
Abstract
Hulless barley sheath rot is a spike disease caused by Dactylobotrys graminicola. In recent years, it has generally occurred in hulless barley growing areas in China, resulting in reduced hulless barley yields. In this study, primers and probes were designed based on conserved genome sequences, and a method was established using recombinant enzyme polymerase amplification-lateral flow burette (RPA-LFD) technology for the rapid diagnosis of sheath rot in hulless barley. The method can be successfully established in five minutes at a constant temperature of 39℃, and the results are consistent with those of normal PCR analysis. The method demonstrated high sensitivity, with a detection limit of 10 fg/µL. Furthermore, the rapid method was able to successfully detect D. graminicola in hulless barley during field incubation, which highlighted the significant advantage of the method in practical applications. In conclusion, the RPA method established in this study exhibited several advantageous characteristics, including high efficiency, simplicity, rapidity and practicality, which provide a theoretical basis for the early detection and prevention of D. graminicola.
Similar content being viewed by others

Introduction
Hulless barley sheath rot is a disease of barley spike caused by the infestation of D. graminicola1. The disease was first discovered in the high altitude (2400 ~ 3200 m) area of central western and southern Gansu Province in China in 2009. Subsequently, the disease was found in hulless barley planting areas such as Shigatse in Tibet, Qinghai Province, Gannan Prefecture in Gansu Province, and Ganzi Prefecture in Sichuan Province2. In the field, the diseased spikes were withered, and the young spikes were partly withered or completely necrotic. The rate of diseased spikes in a few seriously diseased fields could reach 20%~30%3. The pathogen has a wide host range and can infect cereal crops and some gramineous weeds. The disease is widespread in hulless barley-producing areas in China and has a trend of increasing yearly, seriously affecting the quality and yield of hulless barley.
Rapid detection of field diseases is beneficial for assessing and predicting disease occurrence and prevalence, facilitating disease prevention and control. Plant pathogenic fungi are identified primarily through the conventional tissue separation method to separate the disease samples collected in the field. These samples are then subjected to verification by Koch’s rule. The advent of rapid diagnosis technology for plant diseases has provided a crucial technical foundation for the timely prevention and control of diseases. At present, the detection methods of hulless barley sheath rot reported in the literature include the tissue separation method2and the polymerase chain reaction (PCR) method based on the internal transcribed spacer (ITS) region of the ribosomal deoxyribonucleic acid (rDNA) sequence interval4. However, these techniques are complex and require expensive materials and equipment, which are difficult to apply to field detection directly.
Recombinase polymerase amplification (RPA) is a rapid, reliable, and portable isothermal nucleic acid amplification technology. It exhibits the following characteristics: high sensitivity, low sample demand, fast amplification speed, low reaction temperature, and no initial heating5. The technology is straightforward to operate and is not constrained by certain environmental factors6. The naked eye can discern the results of the reaction, and a considerable number of samples can be identified in a brief period without the necessity for additional equipment or instruments. It has considerable potential for application in port and field detection7,8. Loop-mediated isothermal amplification (LAMP) is also a new type of nucleic acid amplification technology. However, compared with RPA detection technology, RPA amplification is faster and the reaction temperature is lower. LAMP detection needs to design four specific primers to identify the target DNA sequence. Six different regions identify the six regions of the target sequence, while RPA only needs to design two specific primers to complete the reaction9.The advantages of this method permit the successful application of RPA to the detection of various pathogens, including viruses10,11, bacteria12,13and parasites14. At present, RPA is primarily employed in detecting pathogens in humans and animals, with the detection of plant pathogenic fungi still in its infancy. However, the RPA-LFD method for detecting hulless barley sheath rot has yet to be reported.
In this study, primers and probes were designed to target a conserved sequence in the D. graminicola genome. This was done to establish a rapid detection system for hulless barley sheath rot using the RPA technology. The rapid visual detection method combined with LFD was realized. The specificity and sensitivity of the detection method were further evaluated, and the detection of suspected samples in the field verified the effect. The detection system established in this study provides a technical foundation for rapid field detection and early prevention and control of sheath rot in hulless barley.
Results
Primer screening and probe design
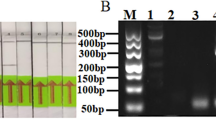
By the principle of RPA primer design, three RPA primer pairs (Table 1) were devised. The genomic DNA of D. graminicola was employed as a template for conventional PCR and detected by 3% agarose gel electrophoresis. The results showed that the specificity of the 2-RPA-F/R and 3-RPA-F/R primers was suboptimal, and other strains also exhibited amplification bands. The 1-RPA-F/R primer is specific for the genomic DNA of D. graminicola, and amplification bands are not observed for different strains. Consequently, the 1-RPA-F/R primers were selected for probe design and subsequent RPA-LFD testing (Fig. 1). Both the PCR and RPA amplified fragments are 153 bp in size.

Electrophoresis results of RPA-LFD primer screening. 1: D. graminicola DGC; 2: D. graminicola YGC; 3: D. graminicola DTC; 4: D. graminicola HTC; 5: D. graminicola BMZ; 6: D. graminicola GMNXC; 7: D. graminicola SJWC; 8: D. graminicola RYX; 9: P. teres; 10: P. graminea; 11: F. graminearum; 12: U. nuda; 13: U. hordei; 14: ddH2O; M: DL500 Marker.
Specificity evaluation of RPA-LFD detection
Agarose gel electrophoresis and LFD were employed to identify the DNA amplification products of the strains in Table 2, respectively. The results demonstrated the DNA amplification bands of D. graminicola DGC were distinct. The LFD exhibited a detection line, indicating a positive outcome (Fig. 2). Conversely, the results for the other strains were negative, consistent with the agarose gel electrophoresis results.

RPA-LFD detection system of D. graminicola. RPA-LFD (A); PCR (B); 1: D.graminicola DGC; 2: P. teres; 3: P. graminea; 4: F. graminearum; 5: U. nuda; 6: U. hordei; 7: ddH2O; M: DL500 Marker; C: control band; T: test band.
Sensitivity evaluation of RPA-LFD detection
The results of the sensitivity test showed (Fig. 3) that strain The DNA concentrations of D. graminicola DGC at 100 ng/µL, 10 ng/µL, 1 ng/µL, 100 pg/µL, and 10 pg/µL were all positive on the LFD test strips. The detection line of DNA with a concentration of 10 pg/µL was weak. DNA concentrations of 1 pg/µL and 100 fg/µL were unable to show the quality control line on the LFD test strip, and the results were negative. Agarose gel electrophoresis demonstrated that the DNA concentration of the D. graminicola strain was 100 ng/µL, 10 ng/µL, and 1 ng/µL. This indicates that the sensitivity of RPA-LFD for D. graminicola DNA detection is significantly greater than that of traditional PCR detection.

RPA-LFD sensitivity detection of D. graminicola. RPA-LFD(A); PCR(B); 1: D. graminicola DGC; 1–7: 100 ng/µL,10 ng/µL,1 ng/µL,100 pg/µL,10 pg/µL,1 pg/µL,100 fg/µL, M: DL500 Marker; C: control band; T: test band.
Optimization of RPA-LFD reaction conditions
A reaction effect test was conducted using 10 ng/µL D. graminicola DGC genomic DNA as a template at six different temperatures (25 °C, 30 °C, 35 °C, 40 °C, 45 °C and 50 °C) to determine the optimal temperature for the RPA reaction system. At temperatures between 35 °C and 45 °C, the LDF test strip exhibited a clear positive detection line. At temperatures of 25 °C to 30 °C and 50 °C, the LDF test strip did not produce a positive detection line (Fig. 4A). Based on the above results, according to the kit instructions and the stability of the reaction, 39 °C was finally determined as the best measurement temperature.

Optimization of the reaction conditions of D.graminicola. RPA-LFD Optimization of the RPA amplification temperature(A)Evaluation of the RPA reaction time(B); NTC: no-template control; C: control band; T: test band.
In order to ascertain the optimal detection time of LF-RPA, 10 ng/µL D. graminicola DGC genomic DNA was employed as a template to test the reaction time of 0, 5, 10, 20, 30, 40, 50, and 60 min at 39 °C. Upon completion of the reaction at 5 min, a faint positive test line was observed, while a clear positive test line was evident at 10 to 50 min (Fig. 4B). The positive detection line was the most distinct at 30 min; thus, this time was determined to be the optimal reaction time.
RPA-LFD detection of natural disease samples in the field
DNA was extracted from the flag leaves, stems, and spikes of plants suspected of being infected in the 6 regions fields and detected by RPA-LFD and gel electrophoresis. In the field sample assay, the selected samples were comprised of diseased and healthy tissues collected from different locations. The results are shown in Fig. 5, where (3, 4, 7, 12, 13, 14, 16, 21, 22, 23, 24, 25, 26, 28, 29, 31, 32, 35, 36) are morbid samples and (1, 2, 5, 6, 8, 9, 10, 11, 15, 17, 18, 19, 20, 27, 30, 33) are healthy samples. The tests were randomized according to location and sample site. The results demonstrated that all healthy samples were negative and all diseased samples were positive. Furthermore, the target pathogens were isolated in all positive samples. Agarose gel electrophoresis demonstrated a positive outcome with the amplified 153 bp band (Fig. 5B). Seven positive samples were identified in flag leaf samples, six in stem samples, and six in spike samples (Table 3). The agarose gel electrophoresis results were consistent with those of RPA-LFD (Fig. 5), with a positive detection rate of 52.8%.

Detection of D. graminicola in infected plant tissues using RPA-LFD(A)and conventional PCR(B). M: DL500Marker; 1–36: Flag leaf, stem and spike samples were collected from diseased and healthy flag leaves, stems and spikes in 6 regions of Qinghai Province.; NTC: no-template control; C: control band; T: test band.
Discussion
In 2009, the sheath rot disease of hulless barley was first observed in Gansu Province. The disease is prevalent in areas where hulless barley is cultivated, and its impact is increasing yearly. This has a great impact on the quality and yield of hulless barley. RPA is a rapid and accurate method for detecting D. graminicola in plants, which enables the determination of whether the disease is present, thus providing an essential guarantee for the prevention and treatment of hulless barley sheath rot. In this study, RPA-specific primers were designed based on the conserved sequence of D. graminicolagenome and combined with LFD to establish an RPA-LFD rapid detection method for sheath rot of hulless barley. The results demonstrated that the optimal detection conditions were 39 °C and 30 min, with a sensitivity that was 100 times higher than that of conventional PCR. In the screening test of the best reaction temperature, the temperature gradient and result determination reference Lu et al6. The method is straightforward to execute and the visible results render it suitable for the direct detection of field disease samples.
New nucleic acid detection technologies such as RPA and loop-mediated isothermal amplification (LAMP) are widely used to detect pathogens of hulless barley diseases rapidly. Hu et al. designed specific primers based on the ITS-rDNA sequence of Pyrenophora gramineaand established a LAMP detection system for hulless barley stripe disease. Under the condition of 63 °C, 70 min, calcein and SYBR Green I were used as reaction indicators. The minimum detection limit of the system was 0.3 pg/µL, which was 100 times higher than that of ordinary PCR. It can be used to detect whether the seeds carry the source of bacteria10. Qi et al. established a LAMP detection system based on the ITS-rDNA sequence of barley stem rot fungus (Fusarium avenaceum), with a DNA sensitivity of up to 10 pg/µL and a sensitivity of 40 pathogen spores per gram of soil15. In comparison to conventional PCR reactions, RPA-LFD detection offers a more convenient and rapid approach. For instance, in this particular test, the reaction time is reduced to a mere 5 min, exhibiting a sensitivity that is over 100 times higher than that of traditional PCR. This eliminates the necessity for a PCR instrument, and instead, a water bath is sufficient to complete the detection process, making it particularly well-suited for use in grassroots settings. The RPA-LFD process allows for the visualisation of the results. However, it is easy to cause nucleic acid contamination in the laboratory when observing the results, and sometimes false positives occur in the negative control9,16. Therefore, the preparation and detection process should be carried out in a special room and clean environment, the reaction tube should be carefully opened and closed, and the gloves should be replaced during detection. The combination of MI-IF-RPA and LF-RPA to detect SARS-CoV-2 reduces the contamination risk by integrating the detection strip and the reaction chamber into a single closed microfluidic chip6,17. Developing this disposable closed nucleic acid detection device can improve this problem and be used to detect plant pathogenic microorganisms in the future. False-negative results may result if the amount of pathogen nucleic acids in the sample is below the sensitivity threshold of the assay; the relevant reagents are invalid and outdated; and the sample may lead to degradation or loss of nucleic acids if it is not handled correctly during collection, storage, or transport, which may lead to a false-negative result18.
This study successfully established an RPA-LFD rapid detection method for the pathogen of hulless barley sheath rot. The technique exhibits high specificity and sensitivity, making the operation straightforward and rapid. The method can specifically detect the field samples of barley sheath rot, thereby providing a rapid means of disease detection in the field. This offers technical support for the early diagnosis and epidemic monitoring of the disease. RPA technology represents a novel approach to pathogen detection, yet its practical applications in real-time detection still need to be improved.
Further breakthroughs may be made in the following areas in the future. RPA primer design professional software could be developed to enhance primer specificity and amplification effect. In conjunction with this, ingenious devices, such as heat preservation cups or portable devices, could be created to facilitate the detection process. Furthermore, developing cheaper and more efficient reaction reagents could effectively reduce the cost of the method, thereby promoting the wider adoption and utilization of this simple and efficient detection method. With the development of scientific research, it is anticipated that this technology will become a principal method for rapidly detecting pathogens.
Materials and methods
Test materials and reagents
The information on the tested strains in this study is shown in Table 2, which was provided by the Key Laboratory of Comprehensive Management of Agricultural Pests in Qinghai Province.
Test medium: potato dextrose agar (PDA) medium included potato 200 g, glucose 20 g, agar 20 g, distilled water 1000 mL, autoclaved at 121 °C for 20 min.
The reagents and instruments used in this study are as follows: RPA-nfo amplification kit (TwistAmp®nfo), purchased from Qinghai Baisai Trading Co., Ltd.; mileniaGenLine HybriDetect Kit, Mile-niaBiotec GmbH, Germany; 2×Taq Master Mix, Tiangen Biochemical Technology Co., Ltd.; dL500DNA marker, Biomedical (Beijing) Biotechnology Co., Ltd.; other reagents were domestic analytically pure. TissueLyserII Tissue Grinding Instrument, Kaijie Enterprise Management Co., Ltd.; bIO-RAD electrophoresis instrument, Champ Gel 6000 gel imager, Epps Biosystems Trading Co., Ltd.; ABI 7500 PCR instrument, BIO-RAD, NanoDrop One micro-nucleic acid protein analyzer and SORVALL ST 16R high-speed refrigerated centrifuge; rXZ-280B intelligent artificial climate box.
Extraction of pathogenic fungus DNA
All the tested strains were cultured in a 25 °C constant temperature incubator on a PDA plate, and the marginal hyphae were selected and cultured at a 25 °C constant temperature on a PDA plate for 7 days. The full-grown hyphae on the PDA plate were scraped in a 2 mL centrifuge tube, and 5 small steel balls disinfected with anhydrous ethanol were placed in each tube. After freezing in liquid nitrogen, it was ground into powder in a tissue crusher. After centrifugation, 600 µL DS-Buffer buffer was added to each tube, then 600 µL, 25:24:1 DNA extract was added, and the mixture was turned up and down, and placed at 4 °C for 20 min. After centrifugation for 10 min, the rotation speed was 12,000 rpm; the supernatant of 450 µL to 1.5 mL centrifuge tube, add 450 µL isopropanol, after mixing at -20 °C stand for 2 h; then centrifuged for 10 min, the speed was 12,000 rpm, and the supernatant was removed. The DNA was washed with 1 000 µL anhydrous ethanol 2–3 times and then dried. Add 50 µL sterile enzyme-free water to dilute; the concentration of extracted DNA was detected and frozen in a refrigerator at-20 °C.
Design of the RPA primers and probes
After sequencing the genome of D. graminicola, a coding sequence (CDS) of 3992 bp was assembled (GenBank: JAXCIM000000000), including a 191 bp open reading frame (ORF). Specific primers and probes were designed using this open reading frame sequence. According to the primer and probe design principles provided by TwistDX’s official website (www.twistdx.co.uk), the primer pairs upstream and downstream of RPA were designed by Primer Premier 5.0 software (Table 1). The length of the RPA probe is usually 46–52 bp. According to the designed RPA primer amplification fragment, a probe with a size of 45 bp was designed. The 5 ' end of the probe was added with the FAM fluorescent group, and the 3 ' end was added with the C3-spacer. The base at 30 bp from the 5 ' end of the probe was replaced by tetrahydrofuran (THF). The 5 ' end of the reverse primer for RPA-LFD was labeled with biotin. The sequence fragments used to design primers are shown in Fig. 6. Detection primers and probes were synthesized by Beijing Aoke Dingsheng Biotechnology Co., Ltd.Primers were evaluated using conventional PCR based on sensitivity and specificity detection. The sensitivity was determined using 10-fold serially diluted genomic D. graminicola DNA (ranging from 100 ng to 100 fg). PCR amplification products were visualized using 3% agarose gel electrophoresis.

Sequence and orientation of primers in genome (GenBank: JAXCIM000000000) used to develop RPA and conventional PCR to detect D. graminicola.
The ordinary PCR system
A specific primer of D. graminicola, 3-RPA-F/3-RPA-R, was used for ordinary PCR detection. The reaction system was as follows: MasterMix 12.5 µL, primers (10 µmol/L) 1 µL each, template 1 µL, and ddH2O 9.5 µL. The amplification program was as follows: 94 °C predenaturation for 3 min; 35 cycles of 94 °C denaturation for 30 s, 65 °C annealing for 30 s, and a 72 °C extension for 1 min; extension at 72 °C for 5 min; and storage at 4 °C. A sample of 10 µL was taken for electrophoresis.
Establishment of RPA-LFD reaction system
Gene sequence amplification was performed according to the RPA (test strip) kit. The reaction freeze-dried enzyme powder 1 tube, A Buffer 25 µL, (2 µM) 1-RPA-F and RPABio-R each 2.0 µL, (2 µM) probe RPA-LFD-P 0.6 µL, template DNA 3 µL, ddH2O 14.9 µL, 2.5 µL B Buffer (MgOAc) were added to the reaction tube in turn. The mixture was mixed upside down 5 ~ 6 times, centrifuged at low speed for a short time, and incubated at 39 °C for 30 min. The reaction solution was mixed with 5 µL and 100 µL detection buffer (HybriDetect). The sample detection end of the flow chromatography test strip was vertically immersed in the mixed solution, and the results were observed after standing at room temperature for 5 min. The purple-red bands appeared in the quality control area and the detection area of the test strip, the result was positive, and the sample contained the target strain; when the test strip only had a purplish red band in the quality control area and no band in the detection area, the result was negative.
Detection of specificity and sensitivity of RPA-LFD
The specificity of the RPA-LFD method for D. graminicola was verified, and the DNA of common hulless barley fungal pathogens was selected for specific detection. The test strains were shown in Table 2, and the specificity test of the RPA-LFD detection method was carried out.
The sensitivity of the RPA-LFD method was verified. The concentration of the extracted D. graminicola genomic DNA was adjusted to 100 ng/µL and diluted to 100 ng/µL, 10 ng/µL, 1 ng/µL, 100 pg/µL, 10 pg/µL, 1 pg/µL and 100 fg/µL in a 10-fold gradient. The sensitivity test of the RPA-LFD detection method was carried out by using the above different DNA concentrations as templates. Sterilization ddH2O was negative control. At the same time, the PCR reaction was carried out, and the PCR amplification products were visualized by 3% agarose gel electrophoresis. The above experiments were carried out in three biological replicates under the same conditions.
Optimization of RPA-LFD reaction conditions
In order to determine the optimal RPA reaction temperature, 10 ng/µL D. graminicola DGC genomic DNA was used as a template for determination. The reaction system was incubated at six different temperatures (25 °C, 30 °C, 35 °C, 40 °C, 45 °C and 50 °C) for 30 min. In order to determine the optimal detection time of LF-RPA, 10 ng/µL D. graminicola genomic DNA was used as a template to test the reaction time of 0, 5, 10, 20, 30, 40, 50 and 60 min at 39 °C. Sterilization ddH2O was was negative control. After the reaction, LFD was used to detect the above products. Different reactions were observed, and the clarity and color depth of the detection band were observed. The best reaction condition for RPA-LFD detection was when the band was clear, and the color was dark. The above experiments were carried out in three biological replicates under the same conditions.
RPA-LFD detection of natural disease samples in the field
To evaluate the feasibility of the RPA-LFD detection method in actual production, plants with suspected symptoms of barley sheath rot in the field (Table 4) were collected in 6 regions of Qinghai Province, and DNA was extracted from the flag leaves, stems, and spikes of suspected plants. Using 10 ng/µL DNA as a template and sterilized ddH2O was negative control. as a blank control, the conventional PCR detection method and the optimized RPA-LFD detection system were used for detection, respectively. The RPA-LFD reaction system and procedures are the same as above. PCR amplification products were visualized using 3% agarose gel electrophoresis. The above experiments were carried out in three biological replicates under the same conditions.
Data availability
The genome assembly was available at GenBank under accession numbers JAXCIM000000000 (Bioproject PRJNA1034805 and Biosample SAMN38074485). Information has been added to the body of the article. All data generated or analyzed during this study are included in this published article.
References
He, S. et al. Dactylobotrys graminiaola: A New PhytopatcausingasheathSheath of Hulless Barley and Oats. Mycosystema. 34, 331–340 (2015).
Chen, L. et al. Genetic diversity of Dactylobotrys graminicola and its pathogenicity to Hordeum Vulgare Var. Nudum Seedlings. Scientia Agricultura Sinica. 53, 213–224 (2020).
He, S. et al. A New Diease of Oat and Hulless Barley—Sheath rot diease. Gansu Agricultural Sci. Technol. 62, 3–4 (2010).
Du, C., Zhang, H. & Yao, Q. Genetic Diversity Analysis of Sheath Rot Pathogen of Hulless barley in Qinghai Province. J. Qinghai Univ. (Natural Science). 41, 33–39 (2023).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. Dna Detection using recombination proteins. Plos Biol. 5, e204 (2006).
Lu, X. et al. Rapid and Simple Detection of Phytophthora Cactorum in Strawberry using a coupled recombinase polymerase amplification–lateral Flow Strip Assay. Phytopathol. Res. 3, 12 (2021).
Pang, J. et al. Rapid Visual Detection of Enterocytozoon Hepatopenaei by Recombinase Polymerase Amplification Combined with lateral Flow Dipstick. Front. Mar. Sci. 9, 1–10 (2022).
Mondal, D. et al. Mobile Suitcase Laboratory for Rapid Detection of Leishmania Donovani using recombinase polymerase amplification assay. Parasite Vector. 9, 281 (2016).
Hu, Z. et al. Establishment of lamp detection system of Pyrenophora Graminea. J. Northwest. F Univ. (Natural Sci. Edition). 49, 105–114 (2021).
Babujee, L. et al. Optimization of an Isothermal recombinase polymerase amplification method for real-time detection of Potato Virus Y O and N types in Potato. J. Virol. Methods. 267, 16–21 (2019).
Miao, F. et al. Rapid and sensitive recombinase polymerase amplification combined with lateral Flow Strip for detecting African swine fever virus. Front. Microbiol. 10, 1004 (2019).
Saxena, A., Pal, V., Tripathi, N. K. & Goel, A. K. A recombinase polymerase amplification lateral Flow Assay for Rapid Detection of Burkholderia Pseudomallei, the Causative Agent of Melioidosis. Braz J. Microbiol. 53, 185–193 (2022).
Pang, J. et al. A real-time recombinase polymerase amplification assay for the Rapid detection of Vibrio Harveyi. Mol. Cell. Probe. 44, 8–13 (2019).
DeShields, J. B., Moroz, N., Braley, L. E., Mora-Romero, G. A. & Tanaka, K. Recombinase polymerase amplification (RPA) for the Rapid Isothermal Detection of Spongospora Subterranea F. Sp. Subterranea and Potato Mop-Top Virus. Am. J. Potato Res. 96, 617–624 (2019).
Qi, Y., Cao, S., Li, X. & Li, M. Development of Loop-mediated isothermal amplification assay for detection of Fusarium Avenaceum. Acta Agrestia Sinica. 26, 1004–1010 (2018).
Kim, N. et al. Rapid and specific detection of Apple Stem Grooving Virus by Reverse transcription-recombinase polymerase amplification. Plant. Pathol. J. 34, 575–579 (2018).
Liu, D. et al. A microfluidic-integrated lateral Flow recombinase polymerase amplification (Mi-If-Rpa) assay for Rapid Covid-19 detection. Lab. Chip. 21, 2019–2026 (2021).
Mouliou, D. S. & Gourgoulianis, K. I. False-positive and false-negative COVID-19 cases: respiratory Prevention and Management Strategies, Vaccination, and further perspectives. Expert Rev. Respir Med. 15, 993–1002 (2021).
Funding
This study was financially supported by the basic research project of Qinghai, China(2023-ZJ-944 J).
Author information
Authors and Affiliations
Contributions
Z.H.Q. and C.L.Y.F. carried out the experiment. Z.H.W. wrote the manuscript. D.R.F. analyzed the bioinformatics data. Y.Q. and H.L. revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors reviewed the manuscript and agreed to publication.
Competing interests
The authors declare no competing interests.
Ethical approval
Our study complies with relevant institutional, national, and international guidelines and legislation. The use of plants in this study, whether cultivated or wild, and the collection of plant material were in line with the policies of the relevant institutions and with national and international norms and laws.
Authors’ contributions statement
Haiqing Zhang and Liyifan Chen carried out the experiment. Haowen Zheng wrote the manuscript. Ruifang Dong analyzed the bioinformatics data. Qiang Yao and Lu Hou revised the manuscript. All authors reviewed the manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, H., Chen, L., Dong, R. et al. Establishment of recombinase polymerase amplification detection method for Dactylobotrys graminicola. Sci Rep 14, 25079 (2024). https://doi.org/10.1038/s41598-024-76921-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-76921-w
Keywords
This article is cited by
-
Isothermal nucleic acid amplification assays for the detection of porcine stool-associated RNA virus
Scientific Reports (2025)
