
Abstract
Natural compounds constitute a major resource for the development of medicines for multiple diseases. While many natural compounds show strong biological activity, the mechanisms that confer clinical benefits are often elusive and have been attributed to multiple pathways. Periplogenin (PPG), a natural compound isolated from Cortex periplocae, exhibits strong anti-tumor activities in several human cancer cell lines. However, its molecular mode of action remained unclear. In this study, we leveraged a forward genetic screening approach in DU145 prostate cancer cells to uncover the molecular target of PPG using chemical mutagenesis. Next generation sequencing revealed that a single amino acid substitution at amino acid 804 in ATP1A1 (ATPase Na + /K + Transporting Subunit Alpha 1) confers resistance to the cytotoxic activity of PPG. Mechanistically, ATP1A1 T804 forms a hydrogen bond with PPG which is abolished by the T804A substitution in ATP1A1, resulting in resistance to PPG treatment in vitro. Importantly, in vivo, PPG strongly suppressed tumor development in a DU145 xenograft model whereas DU145 xenograft tumors carrying a ATP1A1-T804A mutation were largely unaffected by the treatment. These findings demonstrate that PPG suppresses the growth of DU145 prostate cancer cells in vitro and in vivo by directly binding to ATP1A1 and highlight the power of our unbiased forward genetic screening approach to uncover direct drug target structures at single amino acid resolution.
Similar content being viewed by others

Introduction
Due to the high diversity, natural compounds have gained much attention as drug leads for treatment of diseases1. Periplogenin (PPG) is a natural compound extracted from traditional Chinese herb Cortex Periplocae which is widely used for the treatment of autoimmune diseases2. It has been reported to inhibit histamine release of mast cells in antigen-sensitized rats and can also ameliorate skin lesions and inflammation in psoriasis-like murine models3,4. In addition, PPG has shown antiproliferative activities in various cancer cells4,5,6,7,8,9. PPG inhibits cell growth and triggers apoptosis by induction of reactive oxygen species in human colorectal cancer cells whereas in esophageal squamous cell carcinoma it was reported to directly bind to STAT3 and thereby strongly inhibit its phosphorylation at position Tyr705 to suppress cell growth and induce apoptosis5,6 The findings of anti-tumor activity of PPG highlights the potential applications of PPG for the treatment of other cancer types.
Prostate cancer is one of the most lethal cancer types for males10. It is well established that the initial growth of prostate cancer is dependent on androgens, which makes hormonal therapy with chemical or surgical castration the favorable treatment option11,12. However, most patients eventually progress to a castration-independent phase, and the treatment of patients with metastatic castration-resistant prostate cancer remains a clinical challenge12,13,14. This has led to the urgent search for new drugs and exploration of new anti-cancer targets against prostate cancer.
An unbiased forward genetic screening method was established to investigate the molecular mechanism of anti-tumor drugs, leading to the identification of novel anti-tumor targets15. In this study we used this screening technology to demonstrate that PPG suppresses growth of DU145 prostate carcinoma cells by docking to an ATP1A1 protein pocket and forming a hydrogen bond with T804. This highlights the possible application of PPG as an anti-prostate cancer drug and identifies ATP1A1 as a potential therapeutic target.
Materials and methods
Cell culture
DU145 (human prostate cancer), DLD-1 (human colorectal cancer), U-87MG (human glioblastoma) and MCF-7 (human breast cancer) cell lines were purchased from American Type Culture Collection (ATCC) and cultured with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum and penicillin–streptomycin. All cells were maintained at 5% CO2, 37 °C.
Cell viability assay
Cells were seeded in 96 well plates and cultured for 24 h to 80% confluence. Then, cells were treated with PPG (Desite, DY0084). Dimethyl sulfoxide (DMSO, Sigma, D8418) was used as negative control. Cell viability was measured and quantified by CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7572) in accordance with the manufacturer’s instructions.
Forward genetic screens
The forward genetic screens were carried out as previously described15. Briefly, DU145 cells were trypsinized and treated with 0.1 mg/ml N-ethyl-N-nitrosourea (ENU, Sigma, N3385) for 2 h in medium at room temperature under rotation. The same procedure was followed to the cells without ENU treatment as control. Then, cells were seeded to 15 cm diameter culture dish at 2 × 106 cells/dish and cultured for 24 h. Periplogenin in medium at the indicated final concentrations was applied to the cells for selection of resistant cells. PPG resistance of emerging cell clones was validated by cell viability assay.
Sequencing and data analysis
Genomic DNA of resistant clones was extracted using TIANamp Genomic DNA kit (TIANGEN, DP304), exome-enriched libraries were generated by SureSelect Human All Exon V6 kit (Agilent, 51900-8863) and sequenced using NovaSeq 6000 paired-end 150. Exome sequencing depth of each sample was at least greater than 150X and data were analyzed as previously described15. In brief, clean reads were aligned to the reference genome (hg19) with bwa (v.0.7.15). Reads were converted to bam format with samtools (v.1.3.1) and sorted with Picard (2.25.0). GATK (v.3.8) Realigner TargetCreator was then used to generate a list of positions for GATK IndelRealigner. After indexing, base recalibration was performed with GATK Base Recalibrator and recalibrated reads were printed with GATK PrintReads. Samtools and GATK pileups were then used to identify variants in the respective samples. After discarding indels, the frequency of the mutation sites in the control sample was less than 0.05, the read depth of the mutation sites in each resistant clone was not less than 10, and the gene frequency was not less than 0.2. After variant annotation with snpEff (v.4.2) variants relating to a moderate or high effect in protein coding were kept.
Molecular docking
The full-length amino acid sequence of human ATP1A1 protein (UniPort: P05023) was downloaded from the UniPort database. The three-dimensional (3D) structure of the predicted full-length ATP1A1 protein (AF-P05023-F1) was obtained from the AlphaFold protein Structure Database, the structure of periplogenin was downloaded from PubChem (CID 10574). The PyMol™ Molecular Graphic System (Version 2.3.0) was used to label the mutated amino acid sites on the 3D structure of the protein and to predict the drug-protein binding pocket based on UniPort annotation. The molecular docking process was carried out using AutoDock Vina 1.1.2 following the parameters: center_x = 18.118, center_y = 5.762, center_z = -37.883, size_x = 52.0, size_y = 62.0, size_z = 48.0, exhaustiveness = 64, num_modes = 9.
CRISPR genome editing
The construction of cell lines with ATP1A1 gene point mutation was performed in DU145 with CRISPR/Cas9 technology as previously described16. DNA sequences of donor homology arms and small guide RNAs were designed with Invitrogen™ TrueDesign™ Genome Editor and synthetized in GENEWIZ. Small guide RNAs were further prepared using GeneArtTM Precision gRNA Synthesis Kit (Thermo Fisher, A29377). Prepared small guide RNAs, corresponding homology arm DNA, and TrueCutTM Cas9 protein v2 (Thermo Fisher, A36498) were co-transfected to DU145 cells using Lipofectamine™ CRISPRMAX™ Cas9 Transfection Reagent (Thermo Fisher, CMAX00001). Transfected cells were maintained in medium containing periplogenin for the selection of resistant cell clones. Resistant cell clones were transferred to 12 well plate and subjected to PCR amplification and Sanger sequencing. The primers targeting ATP1A1 specific region used for the amplification were as follows: Forward 5′- AAGATCTTATTCAGATACTG-3′ and Reverse 5′-CACTACTATCGATCTGTGAC-3′. The sequences of synthesized DNA were followed: 804 mutation Donor homology arm DNA 5′-TTCCTGATATTTATTATTGCAAACATTCCACTACCCCTGGGGGCTGTCACCATCCTCTGCATTGACTTGGGCACTGAC-3′, and 804 mutation guide RNA 5′-GCAAACATTCCACTACCACT-3′.
Reverse transcription real-time quantitative PCR (RT-qPCR)
RT-qPRC was performed as previously described with minor changes17,18,19. Briefly, cells were harvested, and RNA was extracted with TRIzol (Thermo Fisher, 15596026CN). The reverse transcription was carried out with Hiscript III Reverse Transcriptase (Vazyme, R302-01). qPCR was carried out using PowerTrack™ SYBR Green master mix (Thermo Fisher, A46112). The GAPDH was used as control for normalization. The primers of GAPDH were: Forward 5′-AGGTCGGAGTCAACGGATTT-3′ and Reverse 5′-AGTTAAAAGCAGCCCTGGTGA-3′. The primers for ATP1A1 were: Forward 5′-AGTATGAGCCTGCAGCTGTT-3′ and Reverse 5’-TGCACGAGCAGATGTTAATCC-3′.
Western blotting
Cells were washed with PBS and harvested. Total protein was extracted with a total protein extraction kit (Epizyme, PC201) and separated with an ExpressPlus™ PAGE gel (Genscript, M42012) and then followed by transferring to a methanol treated polyvinylidene difluoride (PVDF) membrane (Thermo Fisher, 88518). After blocking with 5% milk for 1 h at room temperature, the membrane was incubated with an anti-ATP1A1 antibody (Abways, CY5159) at 4 °C overnight and then probed with a secondary antibody conjugated with horseradish peroxidase (HRP) for 1 h at room temperature.
Measurement of Na+/K+ -ATPase activity
Cell lysates were harvested, and the protein concentrations were determined using BCA Protein Assay Kit (KeyGEN Biotech, KGB2101). Na+ /K+ -ATPase activity was measured according to the manual of the Na+ /K+ -ATPase activity assay kit (Sangon Biotech, D799642).
Animal trial
Male BALB/c nude mice (4–6 weeks old) were purchased from Shanghai Lingchang Biotechnology Co., Ltd. 5 × 106 DU145 cells with T804A point mutation in ATP1A1 (DU145-804MUT group, n = 18) or wild type DU145 cells (DU145 group, n = 18) in a 100 μL 1:1 mixture of PBS and matrigel were subcutaneously injected in the right axillary region of mouse. When the tumor volume reached 100mm3, each group was randomly divided into the following three subgroups (n = 6 per subgroup): DMSO and PPG (10 mg/kg and 20 mg/kg). PPG or DMSO were administered via oral gavage daily.
Statistical analysis
Results were presented as means plus standard deviations. Statistical analysis was done with PRISM 9 (GraphPad Software, USA) using 2-tailed Student’s t test or One-way ANOVA. P-values for scoring genes in the whole exome sequencing data (Fig. 1B) were calculated based on the number of unique mutations identified per gene using cumulative binominal distribution function (CDF). The total number of identified mutations in the dataset was quantified using a threshold of 10 reads and 0.2 allele frequency. The average number of mutations per clone (201.2) was divided by the exome size (30 M) and a p-value to hit a single gene at least once was calculated using CDF, this accounts for larger genes being hit more likely compared to shorter genes. Then for every gene with at least one mutation (1,572 genes), the p-value to find at least a consistent number of each gene's unique mutations in 9 clones was again calculated using CDF. Each experiment was repeated at least three times unless otherwise specified.

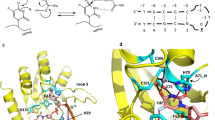
Forward genetic screen identifies ATPAT1 as target of periplogenin. (A) Schematic of the forward genetic screen approach. (B) Forward genetic screen results. The -log10(p-value) of the analysis in the screen was plotted on the y-axis and 1572 candidate genes were organized by chromosome position on x-axis, the size of the dot exhibits the clone numbers containing the identified mutations of corresponding gene. (C, D) Molecular docking of ATP1A1 with periplogenin. (E) The effect of periplogenin treatment on the Na+ /K +-ATPase activity. The cell lysates were derived from wild-type DU145 cells (WT) and CRISPR/Cas9 engineered DU145 cell clones with T804A substitution (T804A). * and ** indicate p values of < 0.05, < 0.01, respectively.
Results
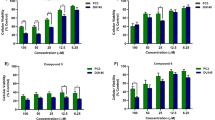
Periplogenin shows anti-tumor activities in multiple cancer cell lines
To uncover the direct molecular target of PPG, we first identified a cell model amenable to subsequent forward genetic target identification screens. Therefore, the cytotoxicity of PPG was determined in DU145, DLD-1, U-87MG, and MCF-7 cells by a cell viability assay. The 90% inhibitory concentration (IC50) values of periplogenin for DU145, DLD-1, U-87MG, and MCF-7 cells were 2.897 µM, 8.712 µM, 3.008 µM, and 20.025 µM, respectively (Supplementary Fig. 1A). The IC90 value of PPG in DU145 was calculated to be lower than to DLD-1, U-87MG, and MCF-7 cells (Supplementary Fig. 1A). Since the applied forward genetic screening approach requires highly effective killing, DU145 cells were further characterized by studying time- and concentration-dependent PPG toxicity to define screening conditions (Supplementary Fig. 1B).
ATP1A1 is identified as target of periplogenin
Next, a forward genetic screen was carried out in the selected DU145 cells. Cells were treated with ENU, a chemical mutagen that causes point mutations throughout the cell genome and facilitates the generation of drug-resistant clones. These clones were then selected for using PPG treatment of 25 µM and 50 µM, respectively (Fig. 1A). Non-mutagenized cells were treated with the same PPG concentration function as a negative control for the screen. After 4 weeks post-treatment, we observed 9 cell clones under selection of PPG in the ENU mutagenized population, while no colonies were observed in the non-mutagenized cell population. The resistance of all cell clones was confirmed by cell viability assay (Supplementary Fig. 2).
To identify the resistance-causing genetic underpinnings, we performed next-generation whole exome sequencing. Our analysis revealed that all examined clones harbor a point mutation in ATP1A1 (Fig. 1B and Supplementary Table 1). The obtained mutations affected the amino acids A108, N129, L800, and T804 (Table 1). Notably, one clone (clone 4) harbored two mutations in the ATP1A1 coding region, A108E and T804I (Table 1).
Identified amino acid positions are essential for PPG activity
To explore the role of the identified amino acid position in PPG activity, we generated the predicted 3D structure of ATP1A1 protein with labeling of the mutated amino acids. Interestingly, although distributed over distinct functional domains, A129, L800, and T804 are located in a potential drug binding pocket (Supplementary Fig. 3A-E). We further assessed the possible binding region of PPG with ATP1A1 by performing molecular docking analysis. The analysis revealed PPG docked to the anticipated ATP1A1 pocket with a binding affinity of − 9.8 kcal/mol, including A129, L800, and T804 (Fig. 1C). Strikingly, PPG was able to form a hydrogen bond (2.2 Å) within the binding pocket of ATP1A1 protein to T804 (Fig. 1D). Since A108 is distant to the potential pocket and clone 4 harbors an additional T804I mutation, it is likely that the A108E alteration is just a bystander mutation without or with limited effect on PPG resistance. To validate the modelling data, we performed CRISPR/Cas9-mediated genome editing to mutate the potential hydrogen bond donor/acceptor by introducing the T804A substitution. Under selection of periplogenin, we observed and confirmed cell clones with T804A substitution using sanger sequencing. We randomly selected two cell clones with T804A substitution for the subsequent experiments.
We performed the in vitro Na+ /K+ -ATPase activity assay to confirm the direct effect of PPG on ATP1A1 protein. The results revealed that PPG inhibits the activity of ATP1A1 in a dose-dependent manner (Supplementary Fig. 5). The T804A mutation of ATP1A1 protein shows no obvious effect on the activity. Importantly, engineered ATP1A1 protein demonstrates better resistance to PPG treatment in the Na+ /K+ -ATPase activity than wild type ATP1A1 protein (Fig. 1E). Meanwhile the treatment of PPG does not lead to expression change of ATP1A1 protein (Supplementary Fig. 4D).
Next, we validated the resistance in two independent T804A engineered clones using a cell viability assay. Both clones are highly resistant to PPG treatment at all tested concentrations (Fig. 2A). The resistance could neither be attributed to altered ATP1A1 expression levels nor to a general effect on cell growth in both clones (Supplementary Fig. 4A-C).

DU145 cells with the T804A substitution are resistant to periplogenin treatment, both in vitro and in vivo. (A) CRISPR/Cas9 engineered DU145 cell clones with T804A substitution (T804A) were treated with periplogenin for 72 h in the indicated dose and compared to wild type DU145 cells (WT). (B) Comparison of relative tumor volumes among all groups, the tumor volume was measured using caliper every 2 days and calculated as length x width2 × 0.5, then was normalized to tumor volume of 0 day post-administration, the relative tumor volumes of the last time point were compared. (C, D) Comparison of tumor weight and size. * and ** indicate p values of < 0.05, < 0.01, respectively, ns, not significant.
Periplogenin suppresses tumor growth in DU145 xenograft tumor model
We next assessed whether the identified single amino acid substitution could ameliorate PPG anti-cancer activities in vivo. To this end, we compared DU145 WT to DU145 ATP1A1 (T804A) cells in xenograft studies. While PPG treatment dramatically reduced the relative tumor volume and tumor weight in DU145 WT xenograft models, DU145 ATP1A1 (T804A) cell-based models were largely insensitive to PPG treatment in this setup (Fig. 2B–D). Notably, none of the mouse groups showed significant changes in body weight (Supplementary Fig. 6).
In conclusion, PPG inhibits cell growth of multiple cancer cell lines including DU145, DLD-1, U-87MG, and MCF-7. The inhibition of PPG to prostate cancer cell DU145 results from direct binding to ATP1A1 protein by forming a hydrogen bond with T804, revealing mode of action at the amino acid level.
Discussion
Natural compounds have been extensively explored as anti-tumor therapeutics. Amongst them, PPG has been reported to show therapeutic effects in various cancer types in vitro and in vivo5,6,20. Mechanistically, PPG was reported to induce reactive oxygen species to inhibit the growth of human colorectal cancer cells and it also directly interacts with STAT3 protein to suppress the growth of esophageal squamous cell carcinoma5,6. Nevertheless, the direct molecular target of PPG, in other cancer models, remained largely elusive.
In the current study, we show that PPG significantly inhibits cell growth in various cancer cells including DU145 (human prostate cancer), DLD-1 (human colorectal cancer), U-87MG (human glioblastoma) and MCF-7 (human breast cancer). Unbiased forward genetic screens reveal that PPG suppresses DU145 prostate cancer cell growth in vitro and in vivo by targeting ATP1A1 via a hydrogen bonding to T804.
The Na+/K+-ATPase is composed of four α-, three β-, and one γ- subunit isoforms and establishes a transmembrane sodium gradient by exchanging three sodium ions with two potassium ions in each pump cycle, by utilizing ATP. It is further described to act as intracellular signal transducer and membrane receptor21,22,23,24,25,26. ATP1A1, α1 subunit, contains the ATP and cation binding sites and is the catalytic subunit of the enzyme27. Mutations in ATP1A1 have been associated with altered cell proliferation and tumorigenesis in aldosterone-producing adenomas28,29,30. In addition, the aberrant expression of ATP1A1 is widely observed in multiple types of tumors. Clinically, the expression of ATP1A1 is significantly decreased in human renal cell carcinoma tissues compared to non-tumor tissues whereas abnormalities in expression are observed in breast cancer samples31,32. All these reports indicate ATP1A1 might play an important role in tumorigenesis.
In this study, we show direct interaction between PPG and ATP1A1 to account for PPG antiproliferative activities, which further indicates ATP1A1 could be a druggable target in anti-tumor treatment. Presumably, PPG binds to ATP1A1’s catalytic subunit and inhibits the enzymatic function leading to a disturbed ion homeostasis and the induction of apoptosis33.
We still observe minimal growth inhibition by PPG when DU145 cells carry an A804T substitution in ATP1A1, both in vitro and in vivo. This could be explained by an attenuated affinity between PPG and ATP1A1 or direct binding of PPG to secondary targets such as STAT3 and others.
In summary, our study demonstrates that PPG exhibits growth inhibitory effects to DU145 cells in vitro and in vivo by directly binding to ATP1A1 and forming a hydrogen bond with T804. Unravelling the molecular mode of action of PPG at amino acid resolution not only allows to induce rationally designed compound modifications, but also to systematically select most relevant indications in the future.
Data availability
The datasets generated during and/or analysed during the current study are available in the Sequence Read Archive (SRA) repository under accession no. PRJNA982409.
References
David, B., Wolfender, J.-L. & Dias, D. A. J. P. R. The pharmaceutical industry and natural products: Historical status and new trends. Phytochem. Rev. 14, 299–315 (2015).
Zhang, W.-J. et al. Periplogenin induces necroptotic cell death through oxidative stress in HaCaT cells and ameliorates skin lesions in the TPA-and IMQ-induced psoriasis-like mouse models. Biochem. Pharmacol. 105, 66–79 (2016).
Gu, W. & Zhao, L. J. Effect of periplogenin on mast cell degranulation and histamine release (1991)
Zhang, H.-Y. et al. Tumor targeted delivery of octreotide-periplogenin conjugate: Synthesis, in vitro and in vivo evaluation. Int. J. Pharm. 502, 98–106 (2016).
Yang, Y. et al. Periplogenin activates ROS-ER stress pathway to trigger apoptosis via BIP-eIF2α-CHOP and IRE1α-ASK1-JNK signaling routes. Anti-Cancer Agents Med. Chem. 21, 61–70 (2021).
Hu, Y. et al. Periplogenin suppresses the growth of esophageal squamous cell carcinoma in vitro and in vivo by targeting STAT3. Oncogene 40, 3942–3958 (2021).
Ye, H. et al. Mechanism of action of periplogenin on nasopharyngeal carcinoma based on network pharmacology and experimental study of vitamin E coupled with periplogenin self-assembled nano-prodrug for nasopharyngeal carcinoma. J. Biomed. Nanotechnol. 16, 1406–1415 (2020).
Han, N. et al. Inhibitory activity of a phytochemically characterized fraction from Streptocaulon juventas on lung cancer in nude mice. Planta Med. 76, 561–565 (2010).
Lohberger, B. et al. Periplocin, the most anti-proliferative constituent of Periploca sepium, specifically kills liposarcoma cells by death receptor mediated apoptosis. Phytomedicine 51, 162–170 (2018).
Gillen, A. D. & McEwan, I. J. Personalised treatment for prostate cancer patients: Are we there yet. AME Med. J. 4 (2019).
Chen, Y., Sawyers, C. L. & Scher, H. I. Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharmacol. 8, 440–448. https://doi.org/10.1016/j.coph.2008.07.005 (2008).
Walczak, J. R. & Carducci, M. A. Prostate cancer: A practical approach to current management of recurrent disease. Mayo Clin. Proc. 82, 243–249. https://doi.org/10.4065/82.2.243 (2007).
Abd Wahab, N. A., Lajis, N. H., Abas, F., Othman, I. & Naidu, R. Mechanism of anti-cancer activity of curcumin on androgen-dependent and androgen-independent prostate cancer. Nutrients https://doi.org/10.3390/nu12030679 (2020).
Chi, K. N. et al. Castration-resistant prostate cancer: From new pathophysiology to new treatment targets. Eur. Urol. 56, 594–605. https://doi.org/10.1016/j.eururo.2009.06.027 (2009).
Horn, M. et al. Unbiased compound-protein interface mapping and prediction of chemoresistance loci through forward genetics in haploid stem cells. Oncotarget 9, 9838–9851. https://doi.org/10.18632/oncotarget.24305 (2018).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. https://doi.org/10.1038/nprot.2013.143 (2013).
Zhang, X., Bilic, I., Marek, A., Glosmann, M. & Hess, M. C-terminal amino acids 471–507 of avian hepatitis E virus capsid protein are crucial for binding to avian and human cells. PLoS ONE 11, e0153723. https://doi.org/10.1371/journal.pone.0153723 (2016).
Zhang, X., Bilic, I., Troxler, S. & Hess, M. Evidence of genotypes 1 and 3 of avian hepatitis E virus in wild birds. Virus Res. 228, 75–78. https://doi.org/10.1016/j.virusres.2016.11.028 (2017).
Feng, J. et al. N(6)-Methyladenosine and reader protein YTHDF2 enhance the innate immune response by mediating DUSP1 mRNA degradation and activating mitogen-activated protein kinases during bacterial and viral infections. mBio 14, e0334922. https://doi.org/10.1128/mbio.03349-22 (2023).
Ye, H. et al. Mechanism of action of periplogenin on nasopharyngeal carcinoma based on network pharmacology and experimental study of vitamin E coupled with periplogenin self-assembled nano-prodrug for nasopharyngeal carcinoma. J. Biomed. Nanotechnol. 16, 1406–1415. https://doi.org/10.1166/jbn.2020.2978 (2020).
Lingrel, J. B. & Kuntzweiler, T. Na+, K(+)-ATPase. J. Biol. Chem. 269, 19659–19662 (1994).
Chen, J. Q. et al. Sodium/potassium ATPase (Na+, K+-ATPase) and ouabain/related cardiac glycosides: A new paradigm for development of anti- breast cancer drugs?. Breast Cancer Res. Treat. 96, 1–15. https://doi.org/10.1007/s10549-005-9053-3 (2006).
Rajasekaran, S. A. et al. Na, K-ATPase beta1-subunit increases the translation efficiency of the alpha1-subunit in MSV-MDCK cells. Mol. Biol. Cell 15, 3224–3232. https://doi.org/10.1091/mbc.e04-03-0222 (2004).
Sakai, H. et al. Up-regulation of Na(+), K(+)-ATPase alpha 3-isoform and down-regulation of the alpha1-isoform in human colorectal cancer. FEBS Lett. 563, 151–154. https://doi.org/10.1016/S0014-5793(04)00292-3 (2004).
Xie, Z. & Cai, T. Na+-K+–ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 3, 157 (2003).
Aperia, A., Akkuratov, E. E., Fontana, J. M. & Brismar, H. Na+-K+-ATPase, a new class of plasma membrane receptors. Am. J. Physiol. Cell Physiol. 310, C491–C495 (2016).
Lingrel, J. B., Williams, M. T., Vorhees, C. V. & Moseley, A. E. Na, K-ATPase and the role of alpha isoforms in behavior. J. Bioenerg. Biomembr. 39, 385–389. https://doi.org/10.1007/s10863-007-9107-9 (2007).
Dutta, R. K. et al. Complementary somatic mutations of KCNJ5, ATP1A1, and ATP2B3 in sporadic aldosterone producing adrenal adenomas. Endocr. Relat. Cancer 21, L1-4. https://doi.org/10.1530/ERC-13-0466 (2014).
Kobuke, K. et al. ATP1A1 mutant in aldosterone-producing adenoma leads to cell proliferation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms222010981 (2021).
Williams, T. A. et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 63, 188–195. https://doi.org/10.1161/HYPERTENSIONAHA.113.01733 (2014).
Bogdanov, A., Moiseenko, F. & Dubina, M. Abnormal expression of ATP1A1 and ATP1A2 in breast cancer. F1000Res. 6, 10. https://doi.org/10.12688/f1000research.10481.1 (2017).
Zhang, D. et al. Downregulation of ATP1A1 promotes cancer development in renal cell carcinoma. Clin. Proteom. 14, 15. https://doi.org/10.1186/s12014-017-9150-4 (2017).
Bejcek, J., Spiwok, V., Kmonickova, E. & Rimpelova, S. Na(+)/K(+)-ATPase revisited: On its mechanism of action, role in cancer, and activity modulation. Molecules 26, 1905. https://doi.org/10.3390/molecules26071905 (2021).
Acknowledgements
We thank colleagues of Angalbio for technical assistance and discussions.
Author information
Authors and Affiliations
Contributions
X. Q. Z and L. Q. Z conceived, designed, supervised, and managed the project. X. Q. Z., T.L. P., M. H., G. M., and M. D. interpreted and analyzed the data. X. Q. Z wrote the manuscript with inputs from all the authors. All the authors read, reviewed, and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Institutional Animal Care and Use Committee at Keygen Biotech reviewed and approved the animal experiment protocol (Approval number: IACUC-20220803). All animal handling and experiments strictly followed the ARRIVE guidelines for animal research and reporting for in vivo experiments. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Pang, T., Zhang, H. et al. The natural compound periplogenin suppresses the growth of prostate carcinoma cells by directly targeting ATP1A1. Sci Rep 14, 20509 (2024). https://doi.org/10.1038/s41598-024-71722-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-71722-7
