
Abstract
Cerebrospinal fluid (CSF) plays an important role in brain tumors, including medulloblastoma (MBL). Recent advancements in mass spectrometry systems and ‘Omics’ data analysis methods enable unbiased, high proteome depth research. We conducted proteomic profiling of the total CSF in MBL patients with the purpose of finding a potential diagnostic biomarker for MBL. We quantified 1112 proteins per CSF sample. Feature selection identified four elevated soluble proteins (SPTBN1, HSP90AA1, TKT, and NME1-NME2) in MBL CSF. Validation with ELISA confirmed that TKT was significantly elevated in MBL. Additionally, TKT-positive extracellular vesicles were significantly enriched in MBL CSF and correlated with the burden of leptomeningeal seeding. Our results provide insights into the proteomics data of the total CSF of MBL patients. Furthermore, we identified the significance of TKT within the total CSF and its presence within circulating EVs in the CSF. We suggest that TKT may serve as a biomarker for MBL.
Similar content being viewed by others


Introduction
Medulloblastoma (MBL) is the most common malignant brain tumor in children and occurs in the cerebellum1,2. Similar to other tumors of the central nervous system, cerebrospinal fluid (CSF) plays an important role in MBL. CSF not only serves as a tumor environment but also acts as a route of dissemination. Leptomeningeal seeding (LMS) is common in MBL and significantly contributes to childhood morbidity and mortality1,3,4. Determining the metastatic stage of MBL is important for risk stratification and treatment. The need for a more adequate method to detect LMS has led to investigations of CSF in MBL patients5,6.
CSF, which directly contacts brain tumor tissue, is an important source of liquid biopsies in primary brain tumors. CSF contains various materials, such as circulating tumor DNA, miRNA, metabolites, proteins, and extracellular vesicles (EVs)7. Materials detected in the CSF of brain tumor patients have higher sensitivity and specificity than those detected in peripheral blood due to the blood–brain barrier8.
CSF proteomics studies have mainly focused on EV proteins rather than the total CSF proteome9. CSF has low protein concentrations (0.2–0.7 mg/mL), which are 50 times lower than those of plasma10,11. However, the protein composition of CSF is as complex as that of plasma. Therefore, it is crucial to apply more efficient proteomic methods to achieve high proteome depth (> 1000 quantified proteins) while minimizing the loss of throughput and operating costs for CSF biomarker discovery.
In the last 10 years, biomarker studies using ‘Omics’ technology with various clinical samples have demonstrated their potential and applicability to clinical research12,13,14,15. Recent advancements in mass spectrometry systems and ‘Omics’ data analysis methods in proteomics have become the main methodologies for a precise molecular-level understanding of organism phenotypes. They have made unbiased biomarker studies more applicable to clinical practice for instance, neurodegenerative diseases and brain tumors16,17,18.
In this study, we performed proteomic profiling of total CSF in patients with MBL using untargeted proteomics by mass spectrometry and ‘Omics’ data analysis. The results of this study provide insights into potential biomarkers and associated biological processes in MBL. In addition, we validated the feature selected proteins using enzyme-linked immunosorbent assay (ELISA) and EV cargo proteins.
Methods
Patient clinical characteristics
In this study, CSF samples from patients who underwent surgery for MBL and hydrocephalus (HC, control group) at our institute between 2016 and 2019, were used. A total of 34 MBL patients and 19 HC patients were included in the study (Suppl. Table S1). There was no significant sex difference between the MBL and HC patients (MBL 1.06 vs. HC 1.11 male:female ratio, p = 1.00). However, the HC group was significantly younger (MBL median age 6.0 years (IQR 3.5–10.5) vs. HC median age 1.1 years (IQR 0.8–2.5), p < 0.01). None of the etiologies for HC patients were tumor associated HC, and the conditions leading to non-tumorous HC occur in younger patients. For MBL molecular subgrouping, the NanoString nCounter element assay was performed as described previously19. Briefly, a total of 100 ng of RNA isolated from snap-frozen tissues was used. Data analysis was performed using nSolver analysis software (Nanostring Technologies, Seattle, WA, USA). Subgrouping using the NanoString assay classified cases into Wingless signaling-activated (WNT, n = 6), Sonic-hedgehog signaling-activated (SHH, n = 3), Group 3 (n = 7), Group 4 (n = 14), and unclassifiable (n = 4) categories. LMS at the initial diagnosis was evaluated by CSF cytology and LMS on craniospinal MRI and was classified according to Chang’s criteria20. The stage of LMS at diagnosis was M0 in 17 patients, M1 in one patient, M2 in three patients, and M3 in 12 patients. CSF cytology at diagnosis was positive in six patients and negative in eight patients. Eight (61.5%) out of 13 patients presented a false-negative result with CSF cytology.
This study protocol was approved by the local Institutional Review Board (IRB No. 1905-121-1035) and was conducted according to the Helsinki Declaration.
Experimental design and statistical rationale
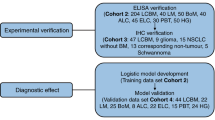
This study comprised two phases: (1) biomarker discovery through proteomic and bioinformatics analyses, and (2) validation of candidate biomarkers using ELISA and EV characterization via ExoView (Fig. 1).

Flow chart illustrating the design of the present study. After discovery proteomics with LC‒MS/MS, one arm represents the process of label-free quantification and DEPs. The other arm represents feature selection with ANOVA and FCBF, with a validation process using ELISA and ExoView methods. Then, enrichment and pathway analyses of the TKT protein were performed, which appears to be a potential biomarker with various methodologies.
In the first phase (discovery), we conducted untargeted mass spectrometry using liquid chromatography-tandem mass spectrometry (LC‒MS/MS) and performed pairwise comparison of MBL and HC to detect differentially expressed proteins (DEPs). Bioinformatics analysis was employed for the identified DEPs, and feature selection was used to identify classifiers for the MBL and HC groups. The top 4 upregulated proteins in the MBL group were selected as biomarker candidates. Initially, a total of 38 CSF samples from 37 patients (MBL = 22, HC = 15) were subjected to LC–MS/MS analysis. However, upon reviewing the medical records, 2 CSF samples from patients with MBL were excluded due to a history of prior treatment, and one sample was utilized as a duplicate for quality control during the sample preparation for LC/MS–MS. As a result, a total of 35 CSF samples from 35 patients (MBL = 21, HC = 14) were included in the LC–MS/MS analysis.
In the second phase (validation), we performed ELISA for 4 biomarker candidates (MBL = 14, HC = 7). Additionally, following an analysis of EV morphology via transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA), ExoView was performed for further validation of TKT protein (MBL = 17, HC = 9). The number of CSF samples used for each test varied, as described above. Since this study was conducted retrospectively, the remaining CSF volume was varied, and we included additional samples for the validation set when there was insufficient CSF remaining after LC–MS/MS analyses. While the sample size was limited to achieve high statistical power, it was adequate for conducting comparative analyses and for assessing both intra-group and inter-group comparisons.
CSF sample collection and preparation for proteomics
CSF collection was done with a standardized operating procedure. A burr hole trephination was made either at the frontal or occipital horn. Ventricle puncture was performed using a ventricular catheter, and CSF was obtained from the lateral ventricle before tumor removal in MBL patients and before the insertion of a shunt device in HC patients. CSF was collected into a plain tube and no more than 10 ml of CSF was drained due to the small volume of lateral ventricle in young children and the risk of CSF over drainage is high in patients with increased intracranial pressure. Blood contamination of the samples was determined based on a gross visual inspection. CSF samples were processed on the same day as they are collected, typically within 4 h. To remove cellular debris, the CSF was centrifuged at 1000×g for 20 min, then at 10,000×g for 30 min at 4 °C. Subsequently, the processed CSF samples were aliquoted into sterile 1.5 ml snap cap micro centrifuge tubes free of protease, with a volume of 0.2–1 ml. They are stored at temperatures of -70 °C, and are used without repeated freezing and thawing during experiments.
For protein digestion, 100–200 μl of each CSF sample was dried using a speed-vac. The dried samples were reconstituted in 50 μl of SDT buffer (2% SDS, 0.1 M dithiothreitol in 0.1 M Tris HCl pH 8.0). After being heated at 95 °C, the denatured proteins were digested by a filter-aided sample preparation method as previously described21 with some modifications. Briefly, protein samples were loaded onto a 30 K amicon filter (Millipore, Billerica, MA, USA), and buffer was exchanges with UA solution (8 M UREA in 0.1 M Tris–HCl pH 8.5) via centrifugation. After three buffer exchange with UA solution, the reduced cysteines were alkylated with 0.05 M iodoacetamide in UA solution for 30 min at room temperature in the dark. Thereafter. UA buffer was exchanged for 40 mM ammonium bicarbonate twice. The protein samples were digested with trypsin/LysC (enzyme to substrate ratio of 1:100) at 37 °C for 16 h. The resulting peptides were collected in new Eppendorf tube via centrifugation, and an additional elution step was performed using 40 mM ammonium bicarbonate and 0.5 M NaCl. Peptides were desalted and fractionated on homemade styrene divinylbenzene reversed-phase sulfonate-StageTips by basic reverse-phase using a stepwise gradient of acetonitrile (40%, 60%, and 80%) in 1% ammonium hydroxide21,22. Fractionated peptides were completely dried with a vacuum dryer and stored at -80 °C.
Establishment of a matching spectral library for LC‒MS/MS
To construct a matching spectral library for matching between runs,23,24 a multiple affinity removal system-14 column (Agilent Technologies, Santa Clara, CA, USA) was used to remove the 14 highest abundance proteins according to the manufacturer’s instructions. After depletion with commercially available normal CSF (991–19-P, Lee Biosolutions, Maryland Heights, MO, USA), samples were digested using the 2-step filter-aided sample preparation as described previously21. Digested peptides were desalted using homemade C18-StageTips. To acquire the in-depth dataset, 25 µg of purified peptides was fractionated using an Agilent 1260 bioinert high performance liquid chromatography (Agilent Technologies, Santa Clara, CA, USA) equipped with an analytical column (4.6 × 250 mm, 5-µm particle). High-pH reversed-phase peptide fractionation was performed at a flow rate of 0.8 mL/min over a 60 min gradient using solvent A (15 mM ammonium hydroxide in water) and solvent B (15 mM ammonium hydroxide in 90% acetonitrile). A total of 96 fractions were collected each minute and noncontiguous pooled into 24 fractions. The fractions were dried in a vacuum centrifuge and stored at -80 °C until LC‒MS/MS analysis.
LC‒MS/MS analysis
LC‒MS/MS analysis was conducted using Quadrupole Orbitrap mass spectrometers, Q Exactive HF-X (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Ultimate 3000 RSLC system (Dionex, Sunnyvale, CA, USA) with a Nano electrospray source as previously described with some modifications25. Peptide samples were separated on the two column setup with a trap column (300 µm I.D. × 0.5 cm, C18, 3 µm, 100 Å) and analytical column (75 µm I.D. × 50 cm, C18 1.9 µm, 100 Å). Prior to sample injection, the dried peptide samples were dissolved in solvent A (2% acetonitrile and 0.1% formic acid). After the samples were loaded onto the Nano LC, a 90-min gradient from 8 to 30% solvent B (100% acetonitrile and 0.1% formic acid) was applied to all samples. The spray voltage was 2.0 kV in positive ion mode and the temperature of the heated capillary was set to 320 °C. Mass spectra were acquired in data-dependent mode using a top 15 method on a Q Exactive. The Orbitrap analyser scanned precursor ions with a mass range of 300–1650 m/z and a resolution of 70,000 at m/z 200. Higher-energy collisional dissociation scans were acquired on the Q Exactive at a resolution of 17,500. Higher-energy collisional dissociation peptide fragments were acquired at a normalized collision energy of 28. The maximum ion injection times for the survey and MS/MS scans were 20 and 120 ms, respectively.
LC‒MS/MS data processing and label-free quantification analysis
Mass spectra were processed with MaxQuant (version 1.6.1.0)23. LC‒MS/MS spectra were searched against the Human UniProt protein sequence database (December 2014, 88,657 entries) using the Andromeda search engine26. Primary searches were performed using a 6 ppm precursor ion tolerance for total protein level analysis. The LC‒MS/MS ion tolerance was set to 20 ppm. Cysteine carbamido-methylation was set as a fixed modification. N-Acetylation of proteins and oxidation of methionine were set as variable modifications. Enzyme specificity was set to full tryptic digestion. Peptides with a minimum length of six amino acids and up to two missed cleavages were considered. The required false discovery rate (FDR) was set to 1% at the peptide, protein, and modification levels. To maximize the number of quantification events across samples, matching between runs was performed using the depleted sample as a library. The expression level of proteins in the CSF was estimated by determining their intensity-based absolute quantification (iBAQ) values calculated using MaxQuant software. Statistical analyses for the data-dependent acquisition data were performed using Perseus software27. Initially, the iBAQ value was imported into Perseus software, and proteins identified only by site, reverse, and contaminants were removed. Because of the skewed distribution of the data, log2 transformation was conducted for these values. Valid values were filtered with proteins with a minimum of 70% quantified values in at least one diagnostic group. Missing values were imputed based on a normal distribution (width = 0.5, downshift = 1.8) to simulate signals of low-abundance proteins. Two-sided t tests were performed for pairwise comparison of MBL and HC groups to detect DEPs. We applied the FDR of 5% to adjust the p values for the statistical significance of DEPs. Protein abundances were subjected to z-normalization followed by hierarchical clustering with Pearson’s correlation distance or Euclidean distance.
Bioinformatics analysis and feature selection
Principal component analysis (PCA) with preprocessed protein expression data was conducted in Perseus software27. Canonical pathways were evaluated by Ingenuity Pathway Analysis (IPA, QIAGEN, Hilden, Germany) based on the annotated DEPs with matched gene names28. The analytical algorithms embedded in IPA use lists of DEPs to predict biological processes and pathways. Canonical pathways with p values < 0.05 and ∣z∣ scores > 0 were considered statistically significant. Protein‒protein interaction (PPI) network data from all of the proteins involved in activated or inhibited canonical pathways in MBL were constructed and exported from the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database29. PPI network data were constructed and visualized using Cytoscape version 3.030. To identify hub proteins, the betweenness centrality of nodes was calculated by CytoNCA (Cytoscape plug-in)31. Dot plots were generated with R software (version 4.0.4; https://www.R-project.org) using the ggplot2 and readr packages. Feature selection based on machine learning algorithms was performed by open source software Orange 3 (https://orange.biolab.si/). Analysis of variance (ANOVA) F value and fast correlation-based filter (FCBF) methods were used for feature selection32.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted using DAVID (https://david.ncifcrf.gov)33,34,35,36,37. The statistical significance of the enriched GO and KEGG pathway terms was determined by Fisher’s exact test.
Enzyme-linked immunosorbent assay (ELISA) performed on CSF samples
CSF protein levels were determined using ELISA kits for spectrin beta nonerthrocytic 1 (SPTBN1, Abbexa, Cambridge, United Kingdom), heat shock protein 90 alpha family class A number 1 (HSP90AA1, Biomatik, Ontario, Canada), transketolase (TKT, Abcam, Waltham, MA, USA), and NME/NM23 nucleoside diphosphate kinase 1 (NME1, Biomatik) according to the manufacturer’s protocol. A 100 μl of CSF sample was used per target and all experiments were conducted in duplicate. Therefore, a total of 800 μl of CSF was used to perform ELISA kits.
Isolation of EVs
EVs, including exosomes, were collected from the CSF using miRCURY™ Exosome Isolation Kits (Qiagen, Hilden, Germany). The morphological characteristics of the exosomes were verified by TEM. NTA using a Malvern Zetasizer Nano ZS90 (Malvern Panalytical, Malvern, United Kingdom) was performed to measure the concentration and size of EVs. The amount of CSF sample used for TEM and NTA was 6–8 ml and 1 ml respectively.
Characterization of EVs in CSF
We validated TEM, NTA, and three EV protein markers according to the three criteria for EV characterization proposed by the International Society for Extracellular Vesicles38,39. TEM was utilized for analysis of EV morphology as described previously19,40 and visualized using TEM (JEOL, Peabody, MA, USA). NTA was employed to detect the size and concentration of EVs using a NanoSight L system (NanoSight Technology Malvern, UK) with NTA software (ver. 2.3) as described previously19,41. The ExoView R100 system (NanoView, Biosciences, Brighton, MA, USA) was carried out according to the manufacturer’s instructions and as described previously19. A 35 μl of CSF sample diluted in solution A (1:5) was incubated with CD81-CF555 (green, NanoView), CD63-CF647 (red, NanoView) and CD9-CF488 (blue, NanoView) from ExoView Tetraspanin standard kit to determine EVs markers. IgG1 (NanoView) was used as a negative control for nonspecific EV binding. To enable access of antibodies to the EVs cargo after affinity capture, EVs were fixed and permeabilized using the Cargo Kit (NanoView) withCD9-CF488 (blue, NanoView), SPTBN1-PE (green, Biorbyt, St Louis, MO, USA), HSP90AA1-PE (green, Biorbyt), TKT-PE (green, Biorbyt), and NME1-PE (green, Biorbyt). After treating the chips, the number of positive particles was detected, quantified using the ExoView R100 reader (nScan 2.8.4, NanoView) and analyzed using NanoViewer 2.8.11 with EV sizing thresholds set to 50–200 nm diameter as previously described19,42.
Statistical analysis
The average protein levels and comparisons between groups were analyzed using Student’s t tests for two groups and ANOVA with the Tukey post hoc method for three or more groups. To determine the sensitivity and specificity of each protein level, receiver operating characteristic (ROC) curves were generated, and the area under the curve (AUC) was calculated using DeLong’s test for confidence intervals. Pearson’s correlation analysis was employed to explore the relationship between LMS and the level of TKT-positive EVs in the MBL CSF. A p value of 0.05 was the cut-off value for significance in all the statistical tests above.
Results
Proteomic profile and DEPs of MBL CSF identified by label-free LC‒MS/MS technique and pairwise comparison
In the LC‒MS/MS process in our study, we employed a robust workflow with high proteome depth using only 100‒200 μl of sample without depletion of highly abundant proteins (Fig. 2A). In total, 2557 protein groups were identified from 38 CSF samples from 37 patients at the protein FDR 1%. After the exclusion of 3 samples as mentioned above, a total of 2230 protein groups from CSF samples of 35 patients were quantified and subjected to statistical analysis (Suppl. Table S2). For comparison of our proteome data to previously published literature, unique gene names were used. Out of 2123 unique gene names, 1841 (86.7%) have been previously described in the brain tissue proteome from MBL patients (Suppl. Fig. S1A)43. Among the 2123 genes, 710 genes were associated with MBL subgroup proteomic features, and 91 genes were related to genomic signatures operative in MBL (Suppl. Fig. S1B-C)44. Seven proteins (FAT1, FLG, SYNCRIP, IDH1, PRKAR1A, CTNNB1, and EPHA7) were identified as driver genes in MBL. On average, we quantified 1112 proteins per CSF sample (Fig. 2B). The number of quantified proteins in each sample ranged from 915 to 1309 in the MBL group and from 1001 to 1318 in the HC group. The number of overlapping proteins was 1877, and the number of unique proteins was 233 in the MBL group and 120 in the HC group (Suppl. Fig. S2A-B). Our data achieved 100% completeness for 379 proteins (17% of 2230 proteins), and 50% completeness for 1088 proteins (49% of 2230 proteins) across 35 samples (Suppl. Fig. S3A). The quantified protein intensities spanned over seven orders of magnitude, with the top ten most abundant proteins contributing 63% of the total protein intensity among all 2230 quantified proteins in the samples (Suppl. Fig. S3B). We assessed intra‐ and inter‐sample group variability in our proteomic analysis, which revealed high reproducibility, with over 1500 proteins having inter‐group coefficients of variation below 10% (Suppl. Fig. S3C). To assess the correlation of protein intensities obtained from label-free LC‒MS/MS, multiscatter plots were generated within and between groups. The mean Pearson correlation coefficient between HC and MBL was 0.71, while within-group correlations for MBL and HC were 0.74 and 0.78, respectively (Suppl. Fig. S4). Blood contamination of the CSF samples were evaluated using the proportion of hemoglobin relative to the total quantified proteins by LC‒MS/MS in each sample (Suppl. Table S2). Although not statistically significant, the percentage of hemoglobin was higher in the MBL group (MBL 3.4% (IQR 2.0–18.6) vs. HC 2.3% (IQR 0.6–4.2), p = 0.083).

CSF proteome characterization and label-free quantitative LC‒MS/MS results. (A) Workflow of proteomic data generation by LC‒MS/MS; (B) Comparison of the average number of quantified proteins in the HC and MBL groups; (C) Principal component analysis score plot generated from quantified proteins in all of the samples; (D) Volcano plot of all of the identified proteins in the HC and MBL groups. The red dots represent upregulated proteins in the MBL group (adjusted p < 0.05). The blue dots represent upregulated proteins in the HC group (adjusted p < 0.05); (E) A heatmap of DEPs between the HC and MBL proteomes.
PCA showed discrimination by PC1 and PC2, indicating that protein expression patterns could differ between the MBL and HC groups. Samples in the HC group were more closely located in the score plot (PC1 and PC2) than those in the MBL group (Fig. 2C).
Next, we performed pairwise comparisons between MBL and HC samples to discover tumor-associated protein signatures with altered expression. First, we extracted quantified proteins that satisfied the replicate rate condition (> 70% valid iBAQ values in at least one group). Out of the extracted proteins, 972 were subjected to t tests. By filtering quantified proteins with an adjusted p value < 0.05, we obtained 273 DEPs between the MBL and HC groups, with 130 proteins upregulated in the MBL group and 143 proteins upregulated in the HC group. (Fig. 2D and Suppl. Table S3). Hierarchical clustering of z score-normalized expression values of DEPs between MBL and HC clearly showed classification into two groups (Fig. 2E).
To further investigate the differences in proteome levels between MBL subgroups and HCs, pairwise comparison analyses were performed (Suppl. Fig. S5A). Pairwise analysis of Group 3 and Group 4 of MBL and HC revealed discrimination between them based on their protein molecular features, but not between Group 3 and Group 4 of MBL (Suppl. Fig. S5B-D and Suppl. Table S4). Next, we examined the protein expression levels of subgroups of MBL and HC divided by the presence of LMS (Suppl. Fig. S6A and Suppl. Table S5). The statistical analysis results revealed that DEPs could clearly distinguish between the LMS (+) and HC groups and the LMS (−) and HC groups. However, there was no clear separation observed between the LMS (+) and LMS (−) groups (Suppl. Fig. S6B-D).
Enrichment analysis of DEPs and canonical pathways revealed hub proteins
We further investigated 273 DEPs extracted from the comparison of MBL and HC through enrichment analysis of biological pathways using IPA software28. Among the 33 enriched canonical pathways that met the criteria (p value < 0.05 and ∣z∣ score > 0), 12 pathways were predicted to be activated in the MBL group, while 11 were predicted to be inhibited. The ‘Glycolysis 1’ pathway was the most highly activated canonical pathway in the MBL group, and the ‘Synaptogenesis Signaling’ pathway was the most highly inhibited (Fig. 3A). Subsequently, all proteins belonging to the activated or inhibited pathways in MBL were merged and subjected to PPI network analysis to identify hub proteins using betweenness centrality analysis. We found proteins with a high number of converged lines, which we referred to as hub proteins. The hub proteins in the activated pathway in the MBL group were GAPDH, CDH2, and PTPRS, while in the inhibited pathway, they were CD44, CDH2, and EFNB3 (Fig. 3B,C). GAPDH was upregulated in the MBL group, while CDH2, CD44 and PTPRS were downregulated (Suppl. Fig. S7A-B and Suppl. Table S3).

Analysis of canonical pathway enrichment. (A) Canonical pathway enrichment analysis using IPA with DEP data. The orange bar indicates a positive Z score, which is an activated pathway in the MBL group. The blue bar indicates a negative Z score, and an inhibited pathway in the MBL group. Color depth represents the degree of the Z score. The horizontal axis indicates −log10 (p value), and the vertical axis represents enriched canonical pathways; (B) PPI network analysis using the STRING database. Proteins for PPI network analysis were extracted from the following enriched canonical pathways: ‘Glycolysis’ and ‘Synatogenesis Signaling Pathway’. Hub proteins for the activated pathway and inhibited pathway in the MBL group are presented. Color depth and node size are expressed by betweenness centrality values.
Feature selection and target protein selection for validation
To identify proteomic signatures for the classification of the two groups, we extracted the top 10 features separately from the upregulated and downregulated DEPs in the MBL groups using two feature selection algorithms, ANOVA and FCBF (Table 1). We selected the top four ranked or overlapping features (excluding immunoglobulin G family proteins) based on the upregulated proteins in the MBL group compared to the HC group as biomarker verification candidates. The selected features, SPTBN1, HSP90AA1, TKT, and NME1-NME2 were taken for further validation. Additionally, the top four features chosen from each set of the top 10 ranked features were positioned within the top 40% abundance level among all quantified proteins (Suppl. Fig. S8).
Confirmation of upregulated TKT in MBL CSF by validation of the feature selected proteins using ELISA
The four feature-selected proteins (SPTBN1, HSP90AA1, TKT, and NME1) all showed significantly elevated normalized iBAQ intensity levels in MBL compared to HC, as observed from the LC‒MS/MS DEP results (Fig. 4A and Suppl. Table S3). ROC analysis demonstrated good performance of all four proteins in discriminating patients with MBL from HCs based on the normalized iBAQ intensity levels from the LC‒MS/MS DEP results (Fig. 4B).

Validation of feature selected proteins in MBL CSF through ELISA. The feature-selected proteins, SPTBN1, HSP90AA1, TKT, and NME1, were selected through ANOVA and FCBF. (A) Dot plots represent the comparison of normalized iBAQ intensity levels from LC‒MS/MS DEP results between the HC and MBL groups; (B) ROC curves show the diagnostic performance of each CSF protein for MBL with normalized iBAQ intensity levels. All four proteins showed significant differences with iBAQ intensity levels; (C) Dot plots represent the comparison of each CSF protein concentration of the HC and MBL groups using ELISA; (D) ROC curves show the diagnostic performance of each CSF protein for MBL at the absolute concentration level with ELISA. Only TKT showed a significant difference between the two groups. Notably, the concentration of TKT was 100 times greater than that of the other three proteins. Each dot represents the CSF protein level of an individual sample and horizontal lines in the dot plots show mean values. *p < 0.05, **p < 0.01, and ***p < 0.001.
To validate the selected proteins obtained through proteomics, ELISA analysis was performed. The concentration level of TKT was significantly higher in MBL than in HC, while SPTBN1, HSP90AA1, and NME1 levels did not show significant differences between MBL and HC (Fig. 4C). ROC analysis showed that TKT exhibited high specificity and sensitivity, with an AUC of 0.989, in discriminating patients with MBL from the HCs (Fig. 4D).
We also investigated whether each selected protein differed depending on the MBL subgroups or the presence of LMS. Three proteins (SPTBN1, HSP90AA1, and TKT) showed significantly elevated normalized iBAQ intensity levels in Group 3 and Group 4 MBL compared to HC, as observed from the LC‒MS/MS results. NME1 showed significant elevation only in Group 4 MBL compared to HC. (Suppl. Fig. S9A). ELISA results revealed that TKT levels were significantly higher only in Group 4 MBL compared to HC (Suppl. Fig. S9B). All four proteins were elevated regardless of LMS status, as indicated by the LC‒MS/MS results. In the ELISA analysis, the TKT concentration was significantly higher in the LMS (−) and the LMS (+) group than in the HC group (Suppl. Fig. S9C-D).
Enrichment of TKT was consistently observed in EV cargo proteins with MBL CSF
In both MBL and HC CSF samples, we observed the typical morphology of EVs, characterized by a round shape with a bilayer membrane. The presence of vesicles with an average diameter and concentration was identified in both groups using NTA (Suppl. Fig. S10 and Suppl. Table S6). CD9, CD63, and CD81 within the CSF were captured by the CD9 antibody were detected in both groups. In summary, there were no significant differences observed in the morphology, size, concentration, or exosome markers of EVs between these two groups.
Next, we identified TKT within EVs in both groups (Fig. 5A). Notably, an increase in staining intensity of TKT and quantitative enrichment were observed within MBL CSF EVs compared to HC (Fig. 5B). To validate the potential of EVs-TKT as a diagnostic marker for MBL CSF, we conducted ROC analysis and obtained an AUC value of 0.9333 (Fig. 5C). Subsequently, we assessed whether protein levels in CSF EVs could inform the subgroups or LMS burden in MBL patients. In subgroups, the number of TKT-positive EVs was significantly higher in Group 3, Group 4, and the unclassifiable group than in the HC group (Fig. 5D). The WNT subgroup was excluded for validation with ExoView since there was no WNT subgroup patient with ELISA validation. Regarding LMS, the number of TKT-positive EVs was significantly enriched in the LMS (+) group, and there was a positive correlation between the M stage and the number of TKT-positive EVs, r(12) = 0.63, p = 0.022 (Fig. 5E,F).

Validation with EV cargo proteins using ExoView demonstrates the significance of TKT in MBL CSF. (A) TEM images of purified EVs isolated from the CSF samples of HCs and MBL patients. EVs captured by the CD9 (blue) antibody were detected and observed simultaneously by green fluorescent tagged TKT (green). More TKT-positive EVs are present in MBL CSF; (B) Dot plot demonstrates that the number of TKT-positive EVs in CSF is significantly higher in the MBL group than in the HC group; (C) ROC curve shows the diagnostic performance of the number of TKT-positive EVs in ExoView; (D) Dot plot by MBL molecular subgroups demonstrates that Group 3, Group 4, and Unclassified MBL samples had significantly higher levels of TKT-positive EVs compared to the HC group; (E,F) Dot plot by LMS status demonstrates that the level of TKT-positive EVs is significantly higher in presence of LMS. Furthermore, there was a positive correlation between the M stage and the number of TKT-positive EVs, r(12) = 0.63, p = 0.022. Each dot represents the CSF EV cargo protein level of an individual sample, and horizontal lines in the dot plots show mean values. *p < 0.05, **p < 0.01, and ***p < 0.001.
Protein interactions and biological pathways of TKT by PPI network of DEPs and enrichment analysis of TKT and its interacting proteins
We first constructed a PPI network of all DEPs extracted from a comparison of the MBL and HC groups using the STRING database and Cytoscape (Fig. 6A). Then, we implemented PPI network analysis of TKT and its 11 interacting proteins (Fig. 6B). All proteins that interacted with TKT were upregulated in the MBL group. To clarify the biological functions of TKT and its 11 proteins, we performed enrichment analysis with GO and KEGG pathway analysis. Metabolic processes related to carbohydrates and nucleotides were the main gene ontology terms enriched from TKT centric PPI data (Fig. 6C and Suppl. Table S7). The main enriched KEGG pathways are the pentose phosphate pathway, gluconeogenesis, glycolysis and fructose and mannose metabolism, which are tightly connected in glucose metabolism (Fig. 6D).

Protein interactions of DEPs and enrichment analysis for TKT and its interacting proteins. (A) PPI network of DEPs extracted from the comparison of the HC and MBL groups. Each node (circle) represents a protein, the red node indicates an upregulated protein in the MBL group, and the blue node indicates a downregulated protein in the MBL group. The degree of fold change is expressed as color depth. The rectangular box highlights the region of TKT and its interacting proteins in whole PPI networks and TKT-centric enrichment analysis; (B) TKT-centric PPI network. PPI network data of TKT and its interacting proteins were extracted from the STRING database (derived from the combined interaction score (> 0.4) of the interaction source: ‘experimentally_determined’, ‘database_annotated’, and ‘coexpression’) and imported into Cytoscape with the fold change of each protein. The thickness of the edge indicates the degree of the combined interaction score value extracted from STRING. The color depth of nodes indicates the degree of fold change; (C) Dot plot of the top 15 GO enrichment results of TKT and its 11 interacting proteins; (D) Dot plot of KEGG pathway enrichment results of TKT and interacting proteins.
Discussion
To discover biomarker candidates, we focused on upregulated DEPs, as they offer advantages in terms of detection and potential targeting45,46. Consequently, four featured proteins from the upregulated DEPs were selected for further analyses. Among these four proteins, TKT was the only significant and quantitatively abundant protein in MBL CSF compared to HC CSF. Notably, TKT also existed in total CSF at a significantly higher concentration, since the scale bar for TKT was 100 times greater than that of the other three proteins. Based on the results of two orthogonal methods, LC‒MS/MS protein expression level and quantitative ELISA, upregulation of TKT was also validated by the CSF EVs of MBL patients. A significantly higher level of TKT-positive CSF EVs was present, especially in the non-WNT/non-SHH group and in the presence of LMS, which can be translated into practicable CSF biomarkers for MBL.
Our approach is the deepest CSF proteome analysis to date obtained by mass spectrometry at MBL, quantifying approximately 1100 proteins per sample. Moreover, our workflow is sensitive, using only 100 μl of CSF for sample preparation without depleting abundant proteins. Since human CSF is characterized by a dynamic range of protein abundance that can prevent the detection of proteins of interest, achieving in-depth CSF analysis previously required extensive fractionation or depletion of abundant proteins often combined with isobaric labeling, which has associated disadvantages47.
CSF EV proteins not only better reflect information derived from tumor cells but also have the potential to participate in specific functions and signal transduction, making them more important than CSF total proteins. In brain tumors, they are reported to play an important role in regulating the metastatic state or communicating with metastasized cells48. Although limited CSF volume prevented us from performing direct exosome isolation and proteomic analysis, which required CSF at milliliter scale, we introduced the ExoView system to examine specific proteins within EVs using minute CSF samples (< 10 μl).
TKT is an important enzyme of the pentose phosphate pathway that affects glycolysis and the production of antioxidants for cancer cells to survive49. The role of TKT in tumor development and progression has also been suggested in other solid tumors50,51. Specifically, TKT has shown a correlation with the invasion and metastasis of tumors. Our group previously mentioned the abundance of TKT in the gene expression of Group 3 and Group 4 MBL tissue samples52. This study is the first to suggest the importance of TKT in MBL CSF. It is noteworthy that upregulation of TKT was consistently observed not only in MBL tissue but also in CSF and CSF EVs. Although the prognostic significance of TKT needs further investigation, the association of TKT with Group 3 and Group 4, as well as LMS, may be related to the higher rate of metastasis at diagnosis in Group 3 and Group 4 MBL, along with their metastatic recurrence patterns53.
From a clinical point of view, the correlation of EVs with TKT and LMS of MBL is a promising result. Detecting LMS in MBL has been challenging due to the low yield of conventional cytology. As a result, liquid biopsy methods such as the detection of circulating tumor DNAs have been suggested in MBL54,55. The results of our data were also consistent in that cytology was negative in many patients with M2 and M3 stage LMS. Detection of EV cargo protein TKTs may be a potential biomarker for earlier, more sensitive diagnosis of LMS during treatment and follow up. However, since CSF was collected only once before the surgery in our study, the usefulness of EV TKT for the diagnosis of LMS could not be inspected. Further studies with CSF obtained preoperatively and during the follow-up period would be necessary to assess tumor control and recurrence status.
The limitation of this study is that due to the small number of CSF samples and the limited amount of CSF, cross-validation was not performed among the samples. As mentioned above, due to the retrospective nature of this study, there was insufficient CSF remaining for validation in some samples. Although the insufficient amount of CSF samples was a limitation of our study, it is also important to note that we performed the analysis with the minimal amount of CSF needed, enabling the use of remaining CSF for additional studies. In addition, differences in age and blood contamination of CSF in our study could introduce bias into the proteomics analysis. As mentioned earlier, the age difference was due to the younger age of patients with non-tumorous HC etiologies. Hemoglobin levels, due to blood contamination, can impact the quantification of proteins in CSF56. In our study, a higher degree of blood contamination was observed in the MBL group. MBL is a tumor prone to hemorrhage. Although no apparent intraventricular hemorrhages were noted, preoperative MRI scans of MBL patients with higher hemoglobin proportions revealed intratumoral micro hemorrhages. Regarding the interpretation of proteomics results, the top four features were positioned within the top 40% abundance levels among all quantified proteins (Suppl. Fig. S8). We cautiously presume that abundant proteins were less affected by blood contamination. However, stricter quality control of blood contamination could lead to more precise quantitative proteomics results.
In conclusion, our proteomics data of the total CSF were able to distinguish between MBL patients and tumor-free HCs. Furthermore, we identified the significance of TKT within the total CSF and its presence within circulating EVs in the CSF. This is the first comprehensive study to show the potential of TKT as a biomarker for MBL.
Data availability
The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD04598257.
References
Phi, J. H. et al. Cerebrospinal fluid M staging for medulloblastoma: Reappraisal of Chang’s M staging based on the CSF flow. Neuro Oncol. 13, 334–344. https://doi.org/10.1093/neuonc/noq171 (2011).
Choi, S. A. et al. In vivo bioluminescence imaging for leptomeningeal dissemination of medulloblastoma in mouse models. BMC Cancer 16, 723. https://doi.org/10.1186/s12885-016-2742-y (2016).
Garzia, L. et al. A hematogenous route for medulloblastoma leptomeningeal metastases. Cell 173, 1549. https://doi.org/10.1016/j.cell.2018.05.033 (2018).
Mumert, M. et al. Functional genomics identifies drivers of medulloblastoma dissemination. Cancer Res. 72, 4944–4953. https://doi.org/10.1158/0008-5472.CAN-12-1629 (2012).
Lin, X. et al. Cerebrospinal fluid circulating tumor cells: A novel tool to diagnose leptomeningeal metastases from epithelial tumors. Neuro Oncol. 19, 1248–1254. https://doi.org/10.1093/neuonc/nox066 (2017).
Im, J. H. et al. Experimental assessment of leptomeningeal metastasis diagnosis in medulloblastoma using cerebrospinal fluid metabolomic profiles. Metabolites 11, 851. https://doi.org/10.3390/metabo11120851 (2021).
Xiao, F. et al. Cerebrospinal fluid biomarkers for brain tumor detection: Clinical roles and current progress. Am. J. Transl. Res. 12, 1379–1396 (2020).
De Mattos-Arruda, L. et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 6, 8839. https://doi.org/10.1038/ncomms9839 (2015).
Thompson, A. G. et al. UFLC-derived CSF extracellular vesicle origin and proteome. Proteomics 18, e1800257. https://doi.org/10.1002/pmic.201800257 (2018).
Reiber, H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin. Chim Acta 310, 173–186. https://doi.org/10.1016/s0009-8981(01)00573-3 (2001).
Yuan, X. & Desiderio, D. M. Proteomics analysis of human cerebrospinal fluid. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 815, 179–189. https://doi.org/10.1016/j.jchromb.2004.06.044 (2005).
Lee, H. et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med. 22, 490–499. https://doi.org/10.1038/s41436-019-0672-1 (2020).
Weiss, N. et al. Cerebrospinal fluid metabolomics highlights dysregulation of energy metabolism in overt hepatic encephalopathy. J. Hepatol. 65, 1120–1130. https://doi.org/10.1016/j.jhep.2016.07.046 (2016).
Zhao, M. et al. A comprehensive analysis and annotation of human normal urinary proteome. Sci. Rep. 7, 3024. https://doi.org/10.1038/s41598-017-03226-6 (2017).
Wang, D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503. https://doi.org/10.15252/msb.20188503 (2019).
Teunissen, C. E. et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement (Amst) 2, 86–94. https://doi.org/10.1016/j.dadm.2015.12.004 (2016).
Thompson, A. G. et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 83, 258–268. https://doi.org/10.1002/ana.25143 (2018).
Wang, H. et al. Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat. Commun. 10, 3718. https://doi.org/10.1038/s41467-019-11661-4 (2019).
Choi, S. A. et al. Extracellular vesicle-associated miR-135b and -135a regulate stemness in Group 4 medulloblastoma cells by targeting angiomotin-like 2. Cancer Cell Int. 20, 558. https://doi.org/10.1186/s12935-020-01645-6 (2020).
Chang, C. H., Housepian, E. M. & Herbert, C. Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93, 1351–1359. https://doi.org/10.1148/93.6.1351 (1969).
Han, D. et al. In-depth proteomic analysis of mouse microglia using a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics 13, 2984–2988. https://doi.org/10.1002/pmic.201300091 (2013).
Rhee, S. J. et al. Comparison of serum protein profiles between major depressive disorder and bipolar disorder. BMC Psychiatry 20, 145. https://doi.org/10.1186/s12888-020-02540-0 (2020).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. https://doi.org/10.1038/nprot.2016.136 (2016).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 13, 2513–2526. https://doi.org/10.1074/mcp.M113.031591 (2014).
Jang, H. N. et al. Mass spectrometry-based proteomic discovery of prognostic biomarkers in adrenal cortical carcinoma. Cancers (Basel) 13, 3890. https://doi.org/10.3390/cancers13153890 (2021).
Cox, J. et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. https://doi.org/10.1021/pr101065j (2011).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. https://doi.org/10.1038/nmeth.3901 (2016).
Krämer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. https://doi.org/10.1093/bioinformatics/btt703 (2014).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612. https://doi.org/10.1093/nar/gkaa1074 (2021).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Tang, Y., Li, M., Wang, J., Pan, Y. & Wu, F. X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 127, 67–72. https://doi.org/10.1016/j.biosystems.2014.11.005 (2015).
Fleuret, F. Fast binary feature selection with conditional mutual information. J. Mach. Learn. Res. 5, 1531–1555 (2004).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216-w221. https://doi.org/10.1093/nar/gkac194 (2022).
Ashburner, M. et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Consortium, T. G. O. et al. The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031. https://doi.org/10.1093/genetics/iyad031 (2023).
Thomas, P. D. et al. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22. https://doi.org/10.1002/pro.4218 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Théry, C. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 7, 1535750. https://doi.org/10.1080/20013078.2018.1535750 (2018).
Lötvall, J. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell Vesicles 3, 26913. https://doi.org/10.3402/jev.v3.26913 (2014).
Comfort, N. et al. Isolation and characterization of extracellular vesicles in saliva of children with asthma. Extracell Vesicles Circ. Nucl. Acids 2, 29–48. https://doi.org/10.20517/evcna.2020.09 (2021).
Bachurski, D. et al. Extracellular vesicle measurements with nanoparticle tracking analysis—An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. J. Extracell Vesicles 8, 1596016. https://doi.org/10.1080/20013078.2019.1596016 (2019).
Silva, A. M. et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J. Extracell Vesicles 10, e12130. https://doi.org/10.1002/jev2.12130 (2021).
Archer, T. C. et al. Proteomics, post-translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 34, 396-410.e398. https://doi.org/10.1016/j.ccell.2018.08.004 (2018).
Hovestadt, V. et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. https://doi.org/10.1038/nature13268 (2014).
Al-Nesf, M. A. Y. et al. Prognostic tools and candidate drugs based on plasma proteomics of patients with severe COVID-19 complications. Nat. Commun. 13, 946. https://doi.org/10.1038/s41467-022-28639-4 (2022).
Park, J. H. et al. Moesin (MSN) as a novel proteome-based diagnostic marker for early detection of invasive bladder urothelial carcinoma in liquid-based cytology. Cancers (Basel) 12, 1018. https://doi.org/10.3390/cancers12041018 (2020).
Günther, R., Krause, E., Schümann, M., Blasig, I. E. & Haseloff, R. F. Depletion of highly abundant proteins from human cerebrospinal fluid: A cautionary note. Mol. Neurodegenerat. 10, 53. https://doi.org/10.1186/s13024-015-0050-7 (2015).
Huang, Z., Keramat, S., Izadirad, M., Chen, Z. S. & Soukhtanloo, M. The potential role of exosomes in the treatment of brain tumors, recent updates and advances. Front. Oncol. 12, 869929. https://doi.org/10.3389/fonc.2022.869929 (2022).
Xu, I. M. et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl. Acad. Sci. USA 113, E725-734. https://doi.org/10.1073/pnas.1508779113 (2016).
Hao, S. et al. The role of transketolase in human cancer progression and therapy. Biomed. Pharmacother. 154, 113607. https://doi.org/10.1016/j.biopha.2022.113607 (2022).
Ricciardelli, C. et al. Transketolase is upregulated in metastatic peritoneal implants and promotes ovarian cancer cell proliferation. Clin. Exp. Metastasis 32, 441–455. https://doi.org/10.1007/s10585-015-9718-1 (2015).
Park, A. K. et al. Subgroup-specific prognostic signaling and metabolic pathways in pediatric medulloblastoma. BMC Cancer 19, 571. https://doi.org/10.1186/s12885-019-5742-x (2019).
Juraschka, K. & Taylor, M. D. Medulloblastoma in the age of molecular subgroups: A review: JNSPG 75th Anniversary Invited Review Article. J. Neurosurg. Pediatrics PED 24, 353–363. https://doi.org/10.3171/2019.5.Peds18381 (2019).
Liu, A. P. Y. et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 39, 1519-1530.e1514. https://doi.org/10.1016/j.ccell.2021.09.012 (2021).
Miller, A. M. et al. Next-generation sequencing of cerebrospinal fluid for clinical molecular diagnostics in pediatric, adolescent and young adult brain tumor patients. Neuro Oncol. 24, 1763–1772. https://doi.org/10.1093/neuonc/noac035 (2022).
Paciotti, S. et al. Cerebrospinal fluid hemoglobin levels as markers of blood contamination: Relevance for α-synuclein measurement. Clin. Chem. Lab Med. 59, 1653–1661. https://doi.org/10.1515/cclm-2020-1521 (2021).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543-d552. https://doi.org/10.1093/nar/gkab1038 (2022).
Acknowledgements
We thank Ji Yoon Lee’s and Young Ki Hong's families for their donation to pediatric brain tumor research at the Seoul National University Children’s Hospital (3020190190 and 3020190180).
Funding
This work was supported by the Mid-career Researcher Program through an NRF grant funded by the Korean government (Ministry of Science and ICT, 2017R1A2B2008422 to SKK and 2022R1A2C1005851 to SAC.), Seoul National University Hospital (04-20200250), and SNUH Lee Kun-hee Child Cancer & Rare Disease Project, Republic of Korea (22A-017-0100).
Author information
Authors and Affiliations
Contributions
J.W.K.: Investigation, Resources, Data Curation, Writing-Original Draft; S.A.C.: Methodology, Validation, Resources, Visualization, Writing-Original Draft; K.D.: Methodology, Software, Formal analysis, Data Curation, Writing-Original Draft; E.J.K., J.H.P., and K.H.K.: Resources, Writing-Review & Editing; S.H.: Methodology, Validation; D.H. and S.K.K.: Conceptualization, Resources, Supervision, Funding acquisition, Writing-Review & Editing. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was approved by the Institutional Review Board of the Seoul National University Hospital (IRB No. 1905-121-1035), and this study was conducted according to the Declaration of Helsinki. The requirement for informed consent was waived by the Institutional Review Board of the Seoul National University Hospital owing to the retrospective nature of this study.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, J.W., Choi, S.A., Dan, K. et al. Proteomic profiling of cerebrospinal fluid reveals TKT as a potential biomarker for medulloblastoma. Sci Rep 14, 21053 (2024). https://doi.org/10.1038/s41598-024-71738-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-71738-z
This article is cited by
-
A Review of Microfluidic Chip-Based Strategies for Extracellular Vesicle Isolation: Bridging Traditional Methods and Clinical Translation
BioChip Journal (2026)
-
MYC promotes group 3 medulloblastoma cell proliferation and alleviates ROS-induced cell death by upregulating transketolase
Acta Neuropathologica Communications (2025)
