
Abstract
Obesity is a serious global health issue. Emerging evidence indicates that the gut microbiota may contribute to the development of obesity, possibly by instigating inflammatory processes. The objective of this research is to conduct a comparative analysis of the gut microbiota composition in obese and normal-weight Uygur adults, while examining the associations with adipocyte-related factors and dietary variables. According to the inclusion and exclusion criteria, twenty-seven Uygur adults with obesity and twenty Uygur adults with normal-weight were recruited from a local community. Anthropometric measurements and blood samples were collected. Gut microbiota composition was analyzed using 16 S rRNA gene sequencing. Adipocyte-related factors were measured using enzyme-linked immunosorbent assay (ELISA). Statistical analyses were performed to compare the gut microbiota composition between the two groups and to identify correlations between gut microbiota and adipocyte-related factors. Compared with the normal-weight group, the obese group exhibited a marked reduction in both diversity and richness of the gut microbiota, alongside a decrease in Ruminococcaceae_UCG_014, Coprococcus_2, and Parabacteroides, and an increase in Megamonas and Lachnoclostridium, implying a potential link to the development of obesity. Individuals with obesity were found to have higher Leptin (LEP), Interleukin-6 (IL-6), and C-reactive protein (CRP) than normal-weight individuals. Obese Uygur adults exhibited a gut microbiota characterized by diminished diversity and richness relative to normal-weight individuals. Parabacteroides, Megamonas, and Lachnoclostridium may play an important role in the development of obesity in Uygur population. Underlying mechanisms need further investigation.
Similar content being viewed by others


Introduction
Obesity has become a serious public health issue throughout the world, with the proportion of overweight and obese people increasing dramatically over the last four decades. According to the latest national prevalence estimates for 2015-19, the overweight and obesity rates among individuals aged 18 years and above were 34.3% and 16.4% in China1. Obesity is associated with an increased risk of noncommunicable diseases such as cardiovascular diseases, diabetes, cancer and so on.
The main cause of obesity is an energy imbalance between energy intake and energy expenditure. Excess energy is stored in the form of adipose tissue when caloric intake surpasses total energy expenditure. Adipose tissue is not only a reservoir for energy, but also an immune organ that secretes a large number of adipokines2. Improper adipose tissue growth causes an inappropriate release of cytokines and adipokines, with an upsurge in tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, leptin (LEP), and resistin (RETN), and a decrease in adiponectin (ADP), resulting in persistent low-grade inflammation3.
In recent years, a large number of studies have shown that the imbalance of gut microbiota is related to obesity4. The gut microbiota encompasses a diverse community of bacteria that carry out various functions influencing the overall health of the host5. Differences in microbiota composition are observed between obese and lean individuals in humans, including a decrease in richness and diversity of gut microbiota and alterations in specific bacterial taxa6. An elevated Firmicutes/Bacteroidetes ratio seems to be characteristic in obese animal models7, Patron et al.8 also reported that obese individuals have a lower Firmicutes/Bacteroidetes ratio compared to normal-weight individuals, suggesting a close association between the Firmicutes/Bacteroidetes ratio in the gut and obesity. Other researchers have reported that obesity may be associated with an increase in Firmicutes and Actinobacteria9.
Gut microbiota is influenced by a variety of variables, including nutrition, lifestyle, and geographical dispersion, among which the influence of diet may be particularly prominent10. Numerous clinical trials have tested the effect of a high-fiber diet in obese individuals, which revealed positive effects in lowering body weight gain and adiposity, improving satiety and glucose tolerance11. Gut bacteria possess the capability of metabolising complex carbohydrates, otherwise undigested by the host, into short-chain fatty acids (SCFAs)5. Mechanistic studies in animal models reveal the positive role of SCFAs, a product of fiber fermentation by the gut microbiota, in maintaining gut barrier health and attenuating inflammation12. On the other hand, obesity is also associated with disruptions in the intestinal barrier function, facilitating the translocation of proinflammatory bacterial constituents, including lipopolysaccharide (LPS) and flagellin, leading to chronic low-grade inflammation related to metabolic dysregulation13. Djuric et al.14 suggested that an imbalance in the gut microbiota of obese individuals, with an increased abundance of potentially pro-inflammatory bacteria (such as Proteobacteria) and a relatively lower abundance of anti-inflammatory strains (such as Faecalibacterium prausnitzii), may be associated with the development of obesity.
The prevalence of overweight and obesity in the Uygur population in Xinjiang was 34.9% and 28.9%, respectively, which was higher than the national level15. Uygur residents have a unique dietary habit, and with the improvement in economic level, the traditional diet and lifestyle of Uygur residents have changed, which may have an impact on the gut microbiota. However, current research on gut microbiota primarily focuses on individuals from industrialized nations, with limited data available from the Uygur population in Xinjiang. A recent study from Pakistan has reported differences in gut microbiota in different countries, this emphasizes the need to consider population-specific differences in gut microbiota and their implications in health16. Additionally, our previous research indicated that the low dietary fiber intake among Uygur residents may be related to the high incidence of obesity, but the specific mechanism remains unclear17. Thus, the aim of this research is to conduct a comparative analysis of the gut microbiota composition in obese and normal-weight Uygur adults, while examining the associations with adipocyte-related factors and dietary variables.
Materials and methods
Study design and participants
Twenty-seven obese Uygur adults were recruited as the sample from Jinglao Road community health service center in Hotan, Southwestern Xinjiang Uygur Autonomous Region of China, from August to September 2019. Meanwhile, twenty people with normal-weight were chosen as controls. The inclusion criteria for sample collection were those aged 25 to 65 years and who lived in this area for at least 5 years. The exclusion criteria were gastrointestinal diseases (diarrhea, abdominal pain, etc.), pregnancy or lactation, consumption of antibiotics within the preceding month, or excessive alcohol consumption, taking weight-loss drugs, antihypertensive drugs, diabetes drugs, or lipid-lowering drugs. Excessive alcohol consumption was defined as drinking 21 or more units of alcohol per week. The study was reviewed and approved by the Ethics Committee of First Affiliated Hospital of Xinjiang Medical University[20170214-150]. The study adhered to pertinent guidelines and regulations, and all participants provided informed consent. The research was carried out in accordance with the Declaration of Helsinki.
Anthropometry measurements and biochemical parameters
Anthropometry measurements (weight, height, waist circumference, and hip circumference) were measured using a standardized protocol. All measurements were carried out by uniformly trained investigators. According to Chinese criteria, a BMI of 18.5–24.0 kg/m2 refers to normal-weight, and BMI ≥ 28.0 kg/m2 refers to obesity18. According to the principles of informed consent, the blood samples (2 ~ 3 mL) of all subjects were collected in the early morning under the condition of fasting for 12 h. After 3000 r/min centrifugation for 10 min, the serum was preserved at -80℃ for subsequent blood lipids and adipocytokines testing. Serum total cholesterol, triglyceride, high-density lipoprotein, and low-density lipoprotein were detected by BS-460 fully automatic biochemical analyzer (Mindray, Shenzhen). Fasting insulin was detected by BS-800 M fully automatic biochemical analyzer (Mindray, Shenzhen). Leptin (LEP), adiponectin (ADP), resistin (RETN), tumor necrosis factor-α(TNF-α), interleukin-6 (IL-6), and C-reactive protein (CRP) were detected by enzyme-linked immunosorbent assay (ELISA), and the kits were all purchased from Elabscience. Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated from fasting glucose and insulin while using the following formula: HOMA-IR = FBG(mmol/L)×FINS(ml/U)/22.5. Dietary intake questionnaire, including a 24 h recall questionnaire for 3 days (2 working days and 1 weekend) was completed by trained investigators. Dietary intake of energy, macronutrients, and micronutrients were analyzed by nutrition calculator software V2.7.5.8 (Beijing Bowen Shixun Technology Co., Ltd.). The protein, carbohydrate, and fat intakes have been appropriately energy-adjusted using the residual method.
Fecal samples collection and DNA extraction
Participants collected fecal samples (approximately 0.5–3 g) at home using the provided sterile stool samplers. After sampling, a DNA protection solution from Guangdong Longsee Biomedical Co., Ltd was added, allowing the fecal samples to be stored at room temperature for 14 days and at 4 ℃ for 60 days. It is advised to deliver the samples to the laboratory within 12 h. The samples were then sent to the laboratory for further tests and stored at -80 °C. QIAamp® DNA Stool Mini Kit (Qiagen, Germany) was used to extract the total DNA of microorganisms from 200 mg stool samples, and NanoDrop was used to detect the concentration and purity of the extracted DNA sample, then 1% agarose gel electrophoresis was used to detect the integrity of the extracted DNA. The DNA samples were sequenced targeting the V3 + V4 hypervariable regions of the bacterial 16 S rRNA gene via the Illumina MiSeq platform according to the manufacturer’s protocols at Guangdong Longsee Biomedical Co., Ltd. (Guangzhou, China).
Bioinformatic analysis
The sequences were processed with FLASH (version1.2.11) to merge paired reads and filter chimeras, following which operational taxonomic units (OTUs) were clustered at 97% similarity using UCHIME (version8.1). Alpha diversity was assessed using the Shannon, Simpson, ACE, and Chao1 indices. Beta diversity measurements, including principal coordinate analysis (PCoA) based on the phylogenetic tree, were computed using Binary-Jaccard, UniFrac distance, and Bray–Curtis distance. PERMANOVA (Permutational Multivariate Analysis of Variance) was employed for the statistical test to evaluate differences between the obese and normal-weight groups. Furthermore, linear discriminant analysis effect size (LEfSe) with a linear discriminant analysis (LDA) score above 2.0 was utilized to identify bacterial taxa with differences between the groups. The significance level was set at P < 0.05, and multiple testing was controlled using false discovery rate (FDR). PICRUSt was used to predict the function of microbial communities, and Welch’s t-test was employed to analyze the differences in KEGG pathways between two groups.
Statistical analysis
SPSS 26.0 statistical software was used for statistical analysis. The normality of the data was tested using the Shapiro-Wilk test. Measurement data following a normal distribution were expressed as mean ± standard deviation (\(\:\stackrel{-}{x}\pm\:S\)), and a two-sample t-test was used for comparison between obese and normal-weight groups. Measurement data that did not follow a normal distribution were presented by median (interquartile range) [M(P25, P75)], and Mann-Whitney U test was employed for comparisons of obese and normal-weight groups. Categorical variables were expressed by relative numbers, and the chi-square test was used for comparison between groups. The correlation analysis of gut microbiota and adipocyte-related factors was assessed using Spearman correlation coefficients. P-values were corrected using the FDR method.
Results
Characteristics of the study population
The anthropometric and biochemical characteristics of the study population are shown in Table 1. Compared with the normal-weight group, the obese group showed higher values in weight, BMI, waist circumference, waist-to-hip ratio, total cholesterol, triglycerides, low-density lipoprotein, fasting blood glucose, fasting insulin, and HOMA-IR, and a lower value in HDL-C (P < 0.01). In addition, the LEP, IL-6, and CRP of the obese group were significantly higher than those of the normal-weight group (P < 0.05), while ADP, RETN, and TNF-α showed no difference between the two groups (P > 0.05). The nutrient intake of participants among the obesity and normal-weight groups is shown in Table 2. Dietary intake of energy, macronutrients, and micronutrients between the obesity and normal-weight groups did not differ significantly.
Gut microbiota
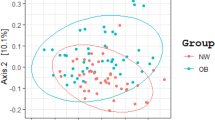
At the phylum level, the main genera in obese and normal-weight groups were Firmicutes (50.37% and 53.02%, respectively), followed by Bacteroidetes (41.62% and 38.55%, respectively) and Proteobacteria (5.80% and 4.92%, respectively) (Fig. 1a). Firmicutes/Bacteroidetes ratio is frequently cited as a marker of obesity, as some studies have described that the gut microbiota of obese animals and humans exhibits higher Firmicutes/Bacteroidetes ratio compared with normal-weight individuals19. In this study, no significant differences were observed in Firmicutes/Bacteroidetes ratio (Fig. 1b). No difference was found at the phylum level between the obese and normal-weight groups. Alpha diversity is used to express species richness among different groups, while beta diversity indicates species turnover and community similarity. The results of alpha diversity analysis showed that the Shannon index of gut microbiota in the obese group was lower than the normal-weight group (P < 0.05). The greater the Shannon value, the higher the community diversity20. The results indicated that the diversity in obese people was lower than that in people with normal-weight. Correspondingly, the Simpson index, ACE index, and Chao1 index showed no significant difference between the two groups (Fig. 2). The results of beta diversity analysis showed that the principal coordinate analysis (PCoA) using Binary-Jaccard distance (Fig. 3a) revealed a significant distinction between the groups (R²=0.043, P = 0.015). Unweighted UniFrac distance also indicated significant differences (R²=0.044, P = 0.033) (Fig. 3b). However, Bray–Curtis distance did not show significant differences (R²=0.027, P = 0.202) (Fig. 3c). LEfSe analysis revealed variations in specific bacterial taxa from the phylum to genus level within each group (Fig. 4a). The results of the LEfSe analysis demonstrated that the obese group exhibited higher abundance of Megamonas, Lachnoclostridium, Adlercreutzia, and Paraprevotella, while the normal-weight group was abundant in Ruminococcaceae_UCG_014, Coprococcus_2, Parabacteroides, Ruminococcaceae_UCG_005, Ruminiclostridium_6, Butyrivibrio, Ruminiclostridium_5, Ruminococcaceae_UCG_009, Anaerostipes, Eubacterium_xylanophilum, Coprobacter, Barnesiella, Coprococcus_1, Christensenellaceae_R_7, CAG_56, Ruminococcus gauvreauiiI, and Izimaplasmatales (Fig. 4b). PICRUSt2 was used for predicting the functional potential of the gut microbiota. As shown in Fig. 5, the top three significantly altered metabolic pathways in the obese group displayed an increase in pathways related to carbohydrate metabolism, membrane transport, and cofactors and vitamins metabolism, while pathways such as translation, replication and repair, and nucleotide metabolism showed a decrease in comparison to the normal weight group.

(a) Relative abundance of top ten bacteria in the phylum level between obese and normal-weight group.; (b) Firmicutes to Bacteroidetes Ratio.

Alpha diversity indexes between obese and the normal-weight group. (a) Shannon index; (b) Simpson index; (c) ACE index and (d) Chao1 index. * Indicates P < 0.05. P-values were corrected using the FDR method.

Microbial principal coordinate analysis (PCoA). (a) Binary-Jaccard distance; (b) UniFrac distance; (c) Bray–Curtis distance.

(a) Cladogram and (b) LDA scores derived from LEfSe analysis.

PICRUSt analysis of the KEGG pathways.
Correlation between gut microbiota, adipocyte-related factors, and dietary variables
A comprehensive correlation analysis was conducted between the abundances of differentially abundant taxa and adipocytokines that distinguish among the research groups, encompassing LEP, IL-6, and CRP, as well as various dietary factors (Fig. 6). Coprobacter was found to be negatively correlated with IL-6 levels; similarly, Coprobacter, Barnesiella, CAG-56, uncultured_bacterium_o_Izimaplasmatales, and the Firmicutes/Bacteroidetes ratio were found to be negatively correlated with CRP.

Correlation heatmap of gut microbiota, adipocyte-related factors, and dietary variables. * Indicates P < 0.05. P-values were corrected using the FDR method.
Discussion
In this study, 16 S high-throughput sequencing technology was used to investigate the microbial diversity of obese Uygur adults. The relative abundance of the top ten bacterial phyla in the obese and normal-weight groups was analyzed. The results showed that the gut microbiota of both groups was primarily composed of five phyla: Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Cyanobacteria, with Firmicutes and Bacteroidetes dominating. Over the last decade, the Firmicutes/Bacteroidetes ratio has been frequently proposed as a potential obesity indicator. However, in this study, there was no significant difference in the Firmicutes/Bacteroidetes ratio between obese individuals and those with normal weight, which may be related to individual dietary habits or other unmeasured factors. The alpha diversity analysis between obese and normal-weight groups was conducted, revealing that the Shannon diversity index of the obese group was found to have significantly declined compared to that of the normal-weight group. Therefore, in this study, the diversity of the obese group significantly decreased. The beta diversity analysis based on PCoA, Binary-Jaccard, unweighted UniFrac, and Bray–Curtis distance were estimated in this study. Jaccard distance considers only the presence or absence of species in the samples, unweighted UniFrac distance is based on the phylogenetic relationships of the species, while Bray-Curtis dissimilarity takes into account both the presence or absence and the relative abundances of the species. Beta diversity analysis using Binary-Jaccard and unweighted UniFrac distances revealed significant differences between the two groups, whereas beta diversity analysis using Bray-Curtis dissimilarity showed no significant differences between the two groups. This may suggest that although the presence or absence of different taxa showed significant differences between the two groups, the changes in relative abundances were not sufficient to serve as the main distinguishing feature between the two groups.
In a Chinese cohort study21, researchers observed that the gut microbiota of Han Chinese and ethnic minority groups from the same sampling sites were more alike than that of the same ethnic minority groups from different sampling sites. However, the microbiota richness of Uygur and Han Chinese from same sites shows a significant difference. This difference could be linked to the dietary habits of the Uygur people, especially their tendency towards consuming refined wheat. Previous research suggested a potential link between low dietary fiber consumption and obesity in Uygur adults17, while the present study revealed no significant difference in dietary fiber intake between the obesity and normal-weight groups. This interpretation may be limited by the small sample size, and it was possible that health concerns may have motivated some obese individuals to consciously reduce their consumption of foods, which could have obscured their actual food intake. In this study, we did not observe a significant association between the gut microbiota and dietary variables, which may be due to the limited sample size or other factors. Further research is needed to investigate whether dietary factors might influence chronic inflammation and, consequently, body weight by altering the abundance of specific gut microbiota. Furthermore, the predicted KEGG pathway analysis reveals an elevation in the carbohydrate metabolism pathways of obese Uygurs, suggesting that these obese individuals may possess the capacity to enrich carbohydrate metabolism in the gut microbiota, potentially leading to increased energy absorption in the body.
Previous studies have indicated that gut microbiota supports the normal functioning of the host organism through a variety of physiological and biochemical activities, including protecting against pathogens, regulating host immunity, shaping the intestinal epithelium, and harvesting energy4. At the genus level, the abundance of Megamonas and Lachnoclostridium was found to be significantly elevated in obese groups, while Ruminococcaceae_UCG_014, Coprococcus_2, and Parabacteroides demonstrated higher levels in the normal-weight group. Ruminococcaceae_UCG_014 and Coprococcus_2 are short-chain fatty acid-producing bacteria. Ruminococcaceae_UCG-014 is a probiotic capable of degrading cellulose to produce butyrate, a short-chain fatty acid (SCFA) that serves as an important energy source for colon cells. SCFAs can affect energy absorption through various mechanisms, such as promoting peptide YY secretion, inhibiting neuropeptide Y in the hypothalamic arcuate nucleus, activating pro-opiomelanocortin (POMC) neurons, influencing appetite and satiety, delaying gastric emptying, and increasing energy expenditure in muscles, thereby controlling weight gain22. The increased presence of Ruminococcaceae_UCG-014 in lean individuals is consistent with previous research23.
Parabacteroides is a genus of Gram-negative bacteria within the Bacteroidetes phylum that has been associated with both pro-inflammatory effects, such as inflammatory bowel disease, and regulation of metabolic diseases. Interestingly, it was found to be significantly lower in the obese group compared to the normal-weight group, suggesting a potential link between this bacterial genus and the development of obesity. Despite its association with inflammation and IBD, specific strains within the Parabacteroides genus, such as Parabacteroides goldsteinii and Parabacteroides distasonis have been linked to potential probiotic effects that can alleviate obesity and related metabolic disorders24. This indicates that the biological effects of Parabacteroides may be strain-specific, warranting further investigation into the role of different strains. Parabacteroides distasonis could convert primary bile acids into secondary bile acids, increasing their levels and potentially directly or indirectly regulating the signaling of FXR and TGR5 receptors through bile acids. This, in turn, may improve glucose and lipid metabolism disorders. Despite these promising findings, more research and population intervention experiments are necessary to further verify the role of Parabacteroides in metabolic diseases and its potential as a therapeutic target.
Megamonas belongs to Firmicutes, which can ferment various carbohydrates, whose end-products are acetic acid, propionic acid, and lactic acid. Ayeni FA et al.25 reported that Megamonas were uniquely present in urban Nigerian population with lower carbohydrate utilization capability, potentially indicating a microbiome signature of urbanization and Western lifestyle. Previous studies have also reported a significant increase in Megamonas abundance in obese children (Mexican26, Nanjing27). Rebiya et al.28 reported a significant increase in Megamonas abundance in obese Uygur patients with impaired glucose regulation. In line with these findings, we found a significant increase in Megamonas in the obese group. Additionally, Megamonas has been shown to exhibit a negative correlation with blood glucose and a positive correlation with apolipoprotein A29, a key protein component of high-density lipoprotein, suggesting its significant role in carbohydrate and lipid metabolism. In our study, however, no significant correlations were found between Megamonas and LEP, IL-6, or CRP. In addition, Megamonas was among the few bacterial genera positively correlated with food addiction30, potentially influencing feeding habits via the gut-brain axis and promoting obesity development31.
Lachnoclostridium has been reported as a characteristic genus of obese patients with polycystic ovary syndrome32. Consistently, our study found a significant increase in Lachnoclostridium in the obese group. However, a three-month Mediterranean diet intervention study observed higher abundances of Lachnoclostridium and Parabacteroides33. In contrast, our results show that Lachnoclostridium is enriched in subjects with obesity, whereas Parabacteroides is enriched in normal-weight subjects. This discrepancy may be attributed to differences in the study populations, dietary patterns, and the duration of the interventions. Our study did not establish a connection between Lachnoclostridium and cytokines or adipokines, which could be attributed to the limited sample size.
Obesity, characterized as a chronic low-grade inflammatory condition, is frequently accompanied by abnormal expression of cytokines and adipokines. Serum levels of IL-6, CRP, and TNF-α levels are substantially lower in normal-weight individuals compared to those with obesity34. In our study, the levels of LEP, IL-6, and CRP in obese group were significantly higher than normal-weight group, reflecting the chronic inflammatory condition with obesity. Emerging evidence suggests that disturbances in composition and functionality of the gut microbiota can compromise gut barrier function, leading to low-grade chronic inflammation35. In healthy conditions, the integrity of the intestinal barrier prevents translocation of the microbiome or its components. However, in obesity, characterized by intestinal dysbiosis and impaired intestinal barrier function, the microbiome or its components such as endotoxins are translocated into the circulation, resulting in low-grade inflammation. Djuric Z et al.14 observed that certain pro-inflammatory bacteria strains, such as some members of Bacteroides, may dominate in obesity, while anti-inflammatory bacteria strains, such as Faecalibacterium Prausnitzii, are less prevalent, potentially contributing to obesity development.
Limitation
There are some limitations in this study. Firstly, the relatively small sample size included in the study may affect the generalizability of the results. Although we planned to expand the sample size, our efforts were delayed due to the impact of COVID-19. We anticipate expanding the sample size in our future research. Additionally, the gut microbiota is influenced by multiple factors, and this study only explores the association between specific food groups and various genera of gut microbiota, without accounting for other potential influencing factors. Furthermore, the correlation between gut microbiota and the level of inflammation was only analyzed using the Spearman method. Future studies may need to consider using other statistical methods for validation.
Conclusions
Overall, compared to the normal-weight group, the diversity and richness of gut microbiota in the obese group are significantly reduced, alongside a decrease in Ruminococcaceae_UCG_014, Coprococcus_2, and Parabacteroides, and an increase in Megamonas and Lachnoclostridium. This suggests that obese individuals may have specific characteristics and metabolic pathways in their gut microbiota. Additionally, our study highlights the significant impact of dietary variables on gut microbiota composition and the potential implications for inflammation and body weight. However, further research is still needed to elucidate the mechanisms underlying these associations and to explore potential dietary interventions to modulate gut microbiota and improve health outcomes.
Data availability
Raw sequencing data have been deposited to the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA1058857.
References
China. TSCIOotPsRo, C. Press briefing for the Report on Chinese Residents’ Chronic Diseases and Nutrition 2020, (2020). http://www.gov.cn/xinwen/2020-12/24/content_5572983.html. Accessed 26 Dec.
Battineni, G. et al. Impact of obesity-Induced inflammation on cardiovascular diseases (CVD). Int. J. Mol. Sci. 22. https://doi.org/10.3390/ijms22094798 (2021).
Aleksandrova, K., Mozaffarian, D. & Pischon, T. Addressing the perfect storm: biomarkers in obesity and pathophysiology of cardiometabolic risk. Clin. Chem. 64, 142–153. https://doi.org/10.1373/clinchem.2017.275172 (2018).
Abenavoli, L. et al. Gut microbiota and obesity: a role for probiotics. Nutrients. 11. https://doi.org/10.3390/nu11112690 (2019).
Al Bander, Z., Nitert, M. D., Mousa, A. & Naderpoor, N. The gut microbiota and inflammation: an overview. Int. J. Environ. Res. Public. Health. 17. https://doi.org/10.3390/ijerph17207618 (2020).
Gerard, P. Gut microbiota and obesity. Cell. Mol. Life Sci. 73, 147–162. https://doi.org/10.1007/s00018-015-2061-5 (2016).
Ley, R., Turnbaugh, P., Klein, S. & Gordon, J. Microbial ecology: human gut microbes associated with obesity. Nature. 444, 1022–1023. https://doi.org/10.1038/4441022a (2006).
Vania, P. et al. Postoperative changes in fecal bacterial communities and fermentation products in obese patients undergoing bilio-intestinal bypass. Front. Microbiol. 7, (2016).
Chakraborti, C. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 6, 110–119. https://doi.org/10.4291/wjgp.v6.i4.110 (2015).
Deschasaux, M. et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531. https://doi.org/10.1038/s41591-018-0160-1 (2018).
Barber, T. M., Kabisch, S., Pfeiffer, A. F. H. & Weickert, M. O. The health benefits of dietary fibre. Nutrients 12. https://doi.org/10.3390/nu12103209 (2020).
Wastyk, H. C. et al. Gut-microbiota-targeted diets modulate human immune status. Cell 184, 4137–4153 e4114. https://doi.org/10.1016/j.cell.2021.06.019. (2021).
Tilg, H., Zmora, N., Adolph, T. E. & Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 20, 40–54. https://doi.org/10.1038/s41577-019-0198-4 (2020).
Djuric, Z. Obesity-associated cancer risk: the role of intestinal microbiota in the etiology of the host proinflammatory state. Transl Res. 179, 155–167. https://doi.org/10.1016/j.trsl.2016.07.017 (2017).
Liu, C. et al. Prevalence on overweight and obesity in Han.Uygur and Kazakh in adults from Xinjiang. Chin. J. Epidemiol.. 10, 1139–1143. https://doi.org/10.3760/cmaoj.issn.0254-6450.2010.10.015 (2010).
Saleem, A. et al. Unique Pakistani gut microbiota highlights population-specific microbiota signatures of type 2 diabetes mellitus. Gut Microbes. 14. https://doi.org/10.1080/19490976.2022.2142009 (2022).
Tie & Ri ge li. Study on the correlation between dietary patterns and obesity in 515 Uygurs subjects. Chi J. Dis. Con Pre .. 21, 461–464. https://doi.org/10.16462/j.cnki.zhjbkz.2017.05.008 (2017).
Zhou, B. F. & Cooperative Meta-Analysis Group of the Working Group. On obesity in, C. Predictive values of body mass index and waist circumference for risk factors of certain related diseases in Chinese adults–study on optimal cut-off points of body mass index and waist circumference in Chinese adults. Biomed. Environ. Sci.: BES. 15, 83–96 (2002).
Magne, F. et al. The Firmicutes/Bacteroidetes ratio: a relevant marker of gut dysbiosis in obese patients? Nutrients. 12. https://doi.org/10.3390/nu12051474 (2020).
Shannon, C. E. The mathematical theory of communication. MD Comput. 14, 306–317 (1997). (1963).
Lu, J. et al. Chinese gut microbiota and its associations with staple food type, ethnicity, and urbanization. Npj Biofilms Microbiomes. 7. https://doi.org/10.1038/s41522-021-00245-0 (2021).
Canfora, E. E. et al. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. 11, 577–591. https://doi.org/10.1038/nrendo.2015.128
Mancabelli, L. et al. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis[J]. FEMS Microbiol. Ecol. 93, fix153. https://doi.org/10.1093/femsec/fix153
Wang, K. et al. Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell. Rep. 26, 222–235e225. https://doi.org/10.1016/j.celrep.2018.12.028 (2019).
Ayeni, F. A. et al. Infant and adult gut microbiome and metabolome in rural bassa and urban settlers from Nigeria. Cell. Rep. 23, 3056–3067. https://doi.org/10.1016/j.celrep.2018.05.018 (2018).
Maya-Lucas, O. et al. The gut microbiome of Mexican children affected by obesity. Anaerobe. 55, 11–23. https://doi.org/10.1016/j.anaerobe.2018.10.009 (2019).
Chen, X. et al. Alteration of the gut microbiota associated with childhood obesity by 16S rRNA gene sequencing. PeerJ. 8, e8317. https://doi.org/10.7717/peerj.8317 (2020).
Nuli, R. et al. Use of Miseq high-throughput sequencing in a preliminary study on the gut microbiota of Uygur patients with IGR. J. Path Bio . 13, 1084–1090. https://doi.org/10.13350/j.cjpb.181007 (2018).
Zhang, C. et al. Dysbiosis of gut microbiota is associated with serum lipid profiles in male patients with chronic traumatic cervical spinal cord injury. Am. J. Transl. Res. 11, 4817–4834 (2019).
Dong, T. S. et al. A distinct brain-gut-microbiome profile exists for females with obesity and food addiction. Obes. (Silver Spring). 28, 1477–1486. https://doi.org/10.1002/oby.22870 (2020).
Martin, C., Osadchiy, V., Kalani, A. & Mayer, E. The brain-gut-microbiome axis. Cell. Mol. Gastroenterol. Hepatol. 6, 133–148. https://doi.org/10.1016/j.jcmgh.2018.04.003 (2018).
Zhou, L. et al. Correlation between fecal metabolomics and gut microbiota in obesity and polycystic ovary syndrome. Front. Endocrinol. (Lausanne). 11, 628. https://doi.org/10.3389/fendo.2020.00628 (2020).
Pagliai, G. et al. Influence of a 3-month low-calorie Mediterranean diet compared to the vegetarian diet on human gut microbiota and SCFA: the CARDIVEG Study. Eur. J. Nutr. 59, 2011–2024. https://doi.org/10.1007/s00394-019-02050-0 (2020).
Zorena, K. et al. Adipokines and obesity. Potential link to metabolic disorders and chronic complications. Int. J. Mol. Sci. 21, 3570. https://doi.org/10.3390/ijms21103570 (2020).
Amabebe, E. et al. Microbial dysbiosis-induced obesity: role of gut microbiota in homoeostasis of energy metabolism. Br. J. Nutr. 123, 1127–1137. https://doi.org/10.1017/s0007114520000380 (2020).
Acknowledgements
We thank the volunteers who participated in the study. We also thank all of the Hotan Jinglao Road Community staff who provide us with assistance.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 82160611) and the 14th Five-Year Plan Distinctive Program of Public Health and Preventive Medicine in Higher Education Institutions of Xinjiang Uygur Autonomous Region.
Author information
Authors and Affiliations
Contributions
DM and JH conceived the experiments. DM wrote the initial draft, which was revised by JH and CRK. DM and ML analyzed the data. DM, ML, MJ, JZ, KZ, GA., AA, and YY. contributed to data collection and database generation. All authors participated in paper drafting and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Muheyati, D., Han, J., Lv, M. et al. Composition of gut microbiota in obese and normal-weight Uygur adults and its association with adipocyte-related factors. Sci Rep 14, 24649 (2024). https://doi.org/10.1038/s41598-024-76351-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-76351-8
This article is cited by
-
Dynamic changes in gut microbiota and identification of inflammation-associated biomarkers in high-fat diet-induced obese mice
Antonie van Leeuwenhoek (2026)
-
Gut Microbiome in Obesity: A Narrative Review of Mechanisms, Interventions, and Future Directions
Probiotics and Antimicrobial Proteins (2025)
