
Abstract
Fecal samples were collected from 640 individuals in Korea, including 523 patients with IBD (223 with Crohn’s disease [CD] and 300 with ulcerative colitis [UC]) and 117 healthy controls. The samples were subjected to cross-sectional gut metagenomic analysis using 16 S rRNA sequencing and bioinformatics analysis. Patients with IBD, particularly those with CD, exhibited significantly lower alpha diversities than the healthy subjects. Differential abundance analysis revealed dysbiotic signatures, characterized by an expansion of the genus Escherichia-Shigella in patients with CD. Functional annotations showed that functional pathways related to bacterial pathogenesis and production of hydrogen sulfide (H2S) were strongly upregulated in patients with CD. A dysbiosis score, calculated based on functional characteristics, highly correlated with disease severity. Markers distinguishing between healthy subjects and patients with IBD showed accurate classification based on a small number of microbial taxa, which may be used to diagnose ambiguous cases. These findings confirm the taxonomic and functional dysbiosis of the gut microbiota in patients with IBD, especially those with CD. Taxa indicative of dysbiosis may have significant implications for future clinical research on the management and diagnosis of IBD.
Similar content being viewed by others


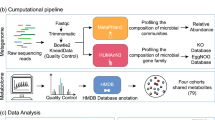
Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic immune-mediated inflammatory disorder of the digestive tract1. Gut microbes play a key role in the development of IBD, with metagenomic and multi-omics studies showing disease-specific signatures and functional profiles of gut microbiota in IBD2,3,4,5. However, inter-study variations, particularly regarding IBD-associated taxa and ecological effect sizes, have limited the ability to translate these metagenomic findings to clinical practice4.
While the prevalence of IBD in traditionally high-incidence regions such as the United States and Europe has stabilized, a marked increase in its incidence has been observed in areas previously considered to have low incidence, including Asia6,7,8. This increase is particularly pronounced in Korea, where both the incidence and prevalence of IBD have escalated significantly in recent decades9. Despite this, prior research has predominantly focused on Western populations. Several studies have examined the characteristics of the gut microbiota in Asian populations with IBD10,11,12; however, previous studies have often been limited by small sample sizes, the absence of healthy controls, or a lack of detailed functional annotations. Thus, region-specific, large-scale comparative studies are crucially required to understand the role of the gut microbiota in the pathogenesis of IBD in Asian populations.
Our systematic, large-scale study aims to fill this gap by characterizing the gut microbiome signature in Korean patients with IBD, comparing it to that of healthy controls matched for ethnicity and lifestyle. Notably, the healthy controls in this study were not merely non-IBD individuals but were rigorously selected healthy individuals who passed comprehensive screening tests, including hematology, urine, blood chemistry, and stool examinations. The patients with IBD who participated in this research were part of a larger cohort of Korean patients with IBD who donated stool, blood, and mucosal tissue samples for a multi-omics study (ClinicalTrials.gov identifier: NCT03589183). Subjects with IBD in a stable condition were carefully selected to minimize the impact of disease activity on the composition of the gut microbiota13.
Diagnosing IBD poses significant challenges for physicians due to the absence of a standardized diagnostic approach, relying instead on a combination of clinical assessments including patient history, clinical features, endoscopy, biopsy, imaging, fecal examination, blood tests, and the physician’s observations14. The difficulty in distinguishing between CD and UC further complicates patient care, leading to cases of “indeterminate colitis” where patients may not receive the most effective treatment, consequently increasing healthcare costs15. To mitigate these diagnostic challenges, biomarkers that can facilitate early and precise IBD diagnosis must be developed. Previous molecular analyses have identified associations between certain microbial taxa and their products and IBD; however, the clinical application of these findings as biomarkers remains to be completely realized16,17,18. Addressing the heterogeneity in disease phenotype and treatment response, our study aimed to identify microbial markers capable of differentiating healthy individuals from those with IBD, and more specifically, distinguishing between CD and UC. Identifying such markers not only has the potential to reduce the incidence of misdiagnosis but also enables the early detection and tailored treatment of these conditions.
The increasing prevalence of IBD in Korea, coupled with the wealth of regional data from joint cohort studies, provides a unique opportunity to explore the epidemiology and regional characteristics of the disease. Our study seeks to leverage this knowledge to pinpoint gut microbial markers that elucidate the pathophysiological distinctions between healthy individuals and patients with IBD (CD or UC). Through a combined analysis of the taxonomic and functional microbial profiles, this study endeavors to fill a critical research void. By identifying specific gut microbial biomarkers, we aim to advance the development of targeted diagnostic and therapeutic strategies, thereby catering to the unique pathophysiological traits of IBD in East Asian populations.
Results
Demographic characteristics of the study population
The baseline characteristics of the study population are summarized in Table 1. The study included 523 patients with IBD (CD, n = 223; UC, n = 300) and 117 healthy subjects, with the latter being healthy subjects selected through strict screening tests (Fig. 1). Of these 171 recruited healthy subjects, 54 were eliminated through the selection criteria, and 117 healthy subjects, of mean age 42.1 years, were finally selected. The 523 patients with IBD were of mean age 41.87 years and a mean disease duration of 8.8 years (Table 1). The proportion of male subjects was 64% in patients with IBD (75.3% in CD; 56% in UC) and 35.9% in healthy subjects. These characteristics reflect the epidemiological characteristics of patients with IBD in the Korean population over the recent decade19. The mean Bristol stool scale (BSS) scores were 4.29 in patients with CD, 4.02 in patients with UC, and 4.00 in healthy subjects. Among patients with CD and UC, 77.1% and 26.3%, respectively, were receiving biologics. The mean Harvey–Bradshaw index (HBI) and Mayo score in patients with CD and UC were 1.93 and 1.76, respectively. Comparisons of their baseline characteristics showed significant differences in age, sex, BMI, height, smoking rate, and BSS between the CD and UC groups. These clinical factors were judged to act as confounding factors in the process of analyzing differential abundant features and were analyzed by applying these factors as confounding variables in multivariable association analysis.

Inclusion and exclusion criteria for selecting healthy subjects and standard values for each test item. Detailed selection criteria for non-IBD healthy subjects are described in the healthy participants subsection of the section on enrollment of study participants in the supplementary text file. AKI, acute kidney injury; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BSS, Bristol stool scale; BUN, blood urea nitrogen; CKD, chronic kidney disease; DBP, diastolic blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; RBC, red blood cell; SBP, systolic blood pressure; WBC, white blood cell.
Overview of bacterial community structure and microbial community diversity
Patients with IBD (CD and UC) had significantly lower species richness, species evenness, and population diversity than healthy subjects (Fig. 2a–c). The reduction in the alpha diversity was more prominent in patients with CD than that in patients with UC (P < 0.05). Sub-categorization of fecal samples according to gender and age showed that the community diversity of gut microbiota was significantly lower in patients with IBD (CD and UC) than that in healthy subjects (Supplementary Fig. 1a–b and Supplementary Table 1). An increase in stool consistency was negatively associated with community diversity in patients with UC (Kruskal-Wallis chi-squared = 12.865, df = 6, P = 0.04523); however, this trend was not observed in healthy subjects and patients with CD (Supplementary Fig. 1c and Supplementary Table 2).

Diversity of the gut microbiota in healthy subjects, in patients with CD, and in patients with UC. (a–c) Comparisons of the alpha-diversity indices among the three patient groups. The intra-individual bacterial diversity within the samples was measured by determining (a) the abundance-based coverage estimator (ACE) for species richness, (b) Shannon’s evenness index for species evenness, and (c) inverse Simpson’s index for community diversity. The alpha-diversity values for each group are presented as box plots. The lines, boxes, and whiskers in the box plots represent the median, 25th and 75th percentiles, and the minimum-to-maximum distributions of replicate values, respectively. (d–e) PCoA, based on the (d) unweighted and (e) weighted UniFrac distance matrix, of the bacterial 16 S rRNA gene sequence data for fecal samples from healthy subjects (n = 117), patients with CD (n = 223), and patients with UC (n = 300). (f–g) Differences in alpha diversity according to disease behavior types (inflammatory, stricturing, and penetrating) in the CD group are presented as (f) richness and (g) evenness. (h) PCoA, based on the weighted UniFrac distance matrix, of the bacterial 16 S rRNA gene sequence data for fecal samples is shown according to the disease behavior types (general vs. advanced types) in patients with CD (n = 223). The data were analysed using the Kruskal-Wallis test (a, b, c, f, and g) and PERMANOVA, with 999 permutations (d–e) to obtain the statistical significance. CD, Crohn’s disease; UC, ulcerative colitis; PCoA, Principal coordinate analysis; CD-Inflammatory, inflammatory type of CD; CD-Stricturing + Penetrating, stricturing and penetrating types of CD.
The gut microbiota of the overall study population belonged mainly to four families (Supplementary Fig. 2a): Bacteroidaceae (29.09%), Ruminococcaceae (18.10%), Lachnospiraceae (15.59%), and Prevotellaceae (14.03%). At the genus level, four genera, Bacteroides, Faecalibacterium, Lachnospira, and Escherichia-Shigella, were predominant (Supplementary Fig. 2b). Principal coordinate analysis (PCoA) of the unweighted and weighted UniFrac distance matrix based on the abundance of ASVs showed distinct bacterial community separation across the study groups (permutational multivariate analysis of variance [PERMANOVA]: P = 0.001, R2 = 0.035 for a comparison of healthy subjects and patients with CD; P = 0.001, R2 = 0.027 for a comparison of healthy subjects and patients with UC; P = 0.001, R2 = 0.016 for a comparison of patients with CD and UC; Fig. 2d and e and Supplementary Fig. 3). Given the complexity of associations between human disease and the gut microbiome, a single factor, such as disease status, cannot explain a large proportion of gut microbiome variation3,4,20. Considering this, the PC1 values (9.63% and 3.47% for unweighted and weighted UniFrac distance) and R squared values (ranging from 1.0 to 3.5%) observed in the current study are comparable to those observed in related studies mentioned previously.
In particular, the alpha diversity tended to decrease as the severity of disease increased (advanced behaviors, stricturing and penetrating types) in patients with CD (Fig. 2f–g and Supplementary Fig. 4b); however, it did not decrease with increased severity (extensive colitis) in patients with UC (Supplementary Fig. 4a, c). In addition, the microbial community differed significantly according to disease behavior in patients with CD (PERMANOVA: P = 0.001, R2 = 0.016; Fig. 2h and Supplementary Fig. 5a), whereas not in patients with UC (Supplementary Fig. 5b and c). These findings confirmed that the gut microbial community differed significantly among patients with IBD and healthy subjects and that the gut microbial community differed depending on disease severity, particularly in patients with CD.
Taxonomic signatures of IBD-associated gut dysbiosis
A microbiome multivariable association analysis was performed using the MaAsLin2 model to determine differentially abundant microbial taxa associated with IBD subtypes after adjustment for multiple covariates affecting the composition of gut microbiota. The genera Agathobacter, Alistipes, Blautia, Phascolarctobacterium, Fusicatenibacter, Lachnospira, Subdoligranulum, Ruminococcus, Barnesiella, Paraprevotella, Butyricicoccus, Coprococcus, and Christensenella showed statistically significant negative coefficients (false discovery rate, FDR < 0.01), indicating a tendency to decrease in both the patients with CD and UC compared with that of the healthy control group (Fig. 3a–c and Supplementary Fig. 6). Numerous beneficial bacteria, mostly producers of short-chain fatty acids (SCFAs), were depleted in both patients with CD and UC (Fig. 3c). However, no genus increased significantly in both the CD and UC groups. In the patients with CD, only the genus Escherichia-Shigella showed a significant (FDR < 0.01) increase compared with that in the healthy control group (Fig. 3a, d). In the patients with UC, the genera Faecalibacterium, Bacteroides, Dialister, Flavonifractor, Faecalitalea, Negativibacillus, and Ruminiclostridium significantly increased (FDR < 0.01) compared with those in the healthy control group (Fig. 3b, d). These results indicated that the composition of the gut microbiome differs in patients with IBD and healthy subjects; patients with IBD, especially those with CD, having a pronounced gut dysbiosis, consistent with previous findings that the genus Escherichia-Shigella dominates the gut.

Differentially abundant genera in the healthy subjects, patients with CD, and patients with UC. (a–b) Differentially abundant genera in (a) patients with CD and (b) with UC compared with those in healthy subjects. Only significant taxa (FDR < 0.01) are shown. The FDR values were based on multivariable generalized linear regression model analyses adjusted for stool consistency, age, sex, body mass index, smoking and alcohol consumption, and disease activity of IBD. Numerical values indicate coefficients. (c) Relative abundance of the genera enriched in healthy subjects compared with those in patients with CD and UC. (d) Relative abundance of genera enriched in patients with CD and patients with UC compared with those in healthy subjects. The relative abundances of differentially abundant genera for each group are presented as box plots. The lines, boxes, and whiskers in the box plots represent the median, 25th and 75th percentiles, and the minimum-to-maximum distributions of replicate values, respectively. (c–d) *P < 0.05, **P < 0.01, and ***P < 0.001 using the Kruskal-Wallis test. CD, Crohn’s disease; UC, ulcerative colitis.
Functional signatures of IBD-associated gut dysbiosis
To infer functional differences in microbiota for each group, the abundances of KEGG orthologs (KOs) and enzyme commission categories (EC) in each sample were predicted through the PICRUSt2 pipeline from the 16 S rRNA abundance table. Using the abundance information of functional orthologs (KO and EC) predicted using the taxa abundance profile and genome information, the Jensen–Shannon Distance (JSD) values between samples were calculated and plotted on a second plane using the PCoA (Fig. 4a and b and Supplementary Fig. 7a and b). These results showed that the functional community of the healthy group was concentrated in one place, whereas the functional community of the disease group, especially the CD group, was dispersed (PERMANOVA: P = 0.001 for a comparison of healthy subjects and patients with CD, P = 0.001 for healthy subjects and patients with UC, and P = 0.001 for patients with CD and UC; Fig. 4a). The findings suggest that, although the gut microbial community may exhibit distinct taxonomic diversity amongst healthy subjects, functional community was generally maintained, indicating a degree of functional redundancy within the microbial community units. Conversely, in diseased states, significant shifts occurred not only in the composition of the microbial community but also in the functional dynamics of these community units. Application of the Anna Karenina principle to the above results showed that microbial community composition varied more in dysbiotic individuals than in healthy individuals, resulting in functional dyshomeostasis in the former and indicating that the balance may be broken in disease groups21.

Gut microbial functional pathways and functional feature profiles in healthy subjects, patients with CD, and patients with UC. (a–b) PCoA, showing the abundance of functional genes in each sample by groups based on (a) KEGG orthologs (KOs) and (b) enzyme commission categories (ECs) predicted using taxa abundance profiles and genome information. (c–d) Predicted (c) KEGG and (d) MetaCyc metabolic pathways differing significantly in patients with CD and healthy subjects. The FDR values were based on a multivariable generalized linear regression model adjusted for stool consistency, age, sex, body mass index, smoking and alcohol consumptions, and disease activity of IBD. Only significant pathways (FDR < 0.01) are shown. Numerical values indicate coefficients. (e) Heatmap representing KEGG pathways related to sulfur metabolism in patients with CD and healthy subjects, based on significantly different KO abundances in these two groups. Positive z-scores are shown in red color, and negative z-scores are shown in blue. f Volcano plot representing KOs related to sulfur metabolism and sulfur relay system, based on significantly different KO abundances in patients with CD and healthy subjects. KOs related to pathways for producing cysteine from taurine are shown in red, and KOs related to pathways for producing H2S from cysteine are shown in blue. Statistical significance was determined using PERMANOVA based on 999 permutations (a–b). CD, Crohn’s disease; UC, ulcerative colitis; KEGG, Kyoto Encyclopedia of Genes and Genomes; PCoA, Principal coordinate analysis.
Through the predicted KO and EC distributions, significant differences (FDR < 0.05) in functional features constituting the intestinal microbiome between healthy subjects and patients with each disease (CD and UC) were evaluated using the MaAsLin2 tool. In patients with CD especially, functional pathways related to pathogenesis, such as shigellosis, bacterial invasion of epithelial cells, lipopolysaccharide biosynthesis, biofilm formation of Escherichia coli, and bacterial secretion system were identified as abundant KEGG and MetaCyc metabolic pathways (Fig. 4c and Supplementary Figs. 8 and 9). In healthy subjects, however, MetaCyc metabolic pathways related to SCFA production, such as methanogenesis from acetate, heterolactic fermentation, and Bifidobacterium shunt, were enriched (Fig. 4d).
Recently, hydrogen sulfide (H2S) has been suggested to play a potential role in the etiology of bowel disorders such as IBD, particularly UC, colorectal cancer, and irritable bowel syndrome, likely by delaying transit in constipation-predominant patients22,23,24,25. Until now, the dissimilatory sulfate reduction and oxidation pathway, in which H2S is produced by sulfate-reducing bacteria, has been widely studied in IBD. The present study revealed that pathways related to sulfur metabolism and the sulfur relay system were enriched in patients with CD (Fig. 4e). The pathway in which cysteine is produced from taurine and the four pathways in which H2S is produced from cysteine were enriched in CD patient group, which is not a previously well-known pathway in the IBD study (Fig. 4f)26,27. The taurine to cysteine bacterial pathway was completed in patients with CD, and the taurine to cysteine–associated KOs were significantly enriched in patients with CD compared with that in healthy subjects (Fig. 4f). Meanwhile, at least four different pathways are present for bacterial degradation of cysteine26,27. The bacterial degradation of cysteine pathway was enriched in the patients with CD, and the bacterial degradation of cysteine pathway–associated KOs were significantly enriched in the patients with CD compared with those in healthy subjects (Fig. 4f). These findings suggested that H2S, produced from taurine and cysteine not sulfate or sulfide, is related to the CD phenotype, with functional analyses predicting that these bacterial pathways might be completed in the guts of patients with CD.
Assessment of IBD phenotype using microbial function-based dysbiosis score
The abundance information of functional orthologs (KOs) predicted using the taxa abundance profiles and genome information were used to calculate the JSD values between samples. The resulting JSD distance matrix was used to calculate the dysbiosis score, representing the degree of dysbiosis3. The dysbiosis scores for all samples ranged from 0.09 to 0.47, peaking at 0.18 in the healthy subjects. In this study, dysbiosis was defined as functional dysbiosis in the microbial population for samples with high scores, based on the threshold of 95% of the dysbiosis score in healthy subjects. The functional community of the eubiosis group was concentrated in one place, whereas the functional community of the dysbiosis group was dispersed (PERMANOVA: P = 0.001 for a comparison of the eubiosis and dysbiosis groups; Fig. 5a and b). Samples of functional eubiosis and dysbiosis were respectively matched to taxonomic bacterial profiles, then represented in PCoA plots, which were based on both unweighted and weighted UniFrac distance matrices. The microbial communities of the eubiosis and dysbiosis groups were largely separated (PERMANOVA: P = 0.001; Fig. 5c and d and Supplementary Fig. 10). These results identified a concordant pattern, in which clusters of functional dysbiosis and eubiosis and taxonomic dysbiosis and eubiosis were separated, representing patterns consistent with Anna Karenina effects.

Evaluation of dysbiosis based on functional feature profiles. (a) PCoA schematizing eubiosis and dysbiosis samples based on KEGG ortholog profiles. (b) PCoA schematizing eubiosis and dysbiosis samples based on enzyme commission category profiles. (c–d) PCoA plots according to eubiosis and dysbiosis groups of bacterial 16 S rRNA gene sequences based on (c) unweighted and (d) weighted UniFrac distance matrices. (e) Distribution of dysbiosis and eubiosis samples within groups of healthy subjects, patients with CD, and patients with UC. (f) Distribution of dysbiosis and eubiosis samples according to disease behavior in patients with CD. (g) Distribution of dysbiosis and eubiosis samples according to ANCA positivity in the disease group. (h–i) WBC counts (h) and ((i) fecal calprotectin levels between eubiosis and dysbiosis groups in the disease group. (j–l) Comparisons of alpha-diversity indices in the eubiosis and dysbiosis groups. Intra-individual bacterial diversity within samples was determined by measuring (j) the abundance-based coverage estimator (ACE) for species richness, (k) Shannon’s evenness index for species evenness, and (l) inverse Simpson’s index for community diversity. (m–n) Abundance-based coverage estimator for species richness in the eubiosis and dysbiosis groups of patients with (m) CD and (n) UC. Alpha-diversity values for each group are presented as box plots. The lines, boxes, and whiskers in the box plots represent the median, 25th and 75th percentiles, and minimum-to-maximum distributions of replicate values, respectively. (o Differentially abundant genera in the eubiosis and dysbiosis groups. (p) Predicted KEGG metabolic pathways differing significantly in the eubiosis and dysbiosis groups. Only significant taxa (false discovery rate [FDR] < 0.01) are indicated. Numerical values indicate coefficients. Statistical significance was analyzed using Chi-square tests (e–g), Wilcoxon rank sum pairwise comparison tests (h–n), and PERMANOVA, with 999 permutations (a–d). CD, Crohn’s disease; UC, ulcerative colitis; PCoA, Principal coordinate analysis; Infla, inflammatory type in CD patient group; Penet, penetrating type in CD patient group; Strict, stricturing type in CD patient group; ANCA positivity, positivity rate of perinuclear anti-neutrophil cytoplasmic antibody; WBC, whole blood cell.
Of the 117 healthy subjects, six (5.1%) were assigned to the dysbiosis group (Fig. 5e). Functional dysbiosis was evaluated in 49.8% (111/223) samples from the CD group and 21.7% (65/300) samples from the UC group (Fig. 5e). These findings confirmed that functional bacterial imbalance rarely occurred in healthy subjects, however, was common in patients with CD and UC (chi-square: P < 0.0001; Fig. 5e). Clinical data (disease behavior, disease location, age at diagnosis, prognosis, whole blood cell count [WBC], CRP, and ESR) were subsequently compared in the function-based dysbiosis and eubiosis groups. Dysbiosis has been associated with disease behavior in patients with CD. Analysis of the CD group showed that the rate of dysbiosis was significantly higher in samples from patients with penetrating and stricturing types of CD (advanced behavior type) than that in samples from patients with inflammatory type of CD (chi-square: P = 0.0049; Fig. 5f). Additionally, the odds ratio (OR) of advanced IBD types (CD-stricturing and penetrating types and UC-extensive type) between the eubiosis and dysbiosis groups was assessed. Unlike those of patients with UC, the ORs of patients with IBD and CD with advanced disease phenotypes were higher (> 2) in the dysbiosis group (Supplementary Fig. 11). Subsequently, the perinuclear anti-neutrophil cytoplasmic antibody (ANCA) positivity rate and WBC levels were significantly higher in the dysbiosis group, but fecal calprotectin levels were not significantly different (chi-square: P < 0.0001; Fig. 5g, Wilcoxon rank sum pairwise comparison: P = 0.0085; Fig. 5h, Wilcoxon rank sum pairwise comparison: P = 0.14; Fig. 5i).
Dysbiotic signatures of the concordant gut microbiome in the function-based dysbiosis group and in patients with IBD
Species richness, species evenness, and population diversity were significantly lower in samples from the dysbiosis than those in samples from the eubiosis group in patients with IBD (Fig. 5j–l). Subgroup analysis of patients with CD and UC also showed that species richness and evenness were significantly lower in samples from the dysbiosis than in those from the eubiosis group (Fig. 5m and n). Analysis of differentially abundant taxa showed that the genera Escherichia-Shigella and Klebsiella belonging to the family Enterobacteriaceae and the genus Veillonella belonging to the family Veillonellaceae were significantly (FDR < 10− 5) more abundant in the dysbiosis group than in the eubiosis group (Fig. 5o and Supplementary Fig. 12a). Functional KEGG pathways found to be enriched in the dysbiosis group included pathogenesis-related pathways, such as Shigellosis, bacterial invasion of epithelial cells, and Staphylococcus aureus infection (Fig. 5p and Supplementary Fig. 12b). Contrarily, pathways related to SCFA production through fermentation, such as Bifidobacterium shunt, heterolactic fermentation, and succinate fermentation to butanoate, were depleted in the dysbiosis group (Supplementary Fig. 12b). These results suggested that the taxonomic and functional characteristics of the function-based dysbiosis group align with those of the disease group. Furthermore, these characteristics appear to be closely associated with the severity of disease, especially in patients with CD.
Microbial biomarkers for disease diagnosis
To identify microbial markers that can distinguish healthy subjects from patients with IBD, patients with CD, and patients with UC; patients with CD from patients with UC group; and to evaluate the discriminative power of these markers, significantly differentially abundant genera (DAGs) were selected for each of the four comparisons. Subsequently, age, sex, body mass index, smoking status, alcohol consumption, and BSS were applied as confounding variables using the MaAsLin2 tool and DAGs that differed significantly (FDR < 0.05) in each of the four comparisons were determined (Supplementary Tables 3–6).
From training a classification model using the fraction data for each sample of the genera corresponding to the selected DAG, the optimal number of markers providing the lowest classification errors were estimated to be two each for comparisons of healthy subjects with patients with IBD (Fig. 6a) and with patients with CD (Fig. 6d) and three each for comparisons of healthy subjects with patients with UC (Fig. 6g) and patients with UC with patients with CD (Fig. 6j). To distinguish between the healthy subjects and patients with IBD, Lachnospiraceae_NK4A136_group (16.5%) and Coprococcus_3 (15%) contributed the maximum to classification accuracy (Fig. 6b), with the application of these two genera leading to accurate results in classifying these two groups (AUROC, Area Under Receiver Operating Characteristic curve = 0.845, Fig. 6c). To distinguish between the healthy subjects and patients with CD, Ruminococcaceae_UCG-010 (14.2%) and Ruminococcaceae_UCG-014 (13.6%) contributed the most to classification accuracy (Fig. 6e), and the application of these two genera led to the most accurate results in classifying these two groups (AUROC = 0.860, Fig. 6f). To distinguish between the healthy subjects and patients with UC, Akkermansia (16.5%), Coprobacter (15.6%), and Christensenellaceae_unclassified (14.5%) made the greatest contribution to classification accuracy (Fig. 6h), with application of these three genera showing high classification ability (AUROC = 0.826, Fig. 6i). Additionally, to distinguish between the patients with CD and with UC, Veillonella (37.7%), Enterobacteriaceae_unclassified (20.6%), and Escherichia-Shigella (20.1%) contributed the highest to classification accuracy (Fig. 6k), with the application of these three genera showing high classification ability (AUROC = 0.718; Fig. 6l).

Evaluation of the performance of microbial markers in distinguishing among healthy subjects, patients with CD, and patients with UC using differentially abundant genera. (a) Optimal number of genus markers in a random forest model based on classification errors comparing healthy subjects and patients with IBD. (b) The importance of marker candidates evaluated in the training process of the random forest model in comparisons of healthy subjects and patients with IBD. (c) Classification ability of the random forest model trained with the two genera selected as markers in distinguishing between healthy subjects and patients with IBD. (d) Optimal number of genus markers in a random forest model based on classification errors comparing healthy subjects and patients with CD. (e) The importance of marker candidates evaluated in the training process of the random forest model in comparisons of healthy subjects and patients with CD. (f) Classification ability of the random forest model trained with the two genera selected as markers in distinguishing between healthy subjects and patients with CD. (g) Optimal number of genus markers in a random forest model based on classification errors comparing healthy subjects and patients with UC. (h) The importance of marker candidates evaluated in the training process of the random forest model in comparisons of healthy subjects and patients with UC. (i Classification ability of the random forest model trained with the three genera selected as markers in distinguishing between healthy subjects and patients with UC. (j) Optimal number of genus markers in a random forest model based on classification errors comparing patients with CD group and patients with UC. (k) The importance of marker candidates evaluated in the training process of the random forest model in comparisons of patients with CD and patients with UC. (l) Classification ability of the random forest model trained with the three genera selected as markers in distinguishing between patients with CD and patients with UC. (a, d, g, and j) The red arrows indicate the optimal number of genus markers in the random forest models. (b, e, h, and k) The genera marked with a star in the shaded area indicated by the red dotted line are the taxa contributing to the highest level of classification accuracy. The classification power of the random forest model trained by applying the optimal number of markers was evaluated by calculating AUROC. CD, Crohn’s disease; UC, ulcerative colitis; OOB Error, out-of-bag error; AUROC, Area Under the Receiver Operating Characteristic curve.
Discussion
The present study analyzed microbial marker taxa that can discriminate between healthy individuals and patients with IBD (CD or UC) in an East Asian cohort. This study revealed that the use of only two or three microbial taxa could accurately distinguish between healthy individuals and patients with IBD, including individual groups of patients with CD and UC, as well as between the latter two groups. The presence of specific microbes in the intestinal mucosa or stool samples of Spanish and Chinese patients with IBD and controls, as determined using quantitative polymerase chain reaction (qPCR) assays, showed good classification ability16,28. Numerous bacterial markers for IBD have also been identified using machine learning models based on 16 S rRNA sequences13,17,18,20. Metagenomic sequencing with a higher taxonomic resolution is increasingly applied to identify microbial markers; however, this method is both expensive and inefficient29,30. The potential role of fungi and viruses in diagnosing IBD has not been extensively studied because the amounts of fungal and viral DNA in the gut are much lower than the amount of bacterial DNA, and fungal and viral genomic references are limited31.
Moreover, the above studies predominantly included patient cohorts from Europe and North America, with only a few Asian cohorts represented. The similarities and differences in gut microbes among different races and ethnicities must be determined32,33. However, universal application of the same microbial biomarkers may not be warranted, as the epidemiological and pathological characteristics of diseases vary by race and region, and the scales of clinical metadata collection differ. Universal and region-specific microbial markers should be developed for more accurate diagnoses. Contrary to previous studies, the current population study based on 16 S rRNA sequencing evaluated the predictive accuracy of a small number of microbial markers. Future studies should apply these markers to large-scale datasets, both to verify their performance and to explore their clinical applicability and potential uses.
The fecal calprotectin test, currently used as a diagnostic marker for IBD, is an effective non-invasive test that uses stool samples to reflect the presence of inflammation, and is believed to be particularly useful for early diagnosis and treatment monitoring. One advantage of this test is that it is low-burden and cost-effective for patients. Moreover, reviews and meta-analyses have demonstrated that the test is highly reliable for diagnosing IBD, with a sensitivity of around 83–100% and a specificity of around 60–100%.34,35 An important limitation of fecal calprotectin is that it is useful for differentiating IBD from normal or functional disease, as well as for post-diagnostic monitoring of IBD. However, it is not effective for differentiating IBD subtypes. Given this limitations of the fecal calprotectin test, a strength of our study is the ability to distinguish between CD and UC. Approximately 20% of patients with IBD are classified as IBD unclassified, where CD and UC cannot be differentiated, leading to a lack of clear diagnostic criteria and delays in diagnosis and treatment36. Accurate differentiation of CD from UC is therefore an important area of research. Representative serological markers used for the diagnosis of IBD and its subtypes include pANCA and ASCA autoantibodies37,38. However, the sensitivity and specificity of these tests are limited, especially when used to differentiate CD from UC, and are not recommended due to clear limitations, so the discovery of new diagnostic markers is essential. Although the primary IBD diagnostic potential is inferior to that of fecal calprotectin-based tests, we expect the diagnostic potential of gut (fecal) microbial markers to contribute secondarily to this, as fecal calprotectin testing based on the presence of intestinal inflammation cannot, in principle, differentiate between IBD subclasses, including CD and UC. However, the diagnostic utility of fecal calprotectin is proven and widely used, so we will consider and evaluate it in future studies.
Functional analysis revealed that functional pathways associated with bacterial pathogenesis, such as shigellosis, bacterial invasion of epithelial cells, lipopolysaccharide biosynthesis, biofilm formation of Escherichia coli, and bacterial secretion system, were strongly upregulated in CD patients, in agreement with several recent studies39,40,41. Furthermore, metabolic pathways, including those involved in sulfur metabolism, the sulfur relay system, and cysteine metabolism, were strongly enriched in patients with CD, along with significant enrichment of the genus Escherichia-Shigella. H2S is a highly reactive gas that participates in various physiological activities in mammals, such as inflammatory responses, reduced intestinal motility, gastrointestinal blood flow, ulcer healing, secretory functions (saliva, gastric acid, pancreatic, bile, and intestine), gastroprotection, and the cardiac system42,43. However, exposure to high concentrations of this gas is deleterious to the host. Although sulfate-reducing bacteria are considered principally responsible for H2S production44, the enzymatic desulfhydration of cysteine by heterotrophic bacteria also contributes to the production of H2S45 Several other microbial taxa produce H2S from the breakdown of SO32−, taurine, cysteine and sulfated organic compounds, with some gut microbes using taurine as an electron acceptor for anaerobic respiration46,47,48. Taurine, which is transported into microbial cells through an ATP-dependent uptake system, is degraded via several pathways, resulting in the release of SO32−.49 Intracellular taurine is desulfonated by TauD or SsuD, resulting in the generation of SO32− and aminoacetaldehyde50. SO32− is subsequently converted to H2S by a dissimilatory sulfite reductase that differs structurally from the enzymes used by sulfate-reducing bacteria. The results of the present study confirmed that the bacterial metabolic pathway for production of H2S from taurine was prominently enriched in patients with CD. Bacteria contain four different pathways for the degradation of cysteine, involving the enzymes cysteine desulfhydrase (CDS), cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST), resulting in the production of pyruvate, serine, NH3 and H2S51. Recently, cysteine desulfurase (IscS) was observed to be responsible for the synthesis of H2S from cysteine in E. coli under anaerobic conditions27. The present study confirmed enrichment of bacterial metabolic pathways that produce H2S, not the dissimilatory sulfate reduction pathway, which was found to be abundant in patients with CD.
Élie Metchnikoff suggested that most diseases are thought to begin in the digestive tract, when the “good” bacteria are no longer able to control the “bad” bacteria52. This condition, called “dysbiosis,” implies a disruption in the mutually harmonious gut ecosystem. In contrast, “eubiosis” generally refers to a state in which the microbial community is balanced, although this condition has not been precisely defined. As an example, an eubiotic state of the gut microbiome of a mature mammal is primarily characterized by the predominance of several species belonging to two bacterial phyla (Bacillota and Bacteroidota), with potentially pathogenic species, such as those from the phylum Pseudomonadota, being present in low abundance53. The present study revealed that the gut microbiomes of patients with CD and UC differed significantly from the gut microbiomes of healthy subjects, with particularly large inter-individual variations in patients with IBD. This microbiome variability associated with the disease has been described as an Anna Karenina principle, with dysbiotic individuals exhibiting more variation in microbial community composition than healthy individuals21. This parallels Leo Tolstoy’s dictum that “all happy families are alike; each unhappy family is unhappy in its own way.” The effects of this principle are common to many disturbed animal microbiomes, with important pathogens, environmental stressors, and immune dysfunctions leading to increased stochasticity in the microbiome54,55,56,57,58,59. Collectively, the associations of microbiome instability with many stressors and diseases suggest that microbiome resistance and resilience are hallmarks of healthy physiology.
The present study had several strengths. The control group did not consist simply of non-IBD patients; rather, strict criteria were applied for the selection of the 117 healthy subjects. Moreover, this was a large-scale, meticulously planned investigation conducted in Korea, a country in East Asia with a rapidly increasing prevalence rate of IBD. Fluctuations in IBD activity and/or intensified medications may contribute to intra-individual instability of the gut microbiota over time13. Therefore, to adjust for disease activity as a covariate, this study selected patients with IBD who had maintained a stable condition without any flares during the 12 weeks preceding recruitment. The present study also had several limitations. The initial results reflected the characteristics of the gut microbiota in each patient at a single time point. As the fecal microbiome fluctuates more frequently in patients with IBD than that in healthy individuals, investigation of longitudinal changes in these populations is required5. Nevertheless, this limitation was mitigated by a large number of subjects with IBD and by minimizing potential confounders. Second, due to the small number of healthy individuals recruited to this study, their demographic variables, such as sex ratio, did not match those of subjects with IBD. This study, however, adjusted for the confounding effects of these demographic variables by incorporating them as covariates in multivariate regression analyses. Third, the taxonomic analysis with functional annotation was based on 16 S rRNA sequencing. Whole metagenomic sequencing with fecal metabolomic analysis is required for finer taxonomic determination of specific gut microbiota, as is an independent biological validation of functional activity. Finally, the findings of the present study require validation in an independent cohort, despite being based on substantial sample sizes. Additionally, the absence of validation (e.g., qPCR) in our study represents a limitation, highlighting an area for improvement. However, we are actively addressing this gap through ongoing multi-cohort studies, which aim to validate our initial findings. This approach will not only reinforce the robustness of our results but also incorporate qPCR validation in future research, ensuring a more comprehensive understanding of the role of the gut microbiome in IBD. In recognition of the limitations of this study, we are currently conducting a nationwide, prospective, observational study targeting CD and UC within the Korean population. Over five years, we aim to recruit at least 900 patients with IBD and 200 of their first-degree relatives (FDRs), and 2,244 healthy controls.
In conclusion, the present large-scale metagenomic study successfully showed that the gut microbiota signature in Korean patients with IBD differed from that in a healthy population of the same ethnicity and lifestyle. Differential analysis of abundance revealed that samples from patients with CD were strongly enriched in the genus Escherichia-Shigella, suggesting that this genus is a key microbe in the etiopathogenesis of CD. These observations were verified by subsequent functional analysis, which showed that functional dysbiotic signatures, such as pathogenic and sulfur metabolic pathways, were significantly enriched in patients with CD, along with a high contribution from the genus Escherichia-Shigella.
Methods
Enrollment of study participants
Ethics approval and consent to participate
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was approved by the Institutional Review Board of Kyung Hee University Hospital (IRB File No., KHUH 2018-03-006-018, KHUH 2018-12-004-003). All study participants provided written informed consent before participating in the study.
IBD patient participants
From May 2018 to March 2022, patients with IBD were recruited at their outpatient visits for routine care at Kyung Hee University Hospital in Seoul, South Korea. These patients met the regional and international diagnostic criteria of IBD, based on clinical, serological, radiological results, and endoscopy with histology14. We excluded patients with indeterminate colitis or unstable patients who had suffered a recent IBD flare within 12 weeks preceding recruitment. All study participants were 16 years or older at study enrollment. We have received consent from the guardians to avoid any conflicts with the Convention on the Rights of the Child and the laws of the Republic of Korea.
Healthy participants
Healthy participants were randomly recruited for comparisons from the local community during the same study period as patients with IBD. All study participants were 16 years or older at study enrollment. Among 171 healthy volunteers who agreed to participate in this study, those who met any of the following criteria were excluded from the study: who have continuously taken probiotics for more than one week within the past three months; who have taken drugs that can affect the gastrointestinal tract, such as pain relievers and anti-inflammatory drugs within the past week; who have taken antibiotics within one month; and who have recently complained of acute gastrointestinal symptoms such as diarrhea and abdominal pain. For subjects included after the 1st screening, the past and present medical and family history of the following diseases was checked: diabetes, hypertension, hyperlipidemia, liver diseases, kidney diseases, tumors, and other diseases. After applying study exclusion criteria, participants without significant abnormalities in vital signs, hematology, blood chemistry, urinalysis, and pregnancy tests were finally included in the healthy control group. The following are detailed test items for each test: for vital signs, blood pressure, pulse rate, and temperature were measured; for hematology, WBC, RBC, hemoglobin, hematocrit, and platelet counts were measured; for blood chemistry, glucose, BUN, cholesterol, total protein, albumin, total bilirubin, direct bilirubin, alkaline phosphatase, AST, ALT, creatinine, triglyceride, LDL-cholesterol, and HDL-cholesterol were measured; for the urine test, specific gravity, pH, glucose, ketone, bilirubin, occult blood, urobilinogen, nitrite, microscopy, protein, and leukocytes were measured. The detailed standard values for each test are recorded in Fig. 1. Finally, 117 healthy individuals were included in our present study.
Collecting baseline data
The demographic and related clinical data of IBD were collected at the time of stool collection. The Montreal system was used for classifying IBD phenotypes15. Advanced phenotype was categorized to facilitate the correlation analysis of the metagenomic results and defined as either stricturing (B2) or penetrating (B3) disease in CD and extensive colitis (E3) in UC. Clinical disease activity of IBD was measured using the Mayo score for UC60 and the Harvey-Bradshaw index (HBI) for CD61. Clinical remission was defined as a Mayo score ≤ 2 in UC and an HBI score ≤ 4 in CD. On the day of fecal sample collection, all study participants completed the questionnaire for dietary habits, smoking and alcohol consumption, familial history of IBD or colon cancer, use of over-the-counter medications, and stool consistency using the Bristol stool scale (BSS)62.
Sample collection and storage
Within 12 weeks of stool sampling, all participants were strictly restricted to the following medications: antibiotics, probiotics, nonsteroidal anti-inflammatory drugs, and proton pump inhibitors. Patients with IBD were allowed to take medications, including 5-aminosalicylic acids, immune modulators, biological drugs, and small-molecule inhibitors. Approximately 1 g of fresh stool was collected in a stool nucleic acid collection and preservation tube (NORGEN BioTek, Canada) and stored at -80 °C in the laboratory until DNA extraction.
Sequencing data analysis
For preparation of Illumina MiSeq sequencing, a fragment of the 16 S rRNA gene spanning the hypervariable V3–V4 region was amplified by PCR using the forward primer 341 F and the reverse primer 805R. For Illumina MiSeq sequencing, a 16 S V3–V4 PCR product library was prepared using the Nextera XT Index (Illumina). The library was then sequenced on the Illumina MiSeq platform and using a paired-end 2 × 300 bp reagent kit, according to the manufacturer’s instructions. Raw paired reads were denoised and assembled using the DADA2 program63. The potential artificial sequences and 16 S rRNA gene-specific primer set in each read were removed using the Skewer program64. Chimeric and contaminated reads containing small subunit rRNA gene sequences originating from chloroplasts, mitochondria, eukaryotes, or archaea were removed using the Mothur pipeline v.1.43.065. Cleaned high-quality reads were taxonomically assigned using the classify.seqs function in the Mothur pipeline with the SILVA database66. To normalize the sequence counts among the samples in distance calculation, a minimum sample size (22,056 reads) was applied for rarefying each sample. A total of 37,387,797 clean, high-quality paired-end reads of the bacterial 16 S RNA gene (a mean of 58,418 ± 17,524 reads per sample) were obtained from 640 samples after denoising and chimera removal. The size of all samples was normalized by rarefying to 22,056 reads, which is the read count of the smallest sample. Good’s coverage estimates of 640 samples were > 99.3%, indicating that excellent weightage was obtained to reflect the bacterial community structure of the samples.
Diversity and clustering analysis
The intra-individual bacterial diversity within samples (alpha diversity) was evaluated using the following metrics: number of observed ASVs, abundance-based coverage estimator (ACE) for species richness, Shannon’s evenness index for species evenness, and inverse Simpson’s index for community diversity. The alpha diversity values were calculated using estimators in Mothur and statistically verified using the Kruskal-Wallis test in R language (v.4.0.4). The differences in alpha diversity values among gender, age, or Bristol stool scale categories were tested by the Kruskal-Wallis test and the Wilcoxon rank sum pairwise comparison test after Benjamini-Hochberg adjustment. Weighted and unweighted-UniFrac distances among the samples were calculated using the sequence information and abundance values of amplified sequence variants to compare and cluster the samples67. Unsupervised clustering was performed using clusterSim (v.0.49.2) package in R language68. The optimal number of clusters was estimated using the Calinski–Harabasz (CH) index69. To assess the beta diversity among disease groups, the analysis of similarities (ANOSIM) and PERMANOVA tests were conducted with weighted and unweighted UniFrac distance matrices. The ANOSIM test was performed using anosim function in Mothur package with 10,000 permutations, and PERMANOVA test was performed by using adonis2 function of the vegan package followed by a pairwise post hoc test using the pairwise.adonis2 function of pairwiseAdonis package with 999 permutations. To test multivariable association through the generalized linear regression between taxonomic relative abundance and phenotype groups by controlling age, gender, BMI, smoking, alcohol consumption, and Bristol stool scale as confounding variables, MaAsLin2 (Microbiome Multivariable Association with Linear Models) program was used with the AST (Arc-sine transformation) and LOG (log transformation) options on taxa abundances and functional feature abundances, respectively70. The diversity was visualized using the ggplot2 (V.3.3.5) and ggpubr packages (V.0.4.0) in R language.
Functional feature abundance
The enzyme commission (EC) numbers and Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs (KO) were predicted using PICRUSt2 (phylogenetic investigation of communities by reconstruction of unobserved states, 2.3.0b0)71,72. Through the predicted EC and KO distribution, differentially abundant functional pathways were projected using the KEGG and MetaCyc metabolic databases. The samples were clustered using the Jensen–Shannon distance (JSD) and partitioning around medoid (PAM) clustering with the functional feature abundance data, and the optimal number of clusters estimated using the CH index.
For multivariable association between clinical phenotypes, covariates, and microbial metagenomic features, the Microbiome Multivariable Association with Linear Models (MaAsLin2) program based on a generalized linear regression model was used with the AST (arcsine transformation) and LOG (log transformation) options for the taxa and functional feature abundance, respectively73. This model was developed to control multi-level covariates and FDR while maintaining statistical rules and was validated through applications in the Integrative Human Microbiome Project (iHMP or HMP2) IBD multi-omics dataset73. We adjusted for potential confounding factors, including age, gender, body mass index, smoking and alcohol consumption, stool consistency, and disease activity of IBD. Gut microbial diversity was visualized using the ggplot2 package (v.3.3.5) and ggpubr package (v.0.4.0) in the R language74. To define samples (dysbiotic samples) with unbalanced functional feature abundances, dysbiosis scores were calculated, based on JSD with normalized functional KEGG ortholog profiles by universal single copy genes. To identify the highly dysbiotic samples, the dysbiosis score at the 95th percentile of healthy group samples was used as the threshold value.
Dysbiosis score
To define samples (dysbiotic samples) with unbalanced functional feature abundances, dysbiosis scores were calculated using the JSD with normalized functional KEGG ortholog profiles by universal single copy genes. To identify the highly dysbiotic samples, the dysbiosis score at the 95th percentile of healthy group samples was used as the threshold value3.
Microbial markers
The performances of the differentially abundant taxa between the healthy group vs. patients with IBD, healthy group vs. patients with CD, healthy group vs. patients with UC, and patients with CD vs. patients with UC as microbial markers were evaluated in terms of area under the receiver operating characteristic curve (AUROC). Differentially abundant genera were selected using MaAsLin2 by applying the criteria of FDR < 0.05. The random forest model was trained using the randomForest package in R language. The performances of the selected marker candidates were evaluated using the decision tree model as a simple and intuitive predictive machine learning model in terms of AUROC by applying them independently or together. To apply the random forest model, IBD cohort data were divided into 70% for training and 30% for validation by random selection. In the case of training data, to overcome the imbalance of sample size between comparison groups (healthy : IBD : CD : UC = 81 : 366 : 156 : 210), random resampling was performed according to the size of each large group in the four comparison conditions. As a result, samples were re-sampled at a ratio of healthy : IBD = 357 : 375, healthy : CD = 144 : 168, healthy : UC = 192 : 228, and UC : CD = 228 : 192 for each comparison condition and were used to train a random forest model. The decision tree algorithm of the party package and the random forest algorithm of the randomForest package in R language were used. The pROC package in R language was used to evaluate the predictive performance of the trained decision tree and random forest models in AUROC values. Data were resampled to improve the imbalance in data sizes among the compared groups, and the sample size of all groups was adjusted to be similar to that of the small size group using the ROSE package in R.
Data availability
Data that support the findings of this study are available within the manuscript and supplementary files, as well as from the corresponding author upon reasonable request. The raw sequence data generated in this study have been deposited in the sequence read archive (SRA) at NCBI BioProject under accession No. PRJNA978516.
Code availability
The code used to support the findings of this study is available as Supplementary materials.
Abbreviations
- ACE:
-
Abundance-based coverage estimator
- AKI:
-
Acute kidney injury
- AST:
-
Aspartate aminotransferase
- ASV:
-
Amplicon sequence variant
- ALT:
-
Alanine aminotransferase
- BSS:
-
Bristol stool scale
- BUN:
-
Blood urea nitrogen
- CD:
-
Crohn’s disease
- CH:
-
Calinski–Harabasz index
- CKD:
-
Chronic kidney disease
- DBP:
-
Diastolic blood pressure
- EC:
-
Enzyme commission
- FDR:
-
False discovery rate
- HBI:
-
Harvey-Bradshaw index
- IBD:
-
Inflammatory bowel disease
- HDL:
-
High-density lipoprotein
- JSD:
-
Jensen–Shannon distance
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KO:
-
KEGG orthologs
- LDL:
-
Low-density lipoprotein
- MaAsLin:
-
Microbiome Multivariable Association with Linear Models
- PAM:
-
Partitioning around medoid
- PCoA:
-
Principal coordinate analysis
- RBC:
-
Red blood cell
- SBP:
-
Systolic blood pressure
- SCFA:
-
Short-chain fatty acid
- UC:
-
Ulcerative colitis
- WBC:
-
White blood cell
References
Lee, M. & Chang, E. B. Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology 160, 524–537 (2021).
Yilmaz, B. et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 25, 323 (2019).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655 (2019).
Abdel-Rahman, L. I. H. & Morgan, X. C. Searching for a consensus among inflammatory bowel disease studies: A systematic meta-analysis. Inflamm. Bowel Dis. 29, 125–139 (2023).
Halfvarson, J. et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2 (2017).
Ng, S. C. et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-Pacific Crohn’s and colitis epidemiology study. Gastroenterology 145, 158 (2013).
Molodecky, N. A. et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54 (2012).
Ng, S. C. et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 390, 2769–2778 (2017).
Lee, J. W. & Eun, C. S. Inflammatory bowel disease in Korea: Epidemiology and pathophysiology. Korean J. Intern. Med. (2022).
Eun, C. S. et al. Does the intestinal microbial community of Korean Crohn’s disease patients differ from that of western patients? BMC Gastroenterol. 16 (2016).
Nishino, K. et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 53, 95–106 (2018).
Kitae, H. et al. Gut Microbiota associated with clinical relapse in patients with quiescent ulcerative colitis. Microorganisms 10 (2022).
Clooney, A. G. et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 70, 499–510 (2021).
Maaser, C. et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis 13, 144–164 (2019).
Silverberg, M. S. et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. gastroenterol. 19(Suppl A), 5A-36A (2005).
Lopez-Siles, M. et al. Changes in the abundance of Faecalibacterium prausnitzii Phylogroups I and II in the intestinal mucosa of inflammatory bowel disease and patients with colorectal Cancer. Inflamm. Bowel Dis. 22, 28–41 (2016).
Pascal, V. et al. A microbial signature for Crohn’s disease. Gut 66, 813–822 (2017).
Manandhar, I. et al. Gut microbiome-based supervised machine learning for clinical diagnosis of inflammatory bowel diseases. Am. J. Physiol. Gastrointest. Liver. Physiol. 320, G328–G337 (2021).
Lee, J. W. & Eun, C. S. Inflammatory bowel disease in Korea: Epidemiology and pathophysiology. Korean J. Intern. Med. 37, 885–894 (2022).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell. Host Microbe 15, 382–392 (2014).
Zaneveld, J. R., McMinds, R. & Vega Thurber, R. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 17121 (2017).
Pitcher, M. C. & Cummings, J. H. Hydrogen sulphide: A bacterial toxin in ulcerative colitis? Gut 39, 1–4 (1996).
Attene-Ramos, M. S., Wagner, E. D., Plewa, M. J. & Gaskins, H. R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 4, 9–14 (2006).
Levine, J., Ellis, C. J., Furne, J. K., Springfield, J. & Levitt, M. D. Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 93, 83–87 (1998).
Chassard, C. et al. Functional dysbiosis within the gut microbiota of patients with constipated-irritable bowel syndrome. Aliment. Pharm. Ther. 35, 828–838 (2012).
Barton, L. L., Ritz, N. L., Fauque, G. D. & Lin, H. C. Sulfur cycling and the intestinal microbiome. Digest Dis. Sci. 62, 2241–2257 (2017).
Wang, J. et al. Hydrogen sulfide from cysteine desulfurase, not 3-mercaptopyruvate sulfurtransferase, contributes to sustaining cell growth and bioenergetics in E. coli under anaerobic conditions. Front. Microbiol. 10, 2357 (2019).
Guo, S. H. et al. A simple fecal bacterial marker panel for the diagnosis of Crohn’s disease. Front. Microbiol. 10 (2019).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305 (2019).
Serrano-Gomez, G. et al. Dysbiosis and relapse-related microbiome in inflammatory bowel disease: A shotgun metagenomic approach. Comput. Struct. Biotechnol. J. 19, 6481–6489 (2021).
El Mouzan, M. I. et al. Fungal dysbiosis predicts the diagnosis of pediatric Crohn’s disease. World J. Gastroenterol. 24, 4510–4516 (2018).
Deschasaux, M. et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531 (2018).
Shi, H. Y. et al. Ethnicity influences phenotype and outcomes in inflammatory bowel disease: A systematic review and meta-analysis of population-based studies. Clin. Gastroenterol. Hepatol. 16, 190–197 e111 (2018).
Freeman, K., Willis, B. H., Fraser, H., Taylor-Phillips, S. & Clarke, A. Faecal calprotectin to detect inflammatory bowel disease: A systematic review and exploratory meta-analysis of test accuracy. BMJ Open. 9 (2019).
Bryce, C. & Bucaj, M. Fecal calprotectin for the evaluation of inflammatory bowel disease. Am. Fam. Phys. 104, 303–304 (2021).
Shao, Y. P. et al. Clinical features of inflammatory bowel disease unclassified: A case-control study. Bmc Gastroenterol. 24 (2024).
Peeters, M. et al. Diagnostic value of anti- and antineutrophil cytoplasmic autoantibodies in inflammatory bowel disease. Am. J. Gastroenterol. 96, 730–734 (2001).
Birimberg-Schwartz, L. et al. pANCA and ASCA in children with IBD-unclassified, Crohn’s colitis, and ulcerative colitis—a longitudinal report from the IBD Porto Group of ESPGHAN. Inflamm. Bowel Dis. 22, 1908–1914 (2016).
Darfeuille-Michaud, A. et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004).
Zhilu, X. et al. Association of adherent-invasive Escherichia coli with severe gut mucosal dysbiosis in Hong Kong Chinese population with Crohn’s disease. Gut Microbes 13, 1994833 (2021).
Zhang, J. W. et al. Elucidation of Proteus mirabilis as a key bacterium in Crohn’s disease inflammation. Gastroenterology 160, 317– (2021).
Shatalin, K., Shatalina, E., Mironov, A. & Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 334, 986–990 (2011).
Cirino, G., Szabo, C. & Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 103, 31–276 (2023).
Rey, F. E. et al. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. U.S.A. 110, 13582–13587 (2013).
Dordevic, D., Jancikova, S., Vitezova, M. & Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 27, 55–69 (2021).
Laue, H., Denger, K. & Cook, A. M. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl. Environ. Microbiol. 63, 2016–2021 (1997).
Laue, H., Friedrich, M., Ruff, J. & Cook, A. M. Dissimilatory sulfite reductase (desulfoviridin) of the taurine-degrading, non-sulfate-reducing bacterium Bilophila wadsworthia RZATAU contains a fused DsrB-DsrD subunit. J. Bacteriol. 183, 1727–1733 (2001).
Metaxas, M. A. & Delwiche, E. A. The L-cysteine desulfhydrase of Escherichia coli. J. Bacteriol. 70, 735–737 (1955).
Rohwerder, T. New structural insights into bacterial sulfoacetaldehyde and taurine metabolism. Biochem. J. 477, 1367–1371 (2020).
Eichhorn, E., van der Ploeg, J. R., Kertesz, M. A. & Leisinger, T. Characterization of alpha-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J. Biol. Chem. 272, 23031–23036 (1997).
Barton, L. L., Ritz, N. L., Fauque, G. D. & Lin, H. C. Sulfur cycling and the intestinal microbiome. Dig. Dis. Sci. 62, 2241–2257 (2017).
Hooks, K. B. & O’Malley, M. A. Dysbiosis and its discontents. mBio 8 (2017).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Giongo, A. et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 5, 82–91 (2011).
Dey, N., Soergel, D. A., Repo, S. & Brenner, S. E. Association of gut microbiota with post-operative clinical course in Crohn’s disease. Bmc Gastroenterol. 13, 131 (2013).
Chen, Y. et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572 (2011).
Chen, Y. et al. Gut dysbiosis in acute-on-chronic liver failure and its predictive value for mortality. J. Gastroenterol. Hepatol. 30, 1429–1437 (2015).
Wu, J. et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 10, 2435–2446 (2016).
Kim, H. S. et al. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 12, 161 (2021).
Schroeder, K. W., Tremaine, W. J. & Ilstrup, D. M. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N. Engl. J. Med. 317, 1625–1629 (1987).
Harvey, R. F. & Bradshaw, J. M. A simple index of Crohn’s-disease activity. Lancet 1, 514 (1980).
O’Donnell, L. J., Virjee, J. & Heaton, K. W. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 300, 439–440 (1990).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Jiang, H., Lei, R., Ding, S. W. & Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 15, 182 (2014).
Schloss, P. D. Reintroducing mothur: 10 years later. Appl. Environ. Microbiol. 86 (2020).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–596 (2013).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Dudek, A. & Walesiak, M. The choice of variable normalization method in cluster analysis. In Education Excellence and Innovation Management: A 2025 Vision to Sustain Economic Development during Global Challenges 325–340 (2020).
Kozak, M. A Dendrite Method for Cluster Analysis by Calinski and Harabasz: A classical work that is far too often incorrectly cited. Commun. Stat. Theor. Methods 41, 2279–2280 (2012).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Manor, O. & Borenstein, E. MUSiCC: A marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 16, 53 (2015).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17 (2021).
Villanueva, R. A. M. & Chen, Z. J. ggplot2: Elegant Graphics for Data Analysis, 2nd edition. Meas. Interdiscip. Res. 17, 160–167 (2019).
Acknowledgements
We would like to thank Editage (www.editage.co.kr) for English language editing.
Funding
This work was supported by: The Medical Research Center Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (grant No. 2017R1A5A2014768). A grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant No. HI23C0661). An NRF grant funded by the Korea government (MSIT) (grant No. 2021R1C1C2008556).
Author information
Authors and Affiliations
Contributions
Study design and idea: C.K.L. Data acquisition: C.K.L., J.E.K., S.J.O., and B-H.K. Analysis of data: H.S.K., Y-K.P., B.K.K., J-Y.L., and C.K.L. Writing of the manuscript: H.S.K., J-W.B., and C.K.L. Critical revision of the manuscript for important intellectual content: H.S.K., J-W.B., and C.K.L. All authors have approved the final version of the manuscript. The views expressed are those of the author[s] and not necessarily those of the funder.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, H.S., Oh, S.J., Kim, B.K. et al. Dysbiotic signatures and diagnostic potential of gut microbial markers for inflammatory bowel disease in Korean population. Sci Rep 14, 23701 (2024). https://doi.org/10.1038/s41598-024-74002-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-74002-6
