of BEND3.

Analysis of SIM2 and methylation. (A) The promoter methylation level of SIM2 in BRCA. (B) The correlations between methylation sites of SIM2 and clinical features in BRCA. (C) The methylation information of CpG islands of SIM2.
Discussion
Breast cancer, the most common malignancy in women, is considered potentially curable in early stage, and molecular targeted therapy has been confirmed significantly helpful in BRCA patients. However, only a minority of patients appear to respond. Hence, it is a top priority to find new therapy targets and novel biomarkers. Many studies have reported that ceRNA mechanism is involved in the initiation and development of breast cancer and has a potential to work as therapy targets or prediction factors51.
CD24 is a membrane protein overexpressed in many solid tumors, and has a role in promoting immune evasive via interacting with sialic-acid-binding Ig-like lectin 10 (Siglec-10), which is expressed by tumor-associated macrophages52. In various cancers, CD24 expressed by cancer cells is described as “do not eat me” signal, which is identified associated with tumor growth in vitro and in vivo. What’s more, CD24 also serves as cancer stemness marker53. However, to our best knowledge, the ceRNA network associated with CD24 remains unknown. In this study, we downloaded RNAseq data of BRCA from TCGA database, divided them into CD24high and CD24low groups based on the median expression level of CD24, and performed a difference analysis. Then we constructed a ceRNA network using 21 DElncRNAs, 2 DEmiRNAs and 29 DEmRNAs.
lncRNA is dominant in the ceRNA triple regulatory network. Accordingly, we first analyzed DElncRNAs. Prognostic analysis reflected that RP1-228H13.5, CTD-3247F14.2 and RP11-384L8.1 were the prognostic signatures, among which RP1-228H13.5 was the only lncRNA which was predicted located in cytoplasm. What’s more, RP1-228H13.5 was upregulated in BRCA tumor tissues compared with paired adjacent tissues, and also had higher RNA expression levels in CD24low group compared with CD24high group. RP1-228H13.5 has been studied in some cancers. A study reported that RP1-228H13.5, referred in that study as CRAL, is downregulated in cisplatin-resistance in gastric cancer (GC) cells and contributes to cisplatin-resistant in GC cells via impairing cisplatin-induced DNA damage as well as cell apoptosis through the miR-505/CYLD/AKT axis54. Another study as well reported that the RNA expression level of RP1-228H13.5 is associated with HCC patients’ overall and recurrence-free survival through performing a data mining using RNA sequencing data from TCGA and two microarray datasets from GEO55. However, too little work has been devoted to the role of RP1-228H13.5 in breast cancer.
According to the ceRNA hypothesis, miRNA is the bridge connecting lncRNA and mRNA in various biological processes. In our study, hsa-miR-135a-5p was found downregulated in BRCA tissues and was identified as a protective factor. The role of hsa-miR-135a-5p has been explored in various cancers, including lung cancer, hepatocarcinogenesis, tongue squamous cell carcinoma, and ovarian cancer56,57,58,59. Nevertheless, its function is different in different tumors. For instance, hsa-miR-135a-5p is upregulated in NSCLC tissues and its loss can restrain lung cancer via interacting with LOXL456. On the contrast, hsa-miR-135a-5p is reported downregulated in ovarian cancer tissues and functions as a tumor suppressor in ovarian cancer via binding CCR259. Similar with RP1-228H13.5, the function of hsa-miR-135a-5p in BRCA remains largely under-explored.
Messenger RNA is able to perform specific biological activities via coding proteins. CD24-associated mRNAs were performed GO enrichment analysis and KEGG pathway analysis, and were found highly correlated with channel activity.
BEND3 is a transcriptional repressor which associated with the nucleolar remodeling complex (NoRC) and plays a key role in repressing rDNA transcription. In GO database, BEND3 is annotated by “rDNA heterochromatin assembly”60,61. It is reported that BEND3 is associated with NuRD, NoRC and PRC2 complexes, and plays an important role in the transcription regulatory62. Recently, in acute myeloid leukemia, BEND3 is identified as a key gene whose deletion results in resistance to TAK-243, a first-in-class inhibitor of ubiquitin-like modifier activating enzyme 1, via regulating breast cancer resistance protein (BCRP)63. The impact of BEND3 in breast cancer has been observed as well. In 2017, a study has confirmed that BEND3 is dysregulated in Basal-like breast cancer64. Moreover, BEND3 is identified upregulated in BRCA cancers with visceral organ metastasis65. However, the molecular mechanism of BEND3 in the initiation, development of breast cancer remains unclear. Although our study revealed that methylation had rare influence on BEND3 in BRCA, BEND3 was reported having a huge impact on methylation level of genes via binding CpG island. Recently, researchers found that BEND3 occupies active (H3K4me3) and bivalent promoters (H3K4me3 and H3K27me3), and BEND3-bound regions have lower CpG methylation level compared with the unbound sites66. Although gene transcription level is usually considered to be negatively correlated with DNA methylation level of promoter in the past, more and more studies support that the high methylation level of promoter can induce gene expression. For example, CD24, the promoter of which exhibited high DNA methylation level, was confirmed overexpressed67. Given complex epigenetic regulatory mechanisms, we guess that BEND3 can promote CD24 expression via hypermethylating its promoter, which deserves further research. BEND3 also exhibited high correlation with the infiltration levels of B cell, CD8 + T cell, Neutrophil and Dendritic cell. In addition, the current study explored the correlation between BEND3 and immune check points. We found that BEND3 was highly negatively associated with TGFβ1, TNFSF13, TNFRSF14, CXCL12, whereas was significantly positively correlated with ULBP1, PVR, TNFRSF13c. In summary, BEND3 is a significant gene highly associated with CD24 in breast cancer, and serves as a potential therapeutic target.
SIM2 encodes a transcription factor that is a master regulator of neurogenesis. SIM2 firstly is well-known as a Down’s syndrome critical locus gene, whereas then is found associated with some solid tumors, for which SIM2 is a promising novel treatment target68. As a transcription factor, SIM2 is involved in tumors via diverse molecular mechanisms. For example, in pancreatic cancer, the silence of SIM2-s (the short form of SIM2) significantly induces CAPAN-1 cell death via apoptosis69. SIM2 is identified overexpressed in prostate cancer tissues as well, and works as a potential target for the immunotherapy of prostate cancer70. Many studies reported that SIM2 plays a significant role in breast cancer. KC Scribner et al. confirmed that SIM2s regulate the progression of primary breast ductal carcinoma to invasive breast cancer through epithelial mesenchymal transition (EMT)71. A novel role of SIM2s is demonstrated by a study in 2019, in which SIM2s is found to attenuate the expression of COX2/PTGS2 via mediating NFκB signaling72. In our study, we found the promoter methylation level of SIM2 in primary tumor was upregulated. However, this methylation level still belongs to hypomethylated level, which could not explain the change of SIM2. In addition, many CpG islands of SIM2 were identified. Combined with previous studies, we hypothesize that the methylation of SIM2 is a potential key regulation mechanism in the procession of breast cancer. We also found that SIM2 gene copy numbers were associated with the infiltration level of CD4 + T cells and macrophages. Some immune cells also showed associations with the expression level of SIM2.
Although we explored the role of CD24-related ceRNA regulator network in breast cancer from multiple dimensions, there were limitations to be considered in this study. First, the dataset from TCGA was the only data analyzed in this study, and more datasets from different sources needed to be included in the analysis. Second, key ceRNA interactions need to be validated by experiments in the further study. Finally, the roles of BEND3 and SIM2 in tumor immune microenvironment in breast cancer was obtained using online web tools, which requires more experimental validations.
Conclusion
Using the RNA sequence data and clinical information form TCGA database, we established a CD24-asssociated ceRNA network (RP1-228H13.5/miR-135a-5p/BEND3 and SIM2) and made a comprehensive analysis via a diversity of bioinformatic tools, which shed a new light on the mechanism of BRCA. These novel critical biomarkers were significantly related with prognosis, and had an impact on tumor immune microenvironment (Supplementary information).
Abbreviations
- BRCA:
-
Breast invasive carcinoma
- TNBC:
-
Triple-negative breast cancer
- ncRNA:
-
Non-coding RNA
- circRNA:
-
NcRNAs comprise circular RNA
- lncNRA:
-
Long non-coding NRA
- miRNA:
-
MicroNRA
- piRNA:
-
PIWI interacting RNA
- TCGA:
-
The Cancer Genome Atlas
- DEGs:
-
Differentially expressed genes
- DElncRNAs:
-
Differentially expressed lncRNAs
- DEmiRNAs:
-
Differentially expressed miRNAs
- DEmRNAs:
-
Differentially expressed mRNAs
- GO:
-
Gene Ontology
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GC:
-
Gastric cancer
- TILs:
-
Tumor-infiltrating lymphocytes
- NoRC:
-
Nucleolar remodeling complex
- BCRP:
-
Breast cancer resistance protein
- EMT:
-
Epithelial mesenchymal transition
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249 (2021).
Siegel, R. L. et al. Cancer statistics, 2022. CA Cancer J. Clin. 72(1), 7–33 (2022).
Harbeck, N. & Gnant, M. Breast cancer. The Lancet 389(10074), 1134–1150 (2017).
Prat, A. et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 24(Suppl 2), S26–S35 (2015).
Goldhirsch, A. et al. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 24(9), 2206–2223 (2013).
Ades, F. et al. Luminal B breast cancer: Molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 32(25), 2794–2803 (2014).
Trapani, D., Rajasekar, A. K. A. & Mathew, A. More options for adjuvant treatment of HER2-positive breast cancer: How to choose wisely?. Int. J. Cancer 145(11), 2901–2906 (2019).
Yin, L. et al. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 22(1), 61 (2020).
Bai, X. et al. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel?. Cancer Lett. 497, 100–111 (2021).
Yeo, S. K. & Guan, J. L. Breast cancer: Multiple subtypes within a tumor?. Trends Cancer 3(11), 753–760 (2017).
Waks, A. G. & Winer, E. P. Breast cancer treatment: A review. JAMA 321(3), 288–300 (2019).
Teshome, M. & Kuerer, H. M. Breast conserving surgery and locoregional control after neoadjuvant chemotherapy. Eur. J. Surg. Oncol. 43(5), 865–874 (2017).
Emens, L. A. Breast cancer immunotherapy: Facts and hopes. Clin. Cancer Res. 24(3), 511–520 (2018).
Barzaman, K. et al. Breast cancer immunotherapy: Current and novel approaches. Int. Immunopharmacol. 98, 107886 (2021).
Heeke, A. L. & Tan, A. R. Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 40(2), 537–547 (2021).
Dees, S. et al. Emerging CAR-T cell therapy for the treatment of triple-negative breast cancer. Mol. Cancer Ther. 19(12), 2409–2421 (2020).
Liu, L. et al. Combination immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther. 26(1), 45–55 (2018).
Marra, A., Viale, G. & Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 17(1), 90 (2019).
Mediratta, K. et al. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers (Basel) 12(12), 3529 (2020).
Panagiotou, E. et al. CD24: A novel target for cancer immunotherapy. J. Pers. Med. 12(8), 1235 (2022).
Wu, H. et al. Prospects of antibodies targeting CD47 or CD24 in the treatment of glioblastoma. CNS Neurosci. Ther. 27(10), 1105–1117 (2021).
Li, O. et al. Massive and destructive T cell response to homeostatic cue in CD24-deficient lymphopenic hosts. J. Exp. Med. 203(7), 1713–1720 (2006).
Huth, H. W. et al. Translocation of intracellular CD24 constitutes a triggering event for drug resistance in breast cancer. Sci. Rep. 11(1), 17077 (2021).
Li, L. et al. ZBTB28 inhibits breast cancer by activating IFNAR and dual blocking CD24 and CD47 to enhance macrophages phagocytosis. Cell Mol. Life Sci. 79(2), 83 (2022).
Anastasiadou, E., Jacob, L. S. & Slack, F. J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 18(1), 5–18 (2018).
Yan, H. & Bu, P. Non-coding RNA in cancer. Essays Biochem. 65(4), 625–639 (2021).
Wang, J. et al. ncRNA-encoded peptides or proteins and cancer. Mol. Ther. 27(10), 1718–1725 (2019).
Chan, J. J. & Tay, Y. Noncoding RNA:RNA regulatory networks in cancer. Int. J. Mol. Sci. 19(5), 1310 (2018).
Thomson, D. W. & Dinger, M. E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 17(5), 272–283 (2016).
Braga, E. A. et al. LncRNAs in ovarian cancer progression, metastasis, and main pathways: ceRNA and alternative mechanisms. Int. J. Mol. Sci. 21(22), 8855 (2020).
Qi, X. et al. ceRNA in cancer: Possible functions and clinical implications. J. Med. Genet. 52(10), 710–718 (2015).
Zhang, Z. et al. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J. Exp. Clin. Cancer Res. 38(1), 166 (2019).
Wang, Y. et al. The emerging function and mechanism of ceRNAs in cancer. Trends Genet. 32(4), 211–224 (2016).
Zhang, X. et al. The novel long noncoding RNA CRART16 confers cetuximab resistance in colorectal cancer cells by enhancing ERBB3 expression via miR-371a-5p. Cancer Cell Int. 20, 68 (2020).
Zhang, P. et al. MicroRNA-3064-5p sponged by MALAT1 suppresses angiogenesis in human hepatocellular carcinoma by targeting the FOXA1/CD24/Src pathway. FASEB J. 34(1), 66–81 (2020).
Ni, C. et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73 + gammadelta1 Treg cells. Signal Transduct. Target Ther. 5(1), 41 (2020).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45(6), 580–585 (2013).
Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45(W1), W98–W102 (2017).
Jeggari, A., Marks, D. S. & Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 28(15), 2062–2063 (2012).
McGeary, S. E. et al. The biochemical basis of microRNA targeting efficacy. Science 366(6472), eaav1741 (2019).
Chen, Y. & Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 48(D1), D127–D131 (2020).
Volders, P. J. et al. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 47(D1), D135-d139 (2019).
Cao, Z. et al. The lncLocator: A subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 34(13), 2185–2194 (2018).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77(21), e108–e110 (2017).
Ru, B. et al. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 35(20), 4200–4202 (2019).
Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19(8), 649–658 (2017).
Men, C. et al. Identification of DNA methylation associated gene signatures in endometrial cancer via integrated analysis of DNA methylation and gene expression systematically. J. Gynecol. Oncol. 28(6), e83 (2017).
Koch, A. et al. MEXPRESS: Visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 16(1), 636 (2015).
Koch, A. et al. MEXPRESS update 2019. Nucleic Acids Res. 47(W1), W561–W565 (2019).
Modhukur, V. et al. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10(3), 277–288 (2018).
Lu, G. et al. Long noncoding RNA LINC00511 contributes to breast cancer tumourigenesis and stemness by inducing the miR-185-3p/E2F1/Nanog axis. J. Exp. Clin. Cancer Res. 37(1), 289 (2018).
Barkal, A. A. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572(7769), 392–396 (2019).
Altevogt, P. et al. Novel insights into the function of CD24: A driving force in cancer. Int. J. Cancer 148(3), 546–559 (2021).
Wang, Z. et al. The long noncoding RNA CRAL reverses cisplatin resistance via the miR-505/CYLD/AKT axis in human gastric cancer cells. RNA Biol. 17(11), 1576–1589 (2020).
Cui, H. et al. A comprehensive genome-wide analysis of long noncoding RNA expression profile in hepatocellular carcinoma. Cancer Med. 6(12), 2932–2941 (2017).
Cao, G. et al. Down-regulation of MBNL1-AS1 contributes to tumorigenesis of NSCLC via sponging miR-135a-5p. Biomed. Pharmacother. 125, 109856 (2020).
Van Renne, N. et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 67(5), 953–962 (2018).
Zheng, Y. et al. LncRNA DANCR promotes the proliferation, migration, and invasion of tongue squamous cell carcinoma cells through miR-135a-5p/KLF8 axis. Cancer Cell Int. 19, 302 (2019).
Duan, S. et al. MicroRNA-135a-3p is downregulated and serves as a tumour suppressor in ovarian cancer by targeting CCR2. Biomed. Pharmacother. 107, 712–720 (2018).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25(1), 25–29 (2000).
Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49(D1), D325–D334 (2021).
Khan, A. & Prasanth, S. G. BEND3 mediates transcriptional repression and heterochromatin organization. Transcription 6(5), 102–105 (2015).
Barghout, S. H. et al. A genome-wide CRISPR/Cas9 screen in acute myeloid leukemia cells identifies regulators of TAK-243 sensitivity. JCI Insight 6(5), e141518 (2021).
Milioli, H. H. et al. Basal-like breast cancer: Molecular profiles, clinical features and survival outcomes. BMC Med. Genom. 10(1), 19 (2017).
Savci-Heijink, C. D. et al. A specific gene expression signature for visceral organ metastasis in breast cancer. BMC Cancer 19(1), 333 (2019).
Zhang, J. et al. Highly enriched BEND3 prevents the premature activation of bivalent genes during differentiation. Science 375, 1053–1058 (2022).
Tolkach, Y. et al. DNA promoter methylation and ERG regulate the expression of CD24 in prostate cancer. Am. J. Pathol. 191(4), 618–630 (2021).
Ratan, R. R. Mining genome databases for therapeutic gold: SIM2 is a novel target for treatment of solid tumors. Trends Pharmacol. Sci. 24(10), 508–510 (2003).
DeYoung, M. P., Tress, M. & Narayanan, R. Down’s syndrome-associated Single Minded 2 gene as a pancreatic cancer drug therapy target. Cancer Lett. 200(1), 25–31 (2003).
Arredouani, M. S. et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin. Cancer Res. 15(18), 5794–5802 (2009).
Scribner, K. C., Behbod, F. & Porter, W. W. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s). Oncogene 32(21), 2631–2639 (2013).
Wyatt, G. L. et al. Cross-talk between SIM2s and NFkappaB regulates cyclooxygenase 2 expression in breast cancer. Breast Cancer Res. 21(1), 131 (2019).
Acknowledgements
We appreciate TCGA database for providing detailed RNA sequence data and clinical information.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
T.X., H.L. and B.Y. designed this study. B.Y., Z.Z., Z.C., P.W., B.X., Y.X. and A.L. conducted this study, including data collection, data analysis, manuscript writing. All authors contributed to the final approval of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, B., Zhao, Z., Chen, Z. et al. CD24-associated ceRNA network reveals prognostic biomarkers in breast carcinoma. Sci Rep 13, 3826 (2023). https://doi.org/10.1038/s41598-022-25072-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-022-25072-x
This article is cited by
-
The role of ceRNAs in breast cancer microenvironmental regulation and therapeutic implications
Journal of Molecular Medicine (2025)
