
Abstract
Chronic rhinosinusitis (CRS) is a prevalent inflammatory airway disease affecting over 10% of the global population, leading to considerable socio-economic impacts, especially in developing countries. The pathogenesis of CRS is multifactorial, involving potential contributions from both genetic and environmental factors. While the influence of allergic and autoimmune diseases on CRS has been observed, the causal relationships between these diseases and CRS remain unclear. We extracted data from large-scale genome-wide association studies (GWAS) and utilized a bidirectional two-sample Mendelian randomization (MR) analysis to explore the causal relationships between CRS and ten autoimmune and allergic diseases, including asthma, allergic rhinitis (AR), atopic dermatitis (AD), psoriasis, type 1 diabetes (T1D), hypothyroidism, celiac disease (CeD), multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE). Additionally, we conducted colocalization analysis to determine whether the allergic/autoimmune diseases showing statistical causal relationships with CRS are driven by the same genetic variants. The MR analysis identified that AR (OR = 1.30; 95% CI = 1.21–1.40; P = 3.26E−13), asthma (OR = 1.35; 95% CI = 1.25–1.45; P = 1.35E−14), and AD (OR = 1.17; 95% CI = 1.06–1.30; P = 0.003) were significantly associated with an increased risk of developing CRS. Interestingly, psoriasis (OR = 0.05; 95% CI = 0.01–0.37; P = 0.004) appeared to have a protective effect against CRS. Associations for T1D and hypothyroidism were also suggestive as potential risk factors for CRS. No significant associations in the reverse MR analysis, suggesting a one-directional relationship. Colocalization analysis indicated that asthma (PP.H4 = 0.99) shared the same genetic variant (IL-33 rs3939286) with CRS. In conclusion, our study confirmed the causal relationships between allergic and autoimmune diseases (AR, asthma, AD, and psoriasis) and CRS. Notably, we identified a shared genetic variant, rs3939286 in the IL-33 gene, between asthma and CRS, suggesting that targeting the IL-33 pathway may provide a therapeutic strategy for both diseases.
Similar content being viewed by others

Introduction
Chronic rhinosinusitis (CRS) is a prevalent inflammatory airway disease that significantly affects the quality of life for individuals, impacting over 10% of the global population and imposing a substantial socio-economic burden, particularly in developing countries1. Accurately identifying risk factors for CRS is crucial for its prevention and management. Although existing studies have explored several etiological factors of CRS, potential risk factors identified include viral and bacterial infections, as well as genetic and anatomical predispositions1,2. However, the impact of immune-related diseases on the development of CRS has not been fully investigated. This study aims to elucidate the causal relationships between immune-related diseases (including allergic and autoimmune diseases) and CRS, thereby enhancing our understanding of CRS pathogenesis and contributing to more effective strategies for its management and prevention.
Common allergic diseases include atopic dermatitis (AD), allergic rhinitis (AR), and asthma. Recent epidemiological studies have indicated that the prevalence of allergic diseases is on the rise, now affecting approximately 20% of the global population3. The immune system, composed of various immune cells, serves to protect the body from foreign antigens4. However, in autoimmune diseases, the immune system fails to differentiate between host cells and foreign antigens, leading to an autoimmune response5. The prevalence of autoimmune diseases is around 5%, with common conditions including celiac disease (CeD), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), type 1 diabetes (T1D), multiple sclerosis (MS), psoriasis, and hypothyroidism6,7. Observational studies have previously suggested that AR and asthma might contribute to an increased incidence of CRS, and the use of immunosuppressive medications and autoimmune diseases has also been linked to CRS8,9,10. However, traditional observational studies are often susceptible to confounding factors and reverse causation, which can obscure the true nature of these relationships.
Mendelian randomization (MR) is an innovative epidemiological method that utilizes common genetic variants as proxies for exposures to explore potential causal relationships between exposures and disease outcomes11,12. Unlike traditional observational studies, MR minimizes confounding and avoids reverse causation, leveraging the random allocation of genetic variants at conception, akin to randomization in a randomized controlled trial (RCT)13. MR uses data from large-scale genome-wide association studies (GWAS), offering a cost-effective and timely alternative to RCTs, and can assess the effects of lifelong exposures that RCTs might miss14. Additionally, MR is suitable for studying outcomes where RCTs are impractical or unethical, providing valuable insights into the effects of modifying risk factors and guiding public health policies15. This method significantly enhances our ability to infer causality in disease research, bridging the gap between correlation and causation.
Building on the strengths of MR to overcome the limitations commonly associated with observational studies, our study aims to apply this method rigorously to elucidate the causal relationships between both allergic and autoimmune diseases and CRS. Through leveraging large-scale GWAS data and employing genetic variants as instrumental variables (IVs), we aim to dissect the intricate interactions between these immune-related diseases and CRS. To the best of our knowledge, no MR studies have yet assessed the relationships between allergic, autoimmune diseases, and CRS. This study, therefore, not only advances our understanding of CRS but also contributes significantly to the use of genetic epidemiology in shaping public health strategies and clinical management of airway inflammatory diseases.
Materials and methods
Study design
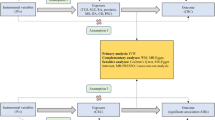
In our study, exposures comprised three prevalent allergic diseases: asthma, AR, and AD; along with seven common autoimmune diseases: psoriasis, T1D, hypothyroidism, CeD, MS, RA, and SLE. We extracted IVs for these exposures from GWAS databases corresponding to each of the ten diseases. Outcome data for CRS were sourced from a Finn Gen database. Subsequently, a bidirectional two-sample MR analysis was conducted, assessing causation between CRS and the ten diseases. The MR analysis adhered to three fundamental assumptions: (1) IVs used in this study must be strongly associated with either allergic or autoimmune diseases; (2) IVs must be independent of confounders affecting allergic diseases, autoimmune diseases, and CRS; (3) IVs affect CRS solely through their impact on autoimmune or allergic diseases. (Fig. 1).

Study design.
Genetic instrumental variables and data sources
Summary data for ten exposures were sourced from the Integrative Epidemiology Unit (IEU) Open GWAS database (https://gwas.mrcieu.ac.uk/) and the UK Biobank (UKB) database. Specifically, T1D, RA, CeD, SLE, MS, asthma, AR, and AD were obtained from various publicly available GWAS datasets (Table 1). Initially, single nucleotide polymorphisms (SNPs) were identified based on their significant association (p < 5 × 10⁻⁸) with a range of allergic and autoimmune diseases, including asthma, AR, AD, psoriasis, T1D, hypothyroidism, CeD, MS, RA, and SLE. These SNPs were subsequently utilized as genetic IVs due to their strong and significant associations with the exposures. Linkage disequilibrium (LD) was calculated based on the 1000 Genomes European reference panel. IVs were carefully selected to ensure no LD: r² < 0.001 and a clumping window exceeding 10,000 kb. We also employed the Steiger test to confirm the absence of any reverse causal relationship between the outcomes and IVs. Additionally, the strength of IVs was assessed by calculating the F-statistic, with IVs exhibiting an F-statistic > 10 considered robust (Table S1).
GWAS summary data of chronic rhinosinusitis
We sourced genetic data for CRS from the publicly available FinnGen biobank analysis (Round 10), with diagnoses based on ICD-10 codes. The cohort including 17,987 cases and 308,457 controls. The FinnGen biobank provides comprehensive genetic data derived from a large, well-characterized cohort of the Finnish population, renowned for its high-quality, population-specific health and genetic information.
Two-sample MR
In our study, MR analyses were performed using four different methods to ensure robust causal estimation: the Inverse Variance Weighted (IVW) model, the weighted median, the MR-Egger method, and the Weighted Mode16. The IVW model aggregates causal estimates from multiple genetic instruments, weighting each instrument inversely proportional to its variance to minimize the overall variance, thereby combining these estimates through a meta-analytical approach for a comprehensive causal estimate. The weighted median estimator provides a robust causal estimate that remains valid even if up to 50% of the information comes from invalid instruments. This method calculates the median of the causal effect estimates, weighted by the inverse variance of each estimate. The advantage of this method is its resilience to outliers and invalid instruments, as long as a majority of the instruments are valid. The MR-Egger method is specifically used to detect and adjust for horizontal pleiotropy, with its regression intercept serving as an indicator of unbalanced pleiotropic effects, where genetic variants affect the outcome through non-exposure pathways. The Weighted Mode method calculates the most common or estimate of the causal relationships, weighted by the inverse variance of each estimate, which provides a robust measure of central tendency that is particularly useful when there is substantial heterogeneity among instrument estimates.
For the sensitivity analyses in our MR study, several methods were utilized to address potential biases and ensure the robustness of our findings. We employed the MR-PRESSO (Mendelian Randomization Pleiotropy RESidual Sum and Outlier) method to detect outliers potentially exhibiting horizontal pleiotropy and to provide corrected estimates after outlier removal. Additionally, the intercept obtained from MR-Egger regression was used to assess the presence of unbalanced pleiotropic effects. Heterogeneity was tested using Cochran’s Q test, and in the presence of heterogeneity, the main results were recalculated using the IVW random effects model. A leave-one-out analysis was conducted to examine if any single SNP disproportionately influenced the significant associations. To minimize the likelihood of false-positive results, Bonferroni correction was applied; results achieving a P-value < 0.005 (corrected from P < 0.05/10) were considered statistically significant, while those with P < 0.05 but not reaching this threshold were deemed suggestive.
Colocalization analysis
We performed colocalization analysis on allergic/autoimmune diseases and CRS with P < 0.005, selecting a 50 kb region around each SNP as the area of analysis. Bayesian colocalization assesses shared genetic variations between traits via posterior probabilities for five hypotheses. This study focused on the posterior probability of hypothesis 4 (PP.H4), indicating both traits are driven by the same variant, with PP.H4 > 0.8 denoting successful colocalization17.
Statistical analysis
To minimize the risk of false positives due to multiple testing, we applied the Bonferroni correction. With this adjustment, a P-value threshold of < 0.005 (0.05/10) was considered statistically significant. Results with P-values < 0.05 that did not meet this threshold were deemed suggestively significant. All Mendelian randomization analyses were conducted in R software (version 4.3.0) using the “TwoSampleMR” and “MendelianRandomization” packages. The MR-PRESSO method was implemented via the “MR-PRESSO” R package (version 1.0), and colocalization analyses were performed using the “coloc” R package (version 5.2.3). We used biorender to draw the study design in Fig. 1.
Results
Causal relationships between allergic diseases and autoimmune diseases with chronic rhinosinusitis
Based on the IVW method, our analysis revealed significant causal relationships between AR (OR = 1.30; 95% CI = 1.21–1.40; P = 3.26E−13), asthma (OR = 1.35; 95% CI = 1.25–1.45; P = 1.35E−14), and AD (OR = 1.17; 95% CI = 1.06–1.30; P = 0.003) with an increased risk of CRS. Interestingly, psoriasis (OR = 0.05; 95% CI = 0.01–0.37; P = 0.004) emerged as a protective factor against CRS. There were suggestive associations of T1D (OR = 1.02; 95% CI = 1.01–1.04; P = 0.007) and hypothyroidism (OR = 2.84; 95% CI = 1.06–7.61; P = 0.037) with CRS. However, there was insufficient evidence to link genetically predicted CeD (OR = 0.95; 95% CI = 0.90–1.01; P = 0.136), MS (OR = 1.04; 95% CI = 1.00–1.08; P = 0.062), RA (OR = 1.04; 95% CI = 0.96–1.12; P = 0.322), and SLE (OR = 0.97; 95% CI = 0.93–1.01; P = 0.115) with CRS (Fig. 2). In the reverse MR analysis, where genetic predisposition to CRS was tested as an exposure with the nine diseases as outcomes (CeD was not feasible to conduct the reverse MR analysis due to the insufficient number of SNPs), there was no association found between the genetic susceptibility of any of these diseases and CRS (Fig. 3).

Two-sample Mendelian randomization estimates of allergic diseases and autoimmune diseases on CRS risk.

Reverse Mendelian randomization estimates of CRS on allergic diseases and autoimmune diseases risk.
To further explore whether the statistical causal relationships identified are due to shared causal SNP variations between the diseases, we conducted colocalization analyses for AR, asthma, AD, psoriasis, and CRS. The colocalization results indicated that asthma (PP.H4 = 0.99) and CRS share the same genetic variant at the IL-33 rs3939286 locus, as detailed in Table 2; Fig. 4. These findings suggest a robust genetic link between asthma and CRS, potentially related to the functionality of the IL-33 gene.

Colocalization analysis results for (A) AR, (B) asthma, (C) AD, (D) Psoriasis and CRS.
Sensitivity analysis
Subsequent sensitivity analyses were conducted to verify the robustness of the IVW results. In the MR-Egger regression analysis, we observed evidence of directional pleiotropy for AD and MS with p-values < 0.05 (Table 3). Cochran’s Q test indicated heterogeneity in the MR results for SLE, RA, AR, T1D, asthma, AD, and hypothyroidism, all significant at p-values < 0.05 (Table 3). The presence of heterogeneity, often inevitable when numerous IVs are involved. We employed a multiplicative random effects IVW as the primary analytical approach, affirming that heterogeneity is acceptable and does not compromise the estimation of causal effects. MR-PRESSO tests identified outliers in the datasets for SLE, AR, T1D, asthma, CeD, and hypothyroidism. Results corrected for outliers are presented in Fig. 2, indicating that removing outliers does not affect the causal relationships suggested by the main IVW results. Leave-one-out analysis demonstrated that all SNPs were uniformly distributed on the zero side. Scatter plots and Leave-one-out plots are provided in Figs. S1 and S2.
Discussion
In this study, we conducted a bidirectional two-sample MR analysis to explore the causal relationships between ten diseases (including allergic and autoimmune disorders) and CRS. Our MR analysis revealed significant causal relationships between AR, asthma, psoriasis, and CRS. To the best of our knowledge, this is the first study to access the causal relationships between allergic and autoimmune diseases and CRS. The findings significantly enhance our understanding of the potential mechanisms underlying CRS and contribute to the development of more effective disease management strategies for CRS.
Asthma is a common allergic inflammatory airway disease characterized by biological and immunological heterogeneity18,19. This heterogeneity allows asthma to be classified into different phenotypes based on the underlying immune mechanisms19. The most prevalent inflammatory phenotype of asthma is Type 2 high inflammation, which is marked by elevated levels of Type 2 inflammatory biomarkers. These include eosinophils, exhaled nitric oxide, IgE, and interleukins such as IL-4, IL-5, and IL-1320. Conversely, Type 2 low inflammation asthma may predominantly involve neutrophil infiltration along with cytokines like IFN-γ and TNF-β21. Additionally, mixed inflammatory asthma presents with co-infiltration of both eosinophils and neutrophils22. CRS and asthma share similar biological and immunological mechanisms in their pathogenesis, underscoring a potential interconnection between the two conditions2. CRS can also be classified based on the heterogeneity of immune mechanisms into different phenotypes: eosinophilic CRS, non-eosinophilic CRS, and mixed inflammatory CRS23,24. Studies have shown that patients with CRS who also have asthma tend to experience more severe symptoms compared to those with CRS alone25,26,27. Furthermore, a observational study indicated that patients with asthma are at an increased risk of developing CRS28. These results align with our findings, suggesting asthma as a risk factor for CRS. The link between these two diseases is likely mediated by shared inflammatory pathways and immune responses.
Furthermore, our colocalization analysis confirmed that asthma and CRS share the pathogenic SNP variation IL-33 rs3939286, implicating the IL-33 gene as a critical mediator in both diseases. IL-33 is an alarmin factor released from airway epithelial cells upon exposure to environmental triggers, driving airway inflammation through Th2 immune responses from various effector cells, which plays a critical role in the pathogenesis of asthma29. The involvement of IL-33 in CRS is equally crucial as it enhances mucosal inflammation. Specifically, IL-33 activates ST2(+) innate lymphoid cells to consistently produce IL-13, a key mediator in regulating both CRS and asthma. IL-13 primarily affects the respiratory tract by promoting mucus secretion and airway hyperreactivity, thus exacerbating the symptoms and pathological changes associated with asthma and CRS30,31. The IL-33-ST2 axis represents an emerging clinical strategy for treating inflammatory diseases. Anti-ST2 therapy has demonstrated potential in reducing airway eosinophilia and controlling acute exacerbations in patients with severe asthma, particularly those unresponsive to conventional treatments32. Furthermore, targeting upstream IL-33 as an attractive pharmacological target, the use of neutralizing antibodies can help reduce inflammation levels in airway epithelial cells and ameliorate epithelial dysfunction, offering a new direction in inflammatory diseases management33. In our findings, this shared genetic foundation via IL-33 rs3939286 suggests that therapeutic targeting of this pathway might be effective for both conditions. Therefore, the IL-33 signaling pathway represents a significant link between asthma and CRS, potentially offering a molecular target for therapeutic intervention to ameliorate symptoms and modify disease progression in patients suffering from both conditions.
AR is a type of allergic upper airway disease primarily characterized by a Type I hypersensitivity reaction. Upon exposure to allergens, plasma cells release IgE antibodies. Concurrently, CD4 + T cells differentiate into allergen-specific Th2 cells. These Th2 cells then prolifically secrete Type 2 inflammatory cytokines such as IL-4, IL-5, and IL-13, which further stimulate B cells to differentiate into plasma cells that produce large amounts of IgE34. Severe asthma is often complicated by coexisting conditions such as AR and CRS. AR and asthma share common inflammatory pathways with CRS, which may contribute to increased airway inflammation in affected individuals35. A study found that patients with AR induced by house dust mites can gradually develop into CRS36. Our MR results have validated that patients with AR are more likely to develop CRS. The pathogenesis of airway diseases is complex and may not solely be driven by individual genetic mutations, suggesting that genetic susceptibility is just one of several contributing factors37. Epigenetic research indicates that AR and asthma share genetic and environmental etiologies, supporting the hypothesis of “one airway, one disease”38. Our findings further substantiate this theory, showing that individuals with AR and asthma have a higher propensity to develop CRS, likely due to shared pathogenic factors. These include disruptions in epithelial barrier functions and accumulations of Th2 cytokines and eosinophils following allergen exposure in the airways39,40,41. Furthermore, AR is characterized by an inflammatory response of the nasal mucosa, which often extends to the adjacent sinus regions, predisposing individuals to CRS. The inflammation associated with AR can lead to obstruction of the sinus openings, impeding normal sinus ventilation and drainage and fostering conditions conducive to bacterial infection and persistent inflammation42. The heightened immune response in individuals with AR results in the release of a plethora of inflammatory mediators, such as histamines and leukotrienes, exacerbating both nasal and sinus inflammation8. However, while our study strengthens the link between AR and asthma with CRS, we did not find evidence to suggest that CRS increases the risk of developing AR or asthma. This aspect of the relationship may require further investigation through more extensive clinical studies and higher-quality GWAS datasets to explore these relationship thoroughly.
Autoimmune diseases are characterized by pathological conditions in which the immune system erroneously targets the body’s own cells as foreign invaders. This misdirected immune response can lead to inflammation and damage to various tissues and organs5. Autoimmune diseases are characterized by their diversity; they can affect nearly any part of the body, including the skin, joints, blood vessels, brain, and internal organs. Psoriasis is a chronic autoimmune skin condition marked by accelerated skin cell turnover, leading to thick, scaly patches that often cause discomfort and itchiness43. The onset of psoriasis typically occurs between the ages of 15 and 25, although it can develop at any age, and impacts both genders equally44,45. The pathogenesis of psoriasis involves a complex interplay of genetic predisposition, immune system dysregulation, and environmental factors. Certain genetic markers, particularly specific HLA alleles, significantly increase susceptibility to psoriasis. The immune system plays a critical role, with T cells, including Th1 and Th17 cells, and dendritic cells driving the condition by overproducing inflammatory cytokines like IL-17, IL-22, and IL-2346. These cytokines disrupt the normal skin cell life cycle, resulting in rapid growth and the formation of the characteristic plaques47. Th1 and Th17 cytokines play a significant role in the pathophysiology of CRS, indicating potential causal links between psoriasis and CRS. However, while several small-scale clinical studies have reported that patients with CRS may have a higher susceptibility to developing psoriasis48,49, our findings suggest a different relationship. We identified psoriasis as a protective factor against CRS, which aligns with an observational study that proposed CRS and psoriasis exhibit mutually exclusive systemic Th1 and Th2 disease patterns, making their co-occurrence relatively rare50. Further supporting this notion, a prospective study showed that patients with CRS treated with dupilumab, an IL-4 and IL-13 pathway monoclonal antibody, exhibited increased Th17 cytokine expression, and three-quarters of these patients subsequently developed symptoms of psoriasis51. IL-17, a primary cytokine from the Th17 cells, is extensively involved in mucosal immunity and chronic inflammation52. Additionally, IL-17 has been shown to enhance the expression of chemokines in gut epithelial cells, bolstering antimicrobial responses and promoting epithelial barrier repair, thus supporting epithelial barrier integrity53. Given that CRS is a disease characterized by airway mucosal dysfunction, it appears that the elevated IL-17 levels in psoriasis may contribute to strengthening airway barriers, which could decrease the susceptibility to CRS in individuals with psoriasis. However, this hypothesis requires confirmation through further large-scale cohort studies to fully understand the dynamics and potential therapeutic implications of these findings in the context of autoimmune and inflammatory diseases. Additionally, we found suggestive evidence that hypothyroidism and T1D may act as risk factors for CRS. These associations could be explained through several immunological and physiological mechanisms which these conditions share with CRS. A study from India confirmed a correlation between hypothyroidism and nasal congestion54. Coincidentally, another recent study found that patient with hypothyroidism may impair the mucociliary clearance in the nasal passages and sinuses55. This impairment can result in a buildup of mucus, creating an environment conducive to bacterial infections and inflammation, common in CRS. Additionally, the altered hormonal levels in hypothyroidism could influence the expression of various cytokines and inflammatory responses, further exacerbating the risk and severity of CRS. T1D, characterized by autoimmune destruction of insulin-producing beta cells in the pancreas, involves complex immune dysregulations that can predispose individuals to various infections and inflammatory diseases56. A Korean cross-sectional study showed that diabetes is significantly associated with the development of CRS57. These results were similar to our research findings. Patient with T1D often experience alterations in immune cell function and heightened inflammatory responses, which can compromise the mucosal barriers of the respiratory tract. Furthermore, the glycosylation of mucosal surfaces in individuals with hyperglycemia can disrupt the normal function of cilia and mucosal defenses, making them more susceptible to sinus infections and prolonged inflammatory responses typical of CRS58. Additionally, the role of reactive oxygen species (ROS) activated by autoimmune responses, targeting glycolytic pathways, could also be a key factor contributing to the dysfunction of nasal mucosal epithelial cells, leading to CRS59,60.
In summary, this study has capitalized on the strengths of MR to explore the complex interplay between autoimmune and allergic diseases and CRS. By utilizing genetic variants as IVs, we have provided more precise insights into the causal relationships, free from the confounding and reverse causation often seen in traditional observational studies. However, our study is has some limitations. The reliance on existing GWAS datasets means our analysis is constrained by the quality and scope of these datasets, which may not fully capture the genetic diversity of the global population. Additionally, MR assumes that the chosen genetic variants are valid instruments that affect the outcome only through the exposure, an assumption that may not always hold true if pleiotropy exists. Further research, particularly involving larger and more diverse populations, and advanced genomic techniques, is needed to confirm our findings and expand upon them, potentially offering new therapeutic targets for CRS in the context of autoimmune and allergic predispositions.
Conclusion
In conclusion, our study has established the causal relationships between allergic and autoimmune diseases and CRS. We have demonstrated significant associations that AR, asthma increase the risk of developing CRS. Notably, We identified a shared genetic variant, rs3939286 in the IL-33 gene, between asthma and CRS, suggesting that targeting the IL-33 pathway may provide a therapeutic strategy for both diseases. These results also supports the concept of “one airway, one disease.” In addition, we found suggestive evidences of hypothyroidism and T1D increase the risk of CRS. Interestingly, our findings also suggest a protective effect of psoriasis against CRS, highlighting the complexity of immune interactions in these conditions. Our findings emphasize the importance of vigilant CRS management in patients with allergic or autoimmune diseases. Continued investigation is required to elucidate the pathophysiological roles of these disorders in CRS development.
Data availability
Data contains in the manuscript are provided within the article. Public data can be found here: (https://www.finngen.fi/en/access_results).
References
Stevens, W. W., Lee, R. J., Schleimer, R. P. & Cohen, N. A. Chronic rhinosinusitis pathogenesis. J. Allergy Clin. Immunol.136, 1442–1453 (2015).
Fokkens, W. J. et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology58, 1–464 (2020).
Tu, J. et al. Causal relationships of metabolites with allergic diseases: a trans-ethnic mendelian randomization study. Respir. Res.25, 94 (2024).
Parkin, J. & Cohen, B. An overview of the immune system. Lancet357, 1777–1789 .
Pisetsky, D. S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol.19, 509–524 (2023).
Miller, F. W. et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J. Autoimmun.39, 259–271 (2012).
Rose, N. R. Nih Names Dr. Noel R. Rose as Chair of Autoimmune Diseases Coordinating Committee (SAGE Publications, 2004).
Bernstein, J. A., Bernstein, J. S., Makol, R. & Ward, S. Allergic rhinitis: a review. JAMA331, 866–877 (2024).
Xu, X. et al. Advances in co-pathogenesis of the united airway diseases. Respir. Med.225, 107580 (2024).
Reddy, S. G. et al. Association of autoimmune disorders with chronic rhinosinusitis in adults. Am. J. Otolaryngol.45, 104177 (2024).
Hingorani, A. & Humphries, S. Nature’s randomised trials. Lancet366, 1906–1908 (2005).
Chen, Y. et al. Genetic insights into therapeutic targets for aortic aneurysms: a mendelian randomization study. EBioMedicine. 83, 104199 (2022).
Zheng, J. et al. Recent developments in mendelian randomization studies. Curr. Epidemiol. Rep.4, 330–345 (2017).
Smith, G. D. & Ebrahim, S. Mendelian randomization: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol.32, 1–22 (2003).
Mokry, L. E., Ahmad, O., Forgetta, V., Thanassoulis, G. & Richards, J. B. Mendelian randomisation applied to drug development in cardiovascular disease: a review. J. Med. Genet.52, 71–79 (2015).
Chen, Y., Sun, Y., Wang, L., Xu, K. & Wang, D. W. Genetic insights into associations of multisite chronic pain with common diseases and biomarkers using data from the UK Biobank. Br. J. Anaesth.132, 372–382 (2024).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R Stat. Soc. Ser. B Stat. Methodol.82, 1273–1300 (2020).
Varricchi, G., Brightling, C. E., Grainge, C., Lambrecht, B. N. & Chanez, P. Airway remodelling in asthma and the epithelium: on the edge of a new era. Eur. Respir. J.63 (2024).
Wenzel, S. E. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med.18, 716–725 (2012).
Fahy, J. V. Type 2 inflammation in asthma–present in most, absent in many. Nat. Rev. Immunol.15, 57–65 (2015).
Moore, W. C. et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J. Allergy Clin. Immunol.133, 1557–1563e1555 (2014).
Ricciardolo, F. L. M., Guida, G., Bertolini, F., Di Stefano, A. & Carriero, V. Phenotype overlap in the natural history of asthma. Eur. Respir. Rev.32 (2023).
Van Nevel, S., Declercq, J., Holtappels, G., Lambrecht, B. N. & Bachert, C. Granulocyte-colony stimulating factor: missing link for stratification of type 2-high and type 2-low chronic rhinosinusitis patients. J. Allergy Clin. Immunol.149, 1655–1665e1655 (2022).
Ryu, G. et al. Age-associated changes in chronic rhinosinusitis endotypes. Clin. Exp. Allergy50, 585–596 (2020).
Staikūniene, J., Vaitkus, S., Japertiene, L. M. & Ryskiene, S. Association of chronic rhinosinusitis with nasal polyps and asthma: clinical and radiological features, allergy and inflammation markers. Medicine (Kaunas)44, 257–265 (2008).
Podwysocka, M., Dąbrowska, K., Fendler, W., Pagacz, K. & Pietruszewska, W. Analysis of the impact of bronchial asthma and hypersensitivity to aspirin on the clinical course of chronic sinusitis with nasal polyps. Otolaryngol. Pol.73, 37–43 (2019).
Ramadan, H. H., Fornelli, R., Ortiz, A. O. & Rodman, S. Correlation of allergy and severity of sinus disease. Am. J. Rhinol.13, 345–347 (1999).
Nabavizadeh, S. H. et al. Epidemiology, Sociodemographic Factors and comorbidity for allergic Rhinitis, Asthma, and Rhinosinusitis among 15 to 65-year-old Iranian patients. Med. J. Islam. Repub. Iran36, 128 (2022).
Hong, H., Liao, S., Chen, F., Yang, Q. & Wang, D. Y. Role of IL-25, IL-33, and TSLP in triggering united airway diseases toward type 2 inflammation. Allergy75, 2794–2804 (2020).
Zhang, M. et al. Hypoxia induces the production of epithelial-derived cytokines in eosinophilic chronic rhinosinusitis with nasal polyps. Int. Immunopharmacol.121, 110559 (2023).
Shaw, J. L. et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am. J. Respir. Crit. Care Med.188, 432–439 (2013).
Kelsen, S. G. et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J. Allergy Clin. Immunol.148, 790–798 (2021).
England, E. et al. Tozorakimab (MEDI3506): an anti-IL-33 antibody that inhibits IL-33 signalling via ST2 and RAGE/EGFR to reduce inflammation and epithelial dysfunction. Sci. Rep.13, 9825 (2023).
Wise, S. K. et al. International consensus statement on allergy and rhinology: allergic rhinitis—2023. Int. Forum Allergy Rhinol. 13, 293–859 (2023).
Heffler, E. et al. The severe Asthma Network in Italy: findings and perspectives. J. Allergy Clin. Immunol. Pract.7, 1462–1468 (2019).
De Marchi, S., Cecchin, E., De Marchi, S. U., Iuri, F. & Sechi, L. A. Subendotyping of dermatophagoides pteronyssinus-induced rhinitis and its impact on respiratory comorbidities. J. Allergy Clin. Immunol. Pract.11, 922–929e922 (2023).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet.49, 1752–1757 (2017).
Grossman, J. One airway, one disease. Chest111, 11s–16s (1997).
Buysschaert, I. D. et al. Genetic evidence for a role of IL33 in nasal polyposis. Allergy65, 616–622 (2010).
Cho, S. H., Hamilos, D. L., Han, D. H. & Laidlaw, T. M. Phenotypes of chronic rhinosinusitis. J. Allergy Clin. Immunol. Pract.8, 1505–1511 (2020).
Schleimer, R. P. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu. Rev. Pathol.12, 331–357 (2017).
Alho, O. P. et al. Subjects with allergic rhinitis show signs of more severely impaired paranasal sinus functioning during viral colds than nonallergic subjects. Allergy58, 767–771 (2003).
Boehncke, W. H. & Schön, M. P. Psoriasis. Lancet386, 983–994 (2015).
Kamiya, K., Kishimoto, M., Sugai, J., Komine, M. & Ohtsuki, M. Risk factors for the development of psoriasis. Int. J. Mol. Sci.20 (2019).
Griffiths, C. E. & Barker, J. N. Pathogenesis and clinical features of psoriasis. Lancet370, 263–271 (2007).
Rendon, A. & Schäkel, K. Psoriasis pathogenesis and treatment. Int. J. Mol. Sci.20 (2019).
Sieminska, I., Pieniawska, M. & Grzywa, T. M. The immunology of psoriasis-current concepts in pathogenesis. Clin. Rev. Allergy Immunol.https://doi.org/10.1007/s12016-024-08991-7 (2024).
Choi, H. G. & Lee, H. J. Increased risk of psoriasis in patients with chronic rhinosinusitis without nasal polyps: a longitudinal follow-up study using Korean national sample cohort. Eur. Arch. Otorhinolaryngol.276, 3105–3111 (2019).
Son, D. S., Cho, M. S. & Kim, D. K. Chronic Rhinosinusitis and the increased incidence of atopic dermatitis. Am. J. Rhinol. Allergy36, 574–582 (2022).
Rashid, R. M., Miller, A., Scianna, J. M. & Stankiewicz, J. A. Chronic rhinosinusitis and psoriasis: do mutually exclusive systemic Th1 and Th2 disease patterns exist? Acta Otolaryngol.127, 780–783 (2007).
Chromy, D. et al. Dupilumab-induced skin-associated side effects in patients with chronic rhinosinusitis with nasal polyposis. J. Dermatol.50, 89–93 (2023).
Amatya, N., Garg, A. V. & Gaffen, S. L. IL-17 signaling: the Yin and the Yang. Trends Immunol.38, 310–322 (2017).
Lee, J. S. et al. Interleukin-23-Independent IL-17 production regulates intestinal epithelial permeability. Immunity43, 727–738 (2015).
U, P. S., Kuruwatti, A., Nasser, A. & Rhinitis, R. B. Nasal obstruction and hypothyroidism: a follow up study with pre-post treatment with levothyroxine. Indian J. Otolaryngol. Head Neck Surg.75, 1394–1398 (2023).
Kulekci Ozturk, S., Sakci, E. & Kavvasoglu, C. Rhinitis in patients with acquired hypothyroidism. Eur. Arch. Otorhinolaryngol.278, 87–92 (2021).
Long, D., Chen, Y., Wu, H., Zhao, M. & Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun.99, 1–14 (2019).
Nam, J. S. et al. Association between diabetes mellitus and chronic rhinosinusitis with nasal polyps: a population-based cross-sectional study. Clin. Otolaryngol.47, 167–173 (2022).
Cohen Atsmoni, S., Brener, A. & Roth, Y. Diabetes in the practice of otolaryngology. Diabetes Metab. Syndr.13, 1141–1150 (2019).
Kohanski, M. A., Tharakan, A., Lane, A. P. & Ramanathan, M. Jr. Bactericidal antibiotics promote reactive oxygen species formation and inflammation in human sinonasal epithelial cells. Int. Forum Allergy Rhinol.6, 191–200 (2016).
Chávez, M. D. & Tse, H. M. Targeting mitochondrial-derived reactive oxygen species in T cell-mediated autoimmune diseases. Front. Immunol.12, 703972 (2021).
Acknowledgements
We want to acknowledge all participants and researchers involved in the GWAS datasets incorporated in this study.
Funding
This study was supported by the National Natural Science Foundation of China (grant nos. 81860182, 82360219), Jiangxi Nutrition and Health Management Medical Research Institute Cultivation Project (2022-PYXM-05) and Central Funds Guiding the Local Science and Technology Development (20221ZDG020066).
Author information
Authors and Affiliations
Contributions
Conception: Junhao Tu. Interpretation or analysis of data: Junhao Tu, Zhiqiang Zhang, Fan jiang, and Jinyang Wen. Preparation of the manuscript: Junhao Tu. Revision for important intellectual content: Junhao Tu and Qing Luo. Supervision: Jing Ye.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This article incorporates human participants’ data obtained from earlier studies. In each corresponding original research, all participants provided their informed consent. The foundation of our study lies in the analysis of large-scale GWAS datasets, rather than individual-level data. Consequently, there was no requirement for ethical approval in this context.
Consent for publication
All authors have read the manuscript and have agreed to its publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tu, J., Zhang, Z., Jiang, F. et al. Causal relationships between allergic and autoimmune diseases with chronic rhinosinusitis. Sci Rep 14, 25406 (2024). https://doi.org/10.1038/s41598-024-77131-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77131-0
