
Abstract
Neovascularization is a prominent cause of irreversible blindness in a variety of ocular diseases. Current therapies for pathological neovascularization are concentrated on the suppression of vascular endothelial growth factors (VEGF). Despite the remarkable efficacy of anti-VEGF drugs, several problems still exist, including ocular complications and drug resistance. Thus, it is still required to design novel drugs for anti-angiogenic treatment. This study aimed to investigate the anti-angiogenic effects of a small molecule multi-target tyrosine kinase inhibitor, DCZ19931, on ocular neovascularization. The results showed that administration of DCZ19931 at the tested concentrations did not cause obvious cytotoxicity and tissue toxicity. DCZ19931 could reduce the size of choroidal neovascularization (CNV) lesions in laser-induced CNV model and suppress ocular neovascularization in oxygen-induced retinopathy (OIR) model. DCZ19931 could suppress VEGF-induced proliferation, migration, and tube formation ability of endothelial cells, exhibiting similar anti-angiogenic effects as Ranibizumab. DCZ19931 could reduce the levels of intercellular cell adhesion molecule-1 (ICAM-1) expression in vivo and in vitro. Network pharmacology prediction and western blots revealed that DCZ19931 exerted its anti-angiogenic effects through the inactivation of ERK1/2-MAPK signaling and p38-MAPK signaling. In conclusion, this study indicates that DCZ19931 is a promising drug for anti-angiogenic therapy for ocular diseases.
Similar content being viewed by others

Introduction
Pathological neovascularization occurs in a wide range of human diseases, including cancers, rheumatoid arthritis, and proliferative retinopathies1. In the eye, pathological neovascularization is a critical feature of ocular diseases, such as age-related macular degeneration (AMD), diabetic retinopathy (DR), and retinopathy of prematurity (ROP)2. Vascular dysfunction would affect ocular microenvironment and cause local ischemia, eventually causing hemorrhage, detachment, fibrosis, and blindness. Thus, inhibiting pathological neovascularization is an effective therapeutic strategy for improving the vision of patients3.
During pathological neovascularization, the balance between angiogenic stimulators and angiogenic inhibitors is often interrupted. A series of pro-angiogenic factors are highly induced in pathological neovascularization, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and matrix metalloproteinases (MMPs)4. Notably, VEGF acts as a pro-angiogenic factor in endothelial cells (ECs), which plays important roles in blood-vessel formation during vascular development and abnormal growth of blood vessels in diseases5. Increased VEGF expression can activate ECs, contributing to EC proliferation, migration, and tube formation. During pathological neovascularization, ECs can degrade the basement membrane through the action of extracellular proteases. Then, activated ECs migrate into extracellular matrix, proliferate induced by angiogenic stimulus, and form the tubules with central lumens. Hence, intervention of VEGF activity is an efficient therapeutic strategy for pathological neovascularization6,7.
At present, anti-VEGF medication is still the primary and first-line modality for treating ocular vascular diseases. These drugs include anti-VEGF antibodies and vascular endothelial growth factor receptor (VEGFR) fragment domain fusion proteins. However, anti-VEGF medication still has several unsatisfactory clinical consequences. Some patients have anti-VEGF drug resistance or have no response for anti-VEGF treatment8. Therefore, in-depth studies for developing new drugs are still required to improve the anti-angiogenic efficiency for ocular vascular diseases9.
Angiogenesis signaling networks are often highly complicated. To obtain a sustained and effective clinical efficiency, it is necessary to target multiple signaling targets synchronously. Multi-target kinase inhibitors (MKIs) are a series of compounds, which can simultaneously restrain the activation of multiple signaling kinases that control cell growth, proliferation, differentiation, motility, and apoptosis. To date, several MKIs have been verified to inhibit tumor growth and tumor angiogenesis10,11,12. MKIs also show great therapeutic efficiency against metabolic and autoimmune disorders, such as myelofibrosis, rheumatoid arthritis, and chronic myeloid leukemia13. For example, sorafenib is a dual aryl urea multi-kinase inhibitor and the first molecule-targeted drug approved for the treatment of hepatocellular carcinoma, which exerts anti-tumor and anti-angiogenic effects via inhibiting multiple targets such as VEGFR, PDGFR-β, and other serine/threonine kinases14. However, there are few reports about the application of MKIs in the treatment of ocular vascular diseases. In addition, although these MKIs have been widely used, several unfavorable side effects still exists during the clinical applications of MKIs. For instance, MKI drugs often have low absorption efficiency in tumors due to poor water solubility, rapid clearance, and metabolic rate. The non-specific uptake of MKIs by normal tissues further aggravate the adverse effects13. In this study, we designed and synthesized a new multi-kinase inhibitor, DCZ19931, and investigated its anti-angiogenic role and anti-angiogenic mechanism in ocular vascular diseases.
Materials and methods
Animals and ethical statement
C57BL/6J (wild-type, WT) mice were bred at constant temperature (25 °C) with alternating 12 h light–dark cycle and had free access to standard chow and clean water. All experiments were approved by the Animal Care and Use Committee of Nanjing Medical University. The experiments were carried out in compliance with the ARRIVE guidelines (https://arriveguidelines.org) and all methods were performed according to relevant guidelines and regulations.
Synthesis of DCZ19931
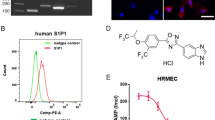
DCZ19931 (MW = 549.5) was designed and synthesized by Shanghai Institute of Materia Medica (Shanghai, China). The synthesis procedure of DCZ19931 was outlined in Fig. 1. Ethyl 2, 2-difluoro-2-(4-hydroxyphenyl) acetate (compound 1), 4-chloro-6,7-bis (2-methoxy) quinazoline (compound 2), triethylenediamine, and triethylamine were dissolved in acetonitrile (5 mL), followed by the reaction at 80 °C for 2 h. The organic solvent was evaporated by rotary evaporation. Compound 3 was obtained by silica gel column chromatography.

Synthesis of DCZ19931.
Compound 3 was dissolved in the mixture of tetrahydrofuran, methanol, and water. After the addition of 1 mol/L sodium hydroxide solution, the solution was stirred at room temperature for 2 h. The reaction solution was neutralized with 1 mol/L hydrochloric acid solution to pH 1 and extracted with ethyl acetate. The organic phases were washed with water and saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and evaporated to remove the solvent to produce compound 4.
Compound 4, 3-methoxy-5-(trifluoromethyl) aniline (Compound 5), N, N-diisopropylethylamine and 2-(7-azobenzotriazole)-N,N,N′,N′-tetramethylurea hexafluorophosphate were dissolved in acetonitrile and stirred at room temperature for 12 h. Then, the reaction mixture was quenched with water and extracted by ethyl acetate. The organic was separated by silica gel column chromatography to produce DCZ19931.
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were obtained from American Type Culture Collection (ATCC). They were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, C11995, USA), supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. They were pre-treated with VEGF (10 ng/mL, R&D System, 293-VE-010, USA) to mimic endothelial angiogenic effects in vitro.
Cell viability assay
Cell viability was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) colorimetric assay. HUVECs were treated with DCZ19931 for 24 h, and then incubated with MTT solution (5 mg/mL, BioFroxx, 1334GR001, Germany) at 37 °C for 3 h. Subsequently, isopropanol was used to dissolve the crystals. The optical density (OD) value of the absorption was detected by a microplate reader (Molecular Devices, FilterMax F5, USA).
Flow cytometry
Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme, A211-01, China) was used to detect cell apoptosis. After the required treatment, HUVECs were digested with 0.05% trypsin solution (EDTA-free) for 20 min and washed with PBS for 3 times. Subsequently, they were suspended in 200 µL binding buffer with Annexin-V-FITC (5 μL) and propidium iodide (PI, 5 μL) for 10 min in dark. Finally, cell apoptosis was detected by a flow cytometer (Beckman Coulter, CytoFLEX, USA).
Calcein-AM/PI staining
Calcein-AM and PI double staining was conducted to examine cell apoptosis. HUVECs were seeded onto 6 well plates and cultivated in 5% CO2 incubator at 37 °C for 24 h. Then, they were incubated with the indicated concentrations of DCZ19931 for 24 h. HUVECs were incubated with the mixture of Calcein-AM and PI to label live cells or apoptotic cells at 37 °C for 30 min. Hoechst staining was used to label cell nuclei. The images were captured by a fluorescence microscope (Olympus, IX73, Japan) and analyzed with Image J software.
Transwell migration assay
Cell migration ability was evaluated by the transwell migration assays. After digestion, HUVECs were re-suspended with the serum-free DMEM and added to the upper chambers of transwell (Millipore, MCEP24H48, USA) and adjusted to 5 × 105/mL. Then, the lower chamber was filled with the complete media (10% FBS in DMEM). The cells were allowed to migrate for 12 h at 37 °C and 5% CO2. Then, the migrated cells were fixed with methanol for 15 min, washed three times with PBS for 5 min, and stained with 0.5% crystal violet for 30 min at room temperature. These non-migrated cells were gently wiped with cotton swabs. The migrated cells were counted in 6 different areas under the optical microscope (Olympus, IX73, Japan) and the average number was calculated.
5-Ethynyl-20-deoxyuridine (EdU) assay
EdU assays were used to detect cell proliferation by BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime, C0071S, China). After the specific treatment, HUVECs were incubated with EdU (10 μM) for 4 h. Then, they were fixed with 4% paraformaldehyde (PFA) for 30 min, permeabilized with 0.3% Triton X-100 for 15 min, and rinsed with 3% bovine serum albumin (BSA) in PBS for 3 times. Finally, HUVECs were incubated with the click reaction solution in dark for 30 min. Cell nuclei were stained by DAPI (1:1500, Beyotime, C1002, China) at room temperature for 30 min. The images were captured by a microscope (Olympus, IX73, Japan).
Tube formation assay
The well of 24-well plate was pre-coated with 50 μL of Matrigel basement membrane Matrix (Corning, 354234, USA) and incubated at 37 °C and 5% CO2 for 1 h to solidify. HUVECs were seeded onto the plated Matrigel (3 × 105 cells per well) in serum-free DMEM medium and incubated at 37 °C. Images of the formation of capillary-like structures were observed after 8-h culture a computer-assisted microscope (Olympus, IX73, Japan).
Laser-induced choroidal neovascularization (CNV) model
Laser-induced CNV models were built using C57BL/6J mice (8-week-old, male). Briefly, the mice were anesthetized with the mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). The pupils were dilated using 0.5% tropicamide. Next, four lesions surrounding the optic discs were induced by laser photocoagulation with the wavelength of 532 nm, the spot size of 50 μm, duration of 100 ms, and power of 120 mW. The lesions were induced at the 3, 6, 9, and 12 o’clock positions, which were 2–3 papillary diameters away from the optic discs. The successful disruption of Bruch’s membrane was confirmed by white bubble formation. Following laser injury, the mice were injected with 10% DMSO (2 μL, Ctrl), DCZ19931 (2 μL, 1 μg/μL), Ranibizumab (2 μL, 10 mg/mL), or 2 μL mixture solution of DCZ19931 and Ranibizumab, respectively. At day 7 after laser injury, the lesions on flat-mounted RPE/choroid were observed by immunofluorescence staining and HE staining.
Oxygen-induced retinopathy (OIR) model
The newborn C57BL/6J mice and their nursing mothers were placed in a 75% oxygen supply chamber from P7 to P12, and then were returned to room air for 5 days. The mice received an intravitreal injection of 1 μL of 10% DMSO (Ctrl), 1 μL of DCZ19931 (1 μg/μL), 1 μL of Ranibizumab (10 mg/mL), or 1 μL mixture solution of DCZ19931 (1 μg/μL) plus Ranibizumab (10 mg/mL) immediately when returning to room air at P12. They were anesthetized and the eyeballs were removed at P17. The retinas were stained with Alexa Fluor 488-conjugated isolectin GS-IB4 (1:100, Invitrogen, I21411, USA).
Choroidal flat-mount and isolectin-B4 staining
At day 7 following laser injury, the eyeballs were enucleated and fixed in 4% PFA for 30 min at room temperature. After the removals of cornea, lens, and retina, the eyecup was cut into 4 petals. Following washing with PBS, the flat-mount choroidal tissue was blocked with 5% BSA for 1 h at 37 °C and incubated with Isolectin-B4 (1:100, L2895, Sigma-Aldrich, USA) to label the neovascular area overnight at 4 °C. The images were photographed by a fluorescence microscope (Olympus, IX73, Japan).
Hematoxylin and eosin (HE) staining
The histopathological change of ocular structure was observed by hematoxylin–eosin staining. Briefly, the sections were dewaxed with xylene and rehydrated with the graded concentrations of ethanol. Subsequently, the sections were successively stained with hematoxylin (1 min) and eosin (30 s). The images were taken to detect the change of ocular structures.
Choroid sprouting assay
Choroidal explants containing RPE/choroid/sclera complexes were cut into 1 mm × 1 mm pieces. They were embedded in 40 μL of Matrigel in 24-well plates. Choroid sprouting was observed at 4 × magnification on day 4, day 5, and day 6. The sprouting area was quantified using Image J software.
Terminal deoxynucleotidyl-transferase-mediated dUTP nick end labeling (TUNEL) staining
The apoptosis was detected by TUNEL assay using the In Situ Cell Detection Kit (Beyotime, C1088, China). After the removal of paraffin with xylene and rehydration in the graded concentrations of ethanol, the tissue sections were digested and permeated with proteinase K (20 μg/mL) for 15 min at 37 °C. Then, 50 μL of labeling reaction mixture was added to the sections for 1 h at 37 °C in dark. The sections were incubated with the recombinant DNase I (Beyotime, C1082, China) as the positive control. The nuclei were labeled with DAPI. The images were photographed by a fluorescence microscope (Olympus, IX73, Japan).
Immunofluorescence staining
The frozen sections of the posterior eyecup were thawed and lyophilized at room temperature for 30 min and fixed with 4% PFA for 20 min. After being permeabilized and blocked with 5% BSA for 30 min, they were incubated with ICAM-1 antibody (1:200, Santa Crus, sc-107, USA) overnight at 4 °C. Then, they were washed with PBS containing 0.1% Tween 20 (PBST) three times followed by the incubation for 2 h with the secondary antibody (1:200, A11001, Invitrogen, USA). The nuclei were stained with DAPI. The images were captured by Olympus IX73.
Cell permeability assay
Evans Blue/BSA solution (0.67 mg/mL, EBA) was prepared by diluting EB solution with BSA solution. After the required treatment, 100 μL of cell suspension was seeded in the upper chamber of transwell and 600 μL of 10% FBS was added to the lower chamber. Then, EBA solution was added to the upper chamber and the lower chamber was filled with 4% BSA solution. The OD value of absorption in the lower chamber was detected by a microplate reader (Molecular Devices, FilterMax F5, USA) after incubation for 1 h.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNAs were isolated from HUVECs or retinal tissues using TRIzol (Invitrogen, 15596018, USA) following the manufacturer’s instructions. The concentration of RNA was measured according to the optical density. The integrity of RNA was estimated based on OD260/280 ratio. Total RNAs were reversely transcribed to cDNAs. qRT-PCRs were performed on PIKOREAL 96 Real-Time PCR System (Thermo Fisher Scientific, PIKOREAL 96, USA) using the PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, A25742, USA). mRNA expression level of ICAM-1 was normalized to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. PCR results were calculated by the relative 2−ΔΔCt method.
Potential targets intersection
In the SwissTargetPrediction database, the species was restricted to “Homo sapiens” and the candidate targets with prediction scores > 0 were selected as the potential targets of DCZ19931. For identifying the targets involved in ocular diseases, the keywords “ocular neovascularization” were searched in three databases including GeneCards (https://www.genecards.org/) database, NCBI Gene (https://www.ncbi.nlm.nih.gov/) database, and OMIM (https://www.omim.org/) database. The species was restricted to “Homo sapiens”. After merging the data, the disease targets were obtained from three databases. Subsequently, Venn diagram was generated using Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny) to obtain the intersectant targets. The common drug-related targets and disease-related targets were input into Cytoscape software (version 3.6.1) to form a network diagram of the “drug- target-disease” interaction.
Construction of PPI network
To determine the interaction between DCZ19931 and ocular neovascularization-related targets, the common targets of DCZ19931-disease were imported into STRING database (https://string-db.org/). The network confidence score ≥ 0.4 was set to construct protein–protein interaction (PPI) network with “Homo sapiens”. Then, Cytoscape software (version 3.6.1) was used to analyze PPI network based on the results of STRING. The color and size of the nodes were adjusted according to the degree value. The degree value presented the importance of the node in the network.
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis
GO enrichment analysis was used for annotating gene function, which contains 3 aspects, including biological process (BP), molecular function (MF), and cellular component (CC). KEGG pathway analysis was used for predicting which pathway a particular gene was enriched, which covered the information resources such as diseases and pathways. The items with the adjusted P value < 0.05 were screened using the “clusterProfiler” package of R version 4.0.2 software to obtain the intersection target enrichment data. GO and KEGG enrichment results were represented by the bubble charts.
Western blot
Total proteins were extracted from choroidal tissues or HUVECs by the Radio Immunoprecipitation Assay (RIPA) buffer (Beyotime, P0013B, China) containing the protease inhibitors (Roche, 04693132001, USA). The concentrations of total proteins were determined by BCA assays (Bio-Rad, 23227, USA). After electrophoresis on SDS-PAGE gels, the proteins were transferred onto the polyvinylidene difluoride (PVDF) membranes. PVDF membranes were blocked with 10% skim milk for 30 min at room temperature. Then, the membranes were incubated with the following primary antibodies at 4 °C overnight, including ERK1/2 (1:1000, Cell Signaling Technology, 9102, USA), p-ERK1/2 (1:1000, Cell Signaling Technology, 4370, USA), p38 (1:1000, Cell Signaling Technology, 9212, USA), p-p38 (1:1000, Cell Signaling Technology, 9215, USA), JNK (1:1000, Cell Signaling Technology, 9252, USA), p-JNK (1:1000, Cell Signaling Technology, 9251, USA), ICAM-1 (1:1000, Abcam, ab171123, USA), and GAPDH (1:1000, Cell Signaling Technology, 2118). Next, the membranes were incubated with the secondary antibody (1:1000, Beyotime, A0208 goat anti-rabbit IgG or A0216 goat anti-mouse IgG, China) for 2 h at room temperature. Finally, the signaling was visualized by an ECL detection kit (Beyotime, P0018S, China).
Statistical analysis
All data are presented as means ± standard error of mean (SEM) of at least four independent experiments. The Student’s t-test and one-way analysis of variance (ANOVA) were performed to determine the differences between multiple groups. Statistical analyses were conducted using GraphPad Prism software. The statistical difference was considered at P < 0.05.
Results
DCZ19931 has no obvious cytotoxicity and tissue toxicity
To evaluate the cytotoxicity of DCZ19931, HUVECs were treated with DCZ19931 (1 nM to 10 μM) for 24 h. MTT assay revealed that DCZ19931 had no obvious cytotoxicity on HUVECs at the test concentrations ranging from 1 nM to 1 μΜ (Fig. 2A). Calcein-AM/propidium iodide (PI) staining and Annexin V/PI staining showed that DCZ19931 did not cause a detectable apoptosis of HUVECs at the tested concentrations ranging from 1 nM to 1 μM (Fig. 2B,C).

DCZ19931 has no obvious cytotoxicity and tissue toxicity. (A–C) HUVECs were incubated with the test concentrations of DCZ19931 (1 nM to 10 μM) or left untreated (Ctrl) for 24 h. Cell viability was measured by MTT assays (A, n = 4). Flow cytometry using Annexin V-FITC/PI double staining (B, n = 4) and Calcein-AM/PI staining (C, n = 4, scale bar, 20 μm) was performed to detect the apoptosis of HUVECs. (D,E) C57BL/6J mice received intravitreal injections of PBS (2 μL, Ctrl), 10% DMSO (2 μL), or DCZ19931 (2 μL, 1 μg/μL) for seven days. H&E staining (D, n = 5 animals per group, scale bar, 50 μm) and TUNEL staining assays were performed to detect retinal histological changes and retinal cell apoptosis (E, n = 5 animals per group, scale bar, 50 μm). DNase I was detected as a positive control in TUNEL staining assays. Statistical significance was evaluated by one-way ANOVA followed by Bonferroni post hoc test. *P < 0.05 indicated significant difference between the marked groups.
To determine the tissue toxicity of DCZ19931, C57BL/6J mice received intravitreal injections of phosphate buffer saline (PBS, Ctrl), 10% DMSO, or DCZ19931. Hematoxylin–eosin (HE) staining indicated that DCZ19931 treatment did not cause marked histopathological changes in retinal structures (Fig. 2D). TUNEL staining demonstrated that cell apoptosis was not observed in the retinas following the administration of DCZ19931 (Fig. 2E). Taken together, these results indicates that DCZ19931 has no obvious toxicity in vitro and in vivo.
DCZ19931 inhibits ocular neovascularization in vivo
To investigate whether DCZ19931 exerted its anti-angiogenic effects in vivo, laser-induced choroidal neovascularization (CNV) model was built through the rupture Bruch’s membrane and RPE by laser photocoagulation in C57BL/6J mice. After laser injury, the mice immediately received intravitreal injections of 10% DMSO (Ctrl), DCZ19931, Ranibizumab, or DCZ19931 plus Ranibizumab. At day 7 following laser photocoagulation, the thickness and areas of CNV lesions were quantified by H&E staining (Fig. 3A). CNV regions were observed by IB4 staining (Fig. 3B). The results showed that the areas of CNV lesions were markedly reduced in DCZ19931-treated group or Ranibizumab-treated group compared with the control group. The area of CNV lesions was smallest in DCZ19931-Ranibizumab group (Fig. 3A,B).

DCZ19931 inhibits ocular neovascularization in vivo. (A,B) C57BL/6J mice received intravitreal injections of 10% DMSO (2 μL, Ctrl), DCZ19931 (2 μL, 1 μg/μL), Ranibizumab (2 μL, 10 mg/mL), Ranibizumab (1 μL, 10 mg/mL) plus DCZ19931 (1 μL, 1 μg/μL) immediately after laser injury. The thickness and areas of neovascular were determined by H & E staining (A, n = 5, scale bar, 50 μm). CNV lesions were visualized by IB4 staining (B, n = 5, scale bar, 100 μm). (C) OIR mouse pups at P12 received intravitreal injections of 10% DMSO (1 μL, Ctrl), DCZ19931 (1 μL, 1 μg/μL), Ranibizumab (1 μL, 10 mg/mL), 1 μL of Ranibizumab (10 mg/mL) plus DCZ19931 (1 μg/μL), respectively. The retinas were harvested on P17 and then stained with GS-IB4 staining to observe retinal vessels. White dashed lines highlighted avascular areas; Yellow area indicated angiogenic regions (n = 5, scale bar, 200 μm). Statistical significance was evaluated by one-way ANOVA followed by Bonferroni post hoc test. *P < 0.05 indicated significant difference between the marked groups.
Oxygen-induced retinopathy (OIR) model was further used to determine the anti-angiogenic effects of DCZ19931 in vivo. The pups of C57BL/6J mice at the postnatal day 7 (P7) with their nursing mothers were placed in 75% oxygen for 5 days (P12) and then returned to room air for another 5 days (P17). The mice received intravitreal injections of 10% DMSO (Ctrl), DCZ19931, Ranibizumab, or DCZ19931 plus Ranibizumab at P12. At P17, the whole‐mount retinas were visualized by GS-IB4 staining. The avascular areas were markedly reduced in DCZ19931-treated group compared with the control group. Few neovascular tufts were observed in DCZ19931-treated group. DCZ19931 plus Ranibizumab showed greater anti-angiogenic effects than DCZ19931 or Ranibizumab alone (Fig. 3C). Collectively, the above-mentioned results indicates that DCZ19931 could inhibit pathological ocular neovascularization in vivo.
DCZ19931 exhibits anti-angiogenic effects in vitro
Endothelial cells play a critical role in pathological angiogenesis5. To determine whether DCZ19931 administration could regulate endothelial angiogenic effects in vitro, HUVECs were treated with VEGF (10 ng/mL) for 12 h, followed by incubation with DCZ19931 (10 nM to 1 μM) for an additional 24 h. MTT assays were carried out to detect cell viability. DCZ19931 administration could decrease cell viability induced by VEGF at the concentration of 500 nM and 1 μM (Fig. 4A). We selected 500 nM as the test concentration for the subsequent experiments. Following exposure to VEGF for 12 h, HUVECs were incubated with DCZ19931, Ranibizumab, DCZ19931 plus Ranibizumab for 24 h. The control group received no treatment. EdU assays demonstrated that DCZ19931 administration significantly decreased cell proliferation induced by VEGF (Fig. 4B). DCZ19931 administration also suppressed VEGF-mediated migration and tube formation ability of HUVECs and reduced the areas of choroidal explant sprouting (Fig. 4C–E). DCZ19931 plus Ranibizumab showed greater anti-angiogenic effects than DCZ19931 or Ranibizumab monotherapy on cell proliferation, migration, and tube formation ability as well as endothelial sprouting ability (Fig. 4B–E).

DCZ19931 exhibits anti-angiogenic effects in vitro. (A) HUVECs were pre-treated with VEGF (10 ng/mL) for 12 h and then treated with DCZ19931 at the concentration of 10 nM to 1 μM for 24 h. Cell viability was determined by MTT assay (n = 4). (B–D) HUVECs were pre-treated with VEGF (10 ng/mL) for 12 h and then treated with DCZ19931 (500 nM), Ranibizumab (250 μg/mL), DCZ19931 (500 nM) plus Ranibizumab (250 μg/mL), or left untreated (VEGF) for 24 h. The group without VEGF treatment was taken as Ctrl group. Cell proliferation was detected by EdU assays (B, n = 4, scale bar, 20 μm). The cells were allowed to migrate for 12 h at 37 °C and 5% CO2 and cell migration ability was assessed by transwell assays (C, n = 4, scale bar, 20 μm). HUVECs were seeded on matrigel matrix and cultured. Tube formation ability was observed under a light microscope at 8 h after seeding (D, n = 4, scale bar, 100 μm). (E) The mouse choroidal sprouting assays were conducted to measure the angiogenic potency of choroidal explants at day 4, day 5, and day 6 after culture. Representative images of the choroidal sprouting areas at indicated time points were shown (n = 4, scale bar, 200 µm). Statistical significance was evaluated by one-way ANOVA followed by Bonferroni post hoc test. For (A–D): *P < 0.05 indicated significant difference between the marked groups. For (E): *P < 0.05 vs. VEGF group, #P < 0.05 vs. DCZ19931 + Ranibizumab group.
DCZ19931 inhibits vascular permeability via downregulation of ICAM-1 expression
The formation of pathological neovascularization is often accompanied by increased vascular permeability. Intercellular cell adhesion molecule-1 (ICAM-1) is an important adhesion molecule that affects vascular permeability by binding to specific receptors on EC surfaces15. Here, we examined the effects of DCZ19931 administration on ICAM-1 expression and vascular permeability.
We first carried out qRT-PCR assays to detect the expression of ICAM-1 mRNA in vitro. DCZ19931 administration markedly down-regulated the levels of ICAM-1 mRNA induced by VEGF treatment in HUVECs (Fig. 5A). Cell permeability assays were performed to determine the effects of DCZ19931 administration on the barrier integrity of HUVECs. Compared with the normal group (WT group), VEGF treatment led to increased cell leakage, while DCZ19931 administration reduced cell leakage induced by VEGF treatment (Fig. 5B). Subsequently, we employed laser-induced CNV model and OIR model to investigate the effects DCZ19931 administration on ICAM-1 expression in vivo. qRT-PCR assays and western blot assays revealed that compared with normal mice (WT group), CNV induction led to up-regulated levels of ICAM-1 expression in choroidal tissues. DCZ19931 administration markedly reduced the levels of ICAM-1 expression in laser-induced CNV model (Fig. 5C,D). Similar results were observed in OIR model. Compared with OIR group (Ctrl group), the levels of ICAM-1 expression were reduced in the OIR groups treated with DCZ19931 (Fig. 5E). Moreover, the anti-angiogenic effects of DCZ19931 plus Ranibizumab treatment were greater than that of single treatment in both CNV model and OIR model (Fig. 5C–E).

DCZ19931 inhibits vascular permeability via downregulation of ICAM-1 expression. (A) HUVECs were cultured with VEGF (Ctrl, 10 ng/mL), VEGF (10 ng/mL) plus DCZ19931 (500 nM), VEGF (10 ng/mL) plus Ranibizumab (250 μg/mL), VEGF (10 ng/mL) plus DCZ19931 (500 nM) and Ranibizumab (250 μg/mL) for 24 h. qRT-PCR assays were performed to detect the expression of ICAM-1 mRNA in HUVECs (n = 4). (B) HUVECs were cultured with VEGF (Ctrl, 10 ng/mL), VEGF (10 ng/mL) plus DCZ19931 (500 nM), VEGF (10 ng/mL) plus Ranibizumab (250 μg/mL), VEGF (10 ng/mL) plus DCZ19931 (500 nM) and Ranibizumab (250 μg/mL), or left untreated (WT) for 24 h. WT indicated normal HUVECs without VEGF induction. EB-transwell assays were conducted to detect cell permeability (n = 4). (C,D) Laser-induced CNV mice received intravitreal injections of 10% DMSO (2 μL, Ctrl), DCZ19931 (2 μL, 1 μg/μL), Ranibizumab (2 μL, 10 mg/mL), Ranibizumab (1 μL, 10 mg/mL) plus DCZ19931 (1 μL, 1 μg/μL) after laser injury. WT indicated normal mice without CNV induction. qRT-PCR assays were performed to detect the expression of ICAM-1 mRNA in choroidal tissues at day 7 after treatment (C, n = 5). Western blots were performed to determine ICAM-1 expression in choroidal tissues. GAPDH was used as the internal control. Representative immunoblots along with the quantitative results were shown (D, n = 5). (E) The pups of OIR mice at P12 received intravitreal injections of 10% DMSO (1 μL, Ctrl), DCZ19931 (1 μL, 1 μg/μL), Ranibizumab (1 μL, 10 mg/mL), 1 μL of Ranibizumab (10 mg/mL) plus DCZ19931 (1 μg/μL) mixture. ICAM-1 immunofluorescence assays were conducted to determine ICAM-1 expression in OIR model. The representative images with the quantitative results were shown (n = 5, scale bar, 50 μm; nuclei, blue; ICAM-1-positive cells, green). Statistical significance was determined by one-way ANOVA followed by Bonferroni post hoc test. *P < 0.05 indicated significant difference between the marked groups.
DCZ19931 inhibits ocular neovascularization through p38-MAPK and ERK1/2-MAPK signaling
We used the SwissTargetPrediction database to predict the targets of DCZ19931. The result showed that a total of 100 targets of DCZ19931 were obtained (Table 1). Taking “ocular neovascularization” as the keyword, a total of 2461 targets, 59 targets, and 200 targets were retrieved from GeneCards database, NCBI database, and OMIM database, respectively. After searching, summarizing, and deleting the duplicates, a total of 2563 targets related to ocular neovascularization were obtained. Then, the targets related to DCZ19931 and ocular neovascularization were input into online Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny), a total of 58 common targets were obtained. The network diagram of “drug-target-disease” interaction was drawn by Cytoscape software (Fig. 6A).

DCZ19931 inhibits ocular neovascularization through p38-MAPK and ERK1/2-MAPK signaling. (A) The drug-target-disease network diagram of DCZ19931 involved in ocular neovascularization. (B) PPI network analysis. (C) GO enrichment analysis. (D) KEGG pathways enrichment analysis. (E) HUVECs were treated with DCZ19931 (500 nM), Ranibizumab (250 μg/mL), DCZ19931 (500 nM) plus Ranibizumab (250 μg/mL), or left untreated (Ctrl). Then, HUVECs were stimulated with or without VEGF (50 ng/mL) for 30 min. The expression levels of total ERK1/2, p38, JNK, p-ERK1/2, p-p38, and p-JNK were detected by western blots (n = 4). (F) HUVECs were treated with DCZ19931 (500 nM) or SB203580 (5 μmol/mL) plus U0126 (5 μmol/mL) for 24 h, and then stimulated with VEGF (50 ng/mL) for 30 min. The group without any treatment was taken as the control (Ctrl). The phosphorylated levels of ERK1/2 and p38 proteins were detected by western blots (n = 4). GAPDH was detected as the internal control. Representative immunoblots along with the densitometric quantitative results were shown. Statistical significance was determined by one-way ANOVA followed by Bonferroni post hoc test. *P < 0.05 indicated significant difference between the marked groups.
The protein interaction network was obtained through STRING database and imported into Cytoscape for further analysis. There were 58 nodes with 211 edges in the network and the protein interaction was enriched (P < 0.001). The average local clustering coefficient was 0.448 and the average node degree was 7.62. The importance of nodes in the network was reflected as degree value. The higher the degree value, the more important the node in the PPI network. The top 10 key targets were MAPK3, MTOR, MAPK1, ESR1, PI3KCA, PTGS2, ATM, APP, KDR, and CDK2 ranked by the degree value (Fig. 6B).
GO terms included biological process (BP), cellular composition (CC), and molecular function (MF). The overlapping genes were enriched into 54 MF, 965 BP, and 51 CC (Fig. 6C). To determine the potential pathways of DCZ19931 involved in ocular neovascularization, a total of 121 signal pathways were predicted by KEGG enrichment analysis. The results were displayed in a bubble diagram (Fig. 6D). These targets were highly enriched in MAPK signaling pathway, VEGF signaling pathway, mTOR signaling pathway, and HIF-1 signaling pathway. Notably, MAPK signaling was ranked as the Top 1 signaling pathway involved in ocular neovascularization. We thus examined the effects of DCZ19931 administration on MAPK signaling activation.
HUVECs were cultured with DCZ19931, Ranibizumab, DCZ19931 plus Ranibizumab for 24 h. Subsequently, HUVECs were stimulated with VEGF for 30 min. Western blot assays demonstrated that the levels of phosphorylated ERK and phosphorylated p38 proteins were markedly decreased following DCZ19931 administration. DCZ19931 plus Ranibizumab had greater inhibitory effects than DCZ19931 or Ranibizumab alone on the expression levels of phosphorylated ERK and phosphorylated p38 (Fig. 6E). We also revealed that DCZ19931 administration could mimic the inhibitory effects of SB203580 (p38 inhibitor) and U0126 (ERK1/2 inhibitor) on MAPK signaling inactivation as shown by decreased expression of phosphorylated ERK and phosphorylated p38 (Fig. 6F). Collectively, the above-mentioned results suggest that DCZ19931 plays its anti-angiogenic role via the inactivation of p38-MAPK and ERK1/2-MAPK signaling pathway.
Discussion
Neovascularization is a critical pathological factor in the progression of ocular diseases. Several pro-angiogenic and anti-angiogenic factors, including VEGF, PDGF, pigment epithelium derived factor (PEDF), could regulate the process of angiogenesis. The imbalance between pro-angiogenic and anti-angiogenic factors could lead to the formation of new blood vessels under pathological condition3,16,17. These hypoplastic vessels often undergo hemorrhages and leakage, leading to retinal structural change18,19. Currently, numerous clinical studies have shown that anti-VEGF agents have achieved good therapeutic effects in inhibiting pathological ocular neovascularization20,21,22. Despite the extensive clinical applications, there were still several deficiencies that have not been filled, such as frequent injection or drug resistance23,24. In this study, we designed and synthesized a new multi-target kinase inhibitor, DCZ19931, which could suppress ocular neovascularization by targeting endothelial angiogenic effects.
Although MKIs have been widely used, there are still some unfavorable side effects. For instance, these drugs have low absorption efficiency due to poor water solubility, rapid clearance, and metabolic rate. In addition, the non-specific uptake of MKIs by normal tissues may lead to a series of adverse effects11,12. Thus, it is required to develop new MKI drugs for anti-angiogenic treatment. Herein, DCZ19931 administration had no significant cytotoxic effects on HUVECs in vitro. HE staining and TUNEL staining revealed that DCZ19931 administration did not cause the structure changes of retinas and did not induce the detectable apoptosis in the retinas. Taken together, DCZ19931 has no obvious toxicity in vitro and in vivo and is a biosafety drug.
Ocular neovascularization contributes to visual impairment in several ocular diseases. Targeting pathological neovessels is an important advancement for treating ocular diseases25,26,27. DCZ19931 treatment could reduce the size of CNV lesions compared with the control group in the laser-induced CNV model. Moreover, the areas of avascular and neovascular were obviously reduced in the retinas of OIR model following DCZ19931 treatment. DCZ19931 suppressed the proliferation, tube formation, and migration of endothelial cells during the process of neovascularization. Notably, we compared the anti-angiogenic effects of DCZ19931 with the existing anti-VEGF drug. Ranibizumab is a monoclonal antibody fragment, which can bind closely to VEGF-A8. DCZ19931 had a similar anti-angiogenic efficiency as Ranibizumab. Importantly, DCZ19931 plus Ranibizumab had greater anti-angiogenic efficiency than Ranibizumab or DCZ19931 alone. The combined therapy may be used to solve the clinical problem of anti-VEGF treatment resistance.
ICAM-1 is a member of the immunoglobulin gene superfamily (IGSF) with low expression levels in endothelial cells under normal condition28,29,30. However, inflammatory stimuli such as interleukin-1β (IL-1β), tumor necrosis factor (TNF-α), and lipopolysaccharide, can significantly increase the expression of ICAM-131,32,33. Abnormal VEGF expression can lead to endothelial angiogenic effects and vascular dysfunction, such as permeability and inflammation. In this study, we showed that VEGF administration could alter endothelial permeability. Endothelial permeability assays revealed that endothelial permeability was higher in VEGF-treated group compared with the normal group. By contrast, DCZ19931 treatment could reduce cell permeability induced by VEGF. Meanwhile, we found that DCZ19931 treatment could effectively inhibit ICAM-1 levels induced by VEGF in endothelial cells. Furthermore, DCZ19931 could suppress the expression levels of ICAM-1 in laser-induced CNV model and OIR model. These results indicate that DCZ19931 treatment could affect endothelial permeability and ICAM-1 expression in vitro and in vivo.
We further explored the molecular mechanism of DCZ19931 on ocular neovascularization based on the network pharmacology analysis. MAPK signaling pathway was predicted as the potential target pathway for DCZ19931 to exert its anti-angiogenic effects. MAPK signaling pathway can transduce extracellular stimulation signals into cells and nuclei through three-level kinase cascade34,35,36. It has been reported to be involved cell proliferation, growth, and apoptosis. There are three subfamilies including ERK1/2, p38, and JNK, which constitute a parallel MAPK signal pathway37,38. In this study, we found that DCZ19931 could inhibit ocular angiogenesis by reducing the phosphorylation levels of p38 and ERK1/2 proteins in MAPK signaling pathway (Supplementary Information).
Conclusions
This study reveals that DCZ19931 presents its anti-angiogenic effects via reducing MAPK signaling pathway in endothelial cells. Importantly, the combined treatment via DCZ19931 plus Ranibizumab is more effective than single treatment. Therefore, DCZ19931 treatment is a promising therapeutic strategy for pathological ocular neovascularization.
Data availability
The datasets generated for this study are available on request to the corresponding authors.
References
Rohlenova, K. et al. Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. 31, 862–877 (2020).
Das, A. & McGuire, P. G. Retinal and choroidal angiogenesis: Pathophysiology and strategies for inhibition. Prog. Retin. Eye Res. 22, 721–748 (2003).
Selvam, S., Kumar, T. & Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 63, 1–19 (2018).
Liu, Y. et al. Repurposing bortezomib for choroidal neovascularization treatment via antagonizing VEGF-A and PDGF-D mediated signaling. Exp. Eye Res. 204, 108446 (2021).
Sene, A., Chin-Yee, D. & Apte, R. S. Seeing through VEGF: Innate and adaptive immunity in pathological angiogenesis in the eye. Trends Mol. Med. 21, 43–51 (2015).
Uemura, A. et al. VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog. Retin. Eye. Res. 84, 100954 (2021).
Lutty, G. A. & McLeod, D. S. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog. Retin. Eye Res. 62, 58–76 (2018).
Mettu, P. S., Allingham, M. J. & Cousins, S. W. Incomplete response to anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 82, 100906 (2021).
Cheung, N., Wong, I. Y. & Wong, T. Y. Ocular anti-VEGF therapy for diabetic retinopathy: Overview of clinical efficacy and evolving applications. Diabetes Care 37, 900–905 (2014).
Scagliotti, G. V. Potential role of multi-targeted tyrosine kinase inhibitors in non-small-cell lung cancer. Ann. Oncol. 18(Suppl 10), x32–x41 (2007).
Shen, G. et al. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 11, 120 (2018).
Böhm, S., Hess, D., Gillessen, S. & Brändle, M. Improved glycemic control with the multi-receptor tyrosine kinase inhibitor pazopanib. Diabetes Care 33, e82 (2010).
Nozal, V. et al. From Kinase inhibitors to multitarget ligands as powerful drug leads for Alzheimer’s disease using protein-templated synthesis. Angew Chem. Int. Ed. Engl. 60, 19344–19354 (2021).
Chu, T. et al. Phase 1b study of sintilimab plus anlotinib as first-line therapy in patients with advanced NSCLC. J. Thorac. Oncol. 16, 643–652 (2021).
Buravkova, L. B., Rudimov, E. G., Andreeva, E. R. & Grigoriev, A. I. The ICAM-1 expression level determines the susceptibility of human endothelial cells to simulated microgravity. J. Cell. Biochem. 119, 2875–2885 (2018).
Robles, J. P. et al. The HGR motif is the antiangiogenic determinant of vasoinhibin: Implications for a therapeutic orally active oligopeptide. Angiogenesis 25, 57–70 (2022).
Rezzola, S. et al. In vitro and ex vivo retina angiogenesis assays. Angiogenesis 17, 429–442 (2014).
Zarkada, G. et al. Specialized endothelial tip cells guide neuroretina vascularization and blood–retina-barrier formation. Dev. Cell 56, 2237–2251 (2021).
Zhang, J. et al. HIF-1alpha and HIF-2alpha redundantly promote retinal neovascularization in patients with ischemic retinal disease. J. Clin. Investig. 131, e139202 (2021).
Sankar, M. J., Sankar, J., Mehta, M., Bhat, V. & Srinivasan, R. Anti-vascular endothelial growth factor (VEGF) drugs for treatment of retinopathy of prematurity. Cochrane Database Syst. Rev. 2, CD009734 (2016).
Hegde, P. S., Wallin, J. J. & Mancao, C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin. Cancer Biol. 52, 117–124 (2018).
Zhao, X. et al. Antiangiogenic nanomicelles for the topical delivery of aflibercept to treat retinal neovascular disease. Adv. Mater. 34, e2108360 (2022).
Sood, S., Mandell, J., Watane, A., Friedman, S. & Parikh, R. Cost of ranibizumab port delivery system vs intravitreal injections for patients with neovascular age-related macular degeneration. JAMA Ophthalmol. 140, 716–723 (2022).
Patil, N. S. et al. Association between visual acuity and residual retinal fluid following intravitreal anti-vascular endothelial growth factor treatment for neovascular age-related macular degeneration: A systematic review and meta-analysis. JAMA Ophthalmol. 140, 611–622 (2022).
Writing Committee for the Diabetic Retinopathy Clinical Research Network. Panretinal photocoagulation vs intravitreous Ranibizumab for proliferative diabetic retinopathy: A randomized clinical trial. JAMA 314, 2137–2146 (2015).
Diabetic Retinopathy Clinical Research Network. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 372, 1193–1203 (2015).
Heier, J. S. et al. Intravitreal aflibercept injection vs sham as prophylaxis against conversion to exudative age-related macular degeneration in high-risk eyes: A randomized clinical trial. JAMA Ophthalmol. 139, 542–547 (2021).
Zhang, W. et al. ICAM-1-mediated adhesion is a prerequisite for exosome-induced T cell suppression. Dev. Cell 57, 329–343 (2022).
Regev, O. et al. ICAM-1 on breast cancer cells suppresses lung metastasis but is dispensable for tumor growth and killing by cytotoxic T cells. Front. Immunol. 13, 849701 (2022).
Gao, Y. et al. Clock upregulates intercellular adhesion molecule-1 expression and promotes mononuclear cells adhesion to endothelial cells. Biochem. Biophys. Res. Commun. 443, 586–591 (2014).
Wang, S. et al. Mechanosensation by endothelial PIEZO1 is required for leukocyte diapedesis. Blood 140, 171–183 (2022).
Glushakova, O. et al. Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J. Am. Soc. Nephrol. 19, 1712–1720 (2008).
HaghayeghJahromi, N. et al. Intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 differentially contribute to peripheral activation and CNS entry of autoaggressive Th1 and Th17 cells in experimental autoimmune encephalomyelitis. Front. Immunol. 10, 3056 (2020).
Van der Noll, R., Leijen, S., Neuteboom, G. H., Beijnen, J. H. & Schellens, J. H. Effect of inhibition of the FGFR-MAPK signaling pathway on the development of ocular toxicities. Cancer Treat. Rev. 39, 664–672 (2013).
Liu, F. et al. Wolfberry-derived zeaxanthin dipalmitate delays retinal degeneration in a mouse model of retinitis pigmentosa through modulating STAT3, CCL2 and MAPK pathways. J. Neurochem. 158, 1131–1150 (2021).
Liu, X. et al. Malvidin and its derivatives exhibit antioxidant properties by inhibiting MAPK signaling pathways to reduce endoplasmic reticulum stress in ARPE-19 cells. Food Funct. 12, 7198–7213 (2021).
Iroegbu, J. D., Ijomone, O. K., Femi-Akinlosotu, O. M. & Ijomone, O. M. ERK/MAPK signalling in the developing brain: Perturbations and consequences. Neurosci. Biobehav. Rev 131, 792–805 (2021).
Kiel, C. & Serrano, L. Challenges ahead in signal transduction: MAPK as an example. Curr. Opin. Biotechnol. 23, 305–314 (2012).
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 82070983 to Q.J., Grant No. 22077131 to B.L., Grant No. 81872797 to WL.Z., and Grant No. 81970909 to B.Y.).
Author information
Authors and Affiliations
Contributions
B.Y., Q.J. and W.L.Z. conceived and designed the project. H.Y.Z., J.J.D., R.Y., Q.Y.Z. and S.G.F. performed experiments. B.L. and Z.J.X. assisted in designing and synthesizing the drug. H.Y.Z., J.J.D., R.Y. and Q.Y.Z. wrote the manuscript. B.Y. revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Li, B., Ding, J. et al. DCZ19931, a novel multi-targeting kinase inhibitor, inhibits ocular neovascularization. Sci Rep 12, 21539 (2022). https://doi.org/10.1038/s41598-022-25811-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25811-0
