
Abstract
Immunotherapy research focuses on reshaping the tumor microenvironment (TME) to enhance its antitumor immune responses, with an emphasis on understanding the impact of RNA methylation in triple-negative breast cancer (TNBC) TME regulation. This study explored the influence of various RNA methyltransferases on TME cells in TNBC and their correlation with prognosis and immunotherapy response. Using non-negative matrix factorization on single-cell RNA-sequencing data, distinct TME cell clusters were identified based on the expression of 30 RNA methyltransferases. Various analyses, including pseudotime, cell communication, transcription factor regulatory network, and gene enrichment, were conducted on these clusters. The roles of RNA methyltransferase-mediated TME clusters in prognosis and immunotherapy response were determined using TNBC bulk RNA-Seq data, and the findings were validated through immunofluorescence analysis of a tissue microarray comprising 87 samples. Spatial transcriptomic analysis further revealed the distribution of TME cell clusters. Different methyltransferase-mediated cell clusters exhibited unique metabolic, immune, transcriptional, and intercellular communication patterns. Survival analysis indicated prognostic significance in specific TME cell clusters, and immunofluorescence analysis confirmed the prognostic value of m6A_WTAP + CD8T + cells. In conclusion, our study illustrated the involvement of these cell subgroups in tumor growth and antitumor immunity modulation, providing insights into the enhancement of TNBC immunotherapy.
Similar content being viewed by others

Introduction
Breast cancer is the most common malignant tumor worldwide, with 2.3 million new cases reported in 20201. Triple-negative breast cancer (TNBC) is the subtype with the poorest prognosis, accounting for 15% of all cases2. Recent research on TNBC biomarkers and the tumor microenvironment (TME) has changed the treatment landscape3. Notably, high PD-L1 expression in TNBC indicates the importance of immune checkpoint inhibitors (ICIs). Clinical trials have shown improvements in pathologic complete response rates and progression-free survival in patients treated with ICIs and neoadjuvant chemotherapy4. However, the main challenge lies in improving the response of patients with TNBC to anti-PD-1/PD-L1 therapy and converting non-responders into responders. Therefore, understanding the landscape of the immune microenvironment in TNBC would be beneficial in exploring more effective therapeutic approaches.
RNA modification is a crucial aspect of epigenetics, affecting cellular biological processes such as RNA synthesis, transport, function, and metabolism5. Methylation modifications include N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), and 7-methylguanosine (m7G)6,7. RNA methylation is mostly catalyzed by methyltransferases (writers) and removed by demethylases (erasers). Partially modified RNAs are recognized by RNA-binding proteins (readers) and help regulate various biological functions8,9,10. N6-adenosine methylation is the predominant RNA modification in eukaryotes, affecting the transcription, maturation, localization, function, and metabolism of different RNAs11,12. m5C is the most abundant in eukaryotic tRNAs and rRNAs13,14 and plays a key role in stabilizing non-coding and coding RNAs and maintaining translational accuracy by regulating the structure and stability of tRNA15,16. m1A modifications share several regulatory factors in tRNAs, rRNAs, mRNAs, and long non-coding RNAs17. Moreover, m1A affects the structure and function of target RNA molecules by influencing RNA base pairing18. Guanosine methylation forms the 5′ cap structure on mature mRNAs; however, m7G modifications also occur in mRNAs, tRNAs, 18 S rRNAs, and precursor microRNAs19.
Breast cancer immunotherapy is highly dependent on the TME and immune regulation20, with TME remodeling being a key research topic. RNA modifications play a crucial role in regulating TME cell functions, with METTL14-deficient macrophages driving CD8 + T cell dysfunction and inhibiting CD8 + T effector cell activation21. N6-adenosine methylation of METTL3 maintains dendritic cell maturation and activation by mediating the N6-adenosine methylation of CD40, CD80, and Toll-like receptor signaling articulator (TIRAP)22. The m7G methyltransferase RNMT may also play a role in T cell activation by regulating ribosome synthesis23. METTL14 deficiency in B cells reduces N6-adenosine methylation and blocks interleukin-7-induced proliferation of pre-B cells24. However, few studies have investigated the effects of different RNA methyltransferases-associated cell subpopulations within the TME on tumor progression and antitumor immunity.
Therefore, in the present study, we aimed to investigate the effects of (i) different RNA methyltransferases on TME cells in TNBC and (ii) different RNA methyltransferase-mediated TME cell clusters on the prognosis and response to immunotherapy in patients with TNBC. This study will contribute to a better understanding of the regulatory role of RNA methylation modifications in the TME and help improve the response to immunotherapy in patients with TNBC.
Methods
Data collection
Single-cell RNA sequence (scRNA-Seq) data from TNBC samples (CID4495, CID44971, CID4515) in GSE176078 (GEO database) were used to analyze 30 RNA methylation enzymes. Cell annotations and raw data were obtained from the original study25. For survival analyses, bulk TNBC transcriptome data were obtained from The Cancer Genome Atlas (TCGA), the METABRIC database, and GEO databases (GSE21653, GSE31519, GSE58812, and GSE135565). The breast cancer immunotherapy transcriptome dataset (GSE173839) was used for immune response analysis. Breast cancer MeRIP-Seq data from GSE217977 were used to compare the number of methylation peaks between the cancer and para-cancer samples. Spatial transcriptome (ST) data were acquired from https://zenodo.org/record/4739739, and annotations of the ST regions were obtained from the original study25. We used a tissue microarray comprising 87 patients with TNBC treated at the Quanzhou First Hospital of Fujian Medical University from January 2013 to April 2020. Detailed clinical data of each patient, including histological tumor grade and clinical stage (defined by the American Joint Committee on Cancer, 8th edition) were collected. Supplementary Table S1 (online) presents follow-up information on recurrence and survival, extracted from electronic medical records. The study was performed under the tenets of the Declaration of Helsinki and was approved by the Medical Ethics Committee of Quanzhou First Hospital, Fujian Medical University (No. 2023-K112). All recruited patients signed informed consent forms.
Single-cell RNA-Seq data processing
The Seurat software package in R (4.1.1) was used to generate Seurat objects for all cells. The top 2,000 variable features were selected, and Seurat’s FindVariableFeatures function was applied to normalize the scRNA-Seq data. Next, we ran the ScaleData and RunPCA functions on the Seurat objects to calculate the number of principal components. The 15 principal components were condensed using uniform manifold approximation and projection (UMAP) dimensionality reduction. TNBC TME cells were annotated and visualized using DimPlot functions, with the incorporation of annotations from a previous study25.
Pseudotime trajectory analysis of RNA methyltransferase in TME cells
The Monocle package in R26 was used to investigate the relationship between RNA methyltransferases and the cellular pseudotime trajectories of TME cell types. Highly variable genes were identified based on the following criteria: mean expression ≥ 0.1 and dispersion_empirical ≥ 1 * dispersion_fit. The DDRTree method was implemented for dimensionality reduction. The plot_pseudotime_heatmap function was used to generate heatmaps to visualize the dynamic expression patterns of different RNA methyltransferases over the pseudotime trajectories of the different TME cell types.
Marker gene identification of different RNA methyltransferase-associated subtypes in TME cells
Thirty unique RNA methyltransferases were obtained from previous studies to investigate their impact on different TME cell types (Supplementary Table S2, online)7. A non-negative matrix factorization (NMF) algorithm, which utilizes a rank value of 10 and the snmf/r method, was employed to identify different cell subtypes based on the RNA methyltransferase expression matrix of all TME cells. The FindAllMarkers function in the Seurat package was used to list marker genes for NMF clusters for each cell type. Cell clusters with RNA methyltransferase logfc threshold > 1 were identified as positively expressing RNA methyltransferase, while unclear clusters with a threshold < 1 were considered unmethylated. The AddModuleScore function was used to calculate scores based on NMF cluster marker genes or cellular subpopulation markers, as well as functional pathway genes from existing studies, and FeaturePlot or DotPlot functions were used to display these scores. The special gene set used for comparison between related clusters27,28,29,30,31,32 is listed online in Supplementary Table S2. The “pheatmap” package was used to display the scaled data of the expression of these genes in various RNA methyltransferase-associated cell clusters.
Cell-cell communication analysis for RNA methyltransferase-associated subtypes
Cell-cell interactions between RNA methyltransferase-associated subtypes were inferred using the CellChat R package33. CellChatDB.human was used to analyze signaling inputs and outputs in RNA methyltransferase-associated cell clusters, and netAnalysis_signalingRole_heatmap was subsequently employed to display differences in outgoing or incoming signals, Moreover, netVisual_bubble was used to display bubble maps of ligand-receptor interactions of RNA methyltransferase-associated clusters.
Single-cell regulatory network inference and clustering (SCENIC) analysis of RNA methyltransferase-associated subtypes
Using the SCENIC R package, the gene regulatory network of transcription factors (TFs) within the RNA methyltransferase-associated isoforms of different TME cell types was investigated34. Transcription start sites and gene regulatory networks were identified in various RNA methyltransferase-associated cell clusters using the gene signature ranking (hg19-tss-centered-10 kbp) from the RcisTarget database. TFs with an adjusted Benjamini–Hochberg false discovery rate (BH-FDR) below 0.05 were selected for analysis.
Functional enrichment analysis of RNA methyltransferase-associated subtypes
Kyoto Gene and Genome Encyclopedia (KEGG) functional enrichment analysis was conducted using the ClusterProfiler R package with marker genes identified in distinct RNA methyltransferase-associated clusters for each TME cell type. Gene sets with p < 0.05 were considered significantly enriched. The scores of different metabolic pathways in various RNA methyltransferase-associated clusters were assessed using the “scMetabolism” R package35.
Deconvolution of scRNA-Seq data for RNA methyltransferase-associated subtypes
The expression matrix of RNA methyltransferase-related cell clusters from the scRNA-Seq data was obtained, and a CIBERSORTx feature matrix was created using the cell types from that data using the “Create Signature Matrix” module (https://cibersortx.stanford.edu/runcibersortx.php)36. The generated feature matrix was used for the CIBERSORTx deconvolution of multiple bulk RNA-Seq cohorts. The inverse convolution scores were used to measure the number of RNA methyltransferase-associated cell clusters in each tumor sample in the cohort.
Analysis of RNA methyltransferase-associated-subtype signatures in public bulk RNASeq datasets
Cox proportional hazard regression was used to investigate the association between RNA methyltransferase-associated subtypes and overall patient survival using survival data obtained from the primary database. The TIDE website (http://tide.dfci.harvard.edu/)37 was used to predict ICI treatment responses in these samples. The dataset GSE173839 from the GEO database, which provides information on breast cancer immunotherapy response, was also used.
Immunofluorescence analysis of RNA methyltransferase-associated cell subtypes
Deparaffinized and antigenically repaired paraffin-embedded TNBC sections were blocked with 5% bovine serum albumin for 30 min. The sections were then incubated overnight at 4 °C with primary antibodies against markers of different RNA methyltransferase-associated cell subtypes and with antibodies diluted in primary antibody dilution buffer (G2025; Servicebio, China). Subsequently, the tissues were washed three times with PBS and incubated with the corresponding secondary antibodies for 50 min at 25 °C. The antibodies were diluted in a secondary antibody dilution buffer (G2009; Servicebio), and the horseradish peroxidase-labeled secondary antibodies were combined with the corresponding Tyramine Signal Amplification dye. The antibodies and dilutions used in this study are listed online in Supplementary Table S3. Subsequently, 4′,6-diamidino-2-phenylindole staining was conducted for 10 min at 25 °C. Fluorescence images were captured using a Nikon ECLIPSE C1 fluorescence microscope.
ST analysis of RNA methyltransferase-associated cell subtypes
The Seurat package was used to normalize and SCT-normalize the ST data and to cluster similar ST points using principal component analysis. To visualize the spatial regional distribution of the ST data, SpatialDimPlot was used. The RCTD package was used to deconvolute RNA methyltransferase-associated cell subtypes at each ST point, allowing us to evaluate the infiltration abundance of different RNA methyltransferase-associated cell subtypes at these points. The scoring of the gene set “h.all.v2022.1.Hs.symbols.gmt” was assessed using the MSigDB database in the tumor region using the ssGSEA algorithm. These scores were then compared with SpatialFeaturePlot to display the levels of cell abundance and gene set scoring in the ST data.
Statistical analysis
Various standard statistical tests were performed. Kruskal-Wallis test was used for comparison of more than two groups of data and Wilcoxon test was used for comparison of two groups of data. The chi-square test was used to compare ratios between different subgroups. All statistical analyses were performed using R version 4.1.1; results with two-sided p < 0.05 were considered statistically significant.
Results
Overview of the RNA methyltransferase pattern of TME cells in TNBC
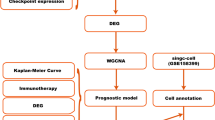
A flowchart of the study is presented in Fig. 1. Initially, we analyzed the differences in the number of methylation peaks between the cancer and paraneoplastic samples in GSE217977. This analysis revealed a high degree of hypermethylation in breast cancer tissues (Supplementary Figure S1 online). Subsequently, we evaluated the mean RNA expression of RNA methyltransferases using the TCGA–TNBC dataset to investigate differential expression in breast cancer samples based on category type (normal vs. tumor), age group (age > 40 vs. young ≤ 40), clinical stage (I, II, III, and IV), and TNM stage (Fig. 2a). We then used the scRNA-Seq dataset of breast cancer to explore the total RNA methyltransferase expression in TME cells. The UMAP and bar plots depicted the clustering of distinct TME cells in TNBC (Fig. 2b). These cell clusters were categorized into 12 groups based on the annotation results of the raw data. The scRNA-Seq dataset revealed distinct expression patterns of various RNA methyltransferases across these 12 breast cancer cell types (Fig. 2c). TME cells exhibited higher expression levels of m6A and m7G methyltransferases than m5C and m1A methyltransferases.

Schematic illustration of this study.

RNA methyltransferase patterns of tumor microenvironment (TME) cells in triple-negative breast cancer (TNBC). (a) The average expression of 30 RNA methyltransferases for the four major RNA methylation types in the TCGA–TNBC dataset was calculated using z-scores according to several clinical variables. (b) UMAP plot of all TME cells of TNBC samples with annotation based on the original dataset. (c) Expression pattern of different RNA methylation transferases in different TME cell types visualized using a heatmap.
Effects of different RNA methyltransferase-associated fibroblasts on the TME
We observed differential expression of various RNA methyltransferases at distinct stages of the developmental trajectory of fibroblasts, as determined via pseudotime analysis (Fig. 3a). We classified cancer-associated fibroblasts (CAFs) were classified into eight clusters through NMF clustering based on the expression of different RNA methyltransferases in fibroblasts (Fig. 3b). Notably, we observed variations in the percentage of CAFs associated with different RNA methyltransferases (Supplementary Figure S2 online). Cell communication analysis revealed varying strengths in the output of cellular signals among different RNA methyltransferase-associated fibroblasts. m5C_NSUN5 + CAF exhibited a stronger secretion intensity of periostin cytokines (Fig. 3c). Moreover, SCENIC analysis indicated diverse patterns of TF regulation among the different RNA methyltransferase-associated fibroblast subtypes (Supplementary Figure S3 online). The expression level analysis of multiple gene sets associated with fibroblast pro-tumorigenic functions revealed substantial differences in functional gene expression within the different RNA methyltransferase-associated fibroblast subtypes (Fig. 3d). Moreover, m5C_NSUN5 + CAF fibroblasts displayed a significantly higher expression of pro-tumorigenic functional genes, as evidenced by functional scores of pathway genes and feature scores of RNA methyltransferase-associated fibroblasts. These scores and subtypes are presented on a UMAP plot in Fig. 3e, showing that CAFs characterized as m5C_NSUN5 + CAF exhibited an enhanced capacity to secrete extracellular matrix (ECM), transforming growth factor β (TGF-β), and matrix metalloproteinases (MMPs); they more closely resembled myofibroblasts (myCAF). The KEGG pathway enrichment analysis highlighted that m5C_NSUN5 + CAF exhibited functions related to adhesion junctions and regulation of the actin cytoskeleton (Fig. 3f).

Effects of different RNA methyltransferases on fibroblasts. (a) Pseudotime trajectory reveals expression patterns of different RNA methyltransferases in CAFs. (b) Markers of different RNA methyltransferase-associated cancer-associated fibroblasts (CAFs) clustered via negative matrix factorization. (c) Cell communication signaling patterns of different RNA methyltransferase-associated CAFs. (d) Different average expression levels of collagens, extracellular matrix (ECM), MMP, matrix metalloprotease (MMPs), tumor growth factor-beta (TGF-β), angiogenic, RAS, and proinflammatory pathway genes in RNA methyltransferase-associated CAFs. (e) UMAP plot for visualizing the feature and function scores as well as the subtype of the CAFs. (f) Activated Kyoto Encyclopedia of Genes and Genomes pathways among different RNA methyltransferase-associated CAF clusters.
Metabolic patterns of tumor-associated macrophages mediated by different RNA methyltransferases
RNA methyltransferase expression levels varied according to the different stages of macrophage development (Supplementary Figure S4 online). Macrophages were categorized into eight groups based on RNA methyltransferase expression levels (Fig. 4a). Similarly, different RNA methyltransferase-associated macrophages accounted for varying proportions (see Supplementary Figure S2 online). In the cell communication analysis, m5C_NSUN5 + Macrophage exhibited strong output signals (Fig. 4b). SCENIC analysis revealed differential activation of potential TFs in different RNA methyltransferase-associated macrophage clusters (Supplementary Figure S3 online). Metabolic pathway analysis revealed variations in the scores of metabolic pathways among different RNA methyltransferase-associated macrophages. The m5C_NSUN5 + Macrophage exhibited improved scores in glucose metabolism pathways, such as glycolysis, gluconeogenesis, and oxidative phosphorylation, as well as in the tricarboxylic acid cycle, the metabolic pathway of fatty acids, and several amino acids (Fig. 4c). Moreover, KEGG enrichment analysis underscored functional distinctions among different RNA methyltransferase-associated macrophages (Fig. 4d). We further analyzed the proportion of lipid-associated macrophages (LAM) within different RNA methyltransferase-associated macrophages in breast cancer, revealing hat LAM constituted a substantial portion of m5C_NSUN5 + Macrophage and m6A_METTL5 + Macrophage (Fig. 4e).

Effects of different RNA methyltransferases on macrophages. (a) Markers of different RNA methyltransferase-associated macrophages clustered by negative matrix factorization. (b) Cell communication signaling patterns of different RNA methyltransferase-associated macrophages. (c) Metabolic activities of different RNA-methyltransferase-associated macrophage clusters. (d) Activated Kyoto Encyclopedia of Genes and Genomes pathways among different RNA methyltransferase-associated macrophage clusters. (e) The proportion of lipid-associated macrophages (LAMs) in different RNA methyltransferase-associated macrophages.
Antigen-presenting abilities mediated by different RNA methyltransferases in DCs
Various RNA methyltransferases displayed differential expression patterns during different developmental stages of DCs, akin to fibroblasts and macrophages (Supplementary Figure S4 online). Using NMF clustering based on the expression of distinct RNA methyltransferases, we categorized DCs into four clusters (Fig. 5a) with different percentages (see Supplementary Figure S2 online). The network analysis further indicated differential activation of TFs among these distinct RNA methyltransferase-associated DC clusters (Supplementary Figure S3 online). Cell communication analysis of RNA methyltransferase-associated DCs revealed that m6A_WTAP + DC exhibited a stronger capacity, resulting in more ligand-receptor linkages between m6A_WTAP + DC and CD8 + T cells (Fig. 5b, c). In addition, the KEGG pathway enrichment analysis suggested that m6A_WTAP + DC was significantly enriched in antigen presentation functions (Fig. 5d). We used heatmaps to display the expression of various major histocompatibility complex (MHC) molecules in various RNA methyltransferase-mediated DCs for a comprehensive view. Notably, the expression of multiple MHC receptors was higher in m6A_WTAP + DC than in the other clusters (Fig. 5e). The AddModuleScore scoring approach, based on DC classification markers from previous studies, demonstrated that m6A_WTAP + DC was associated with classical dendritic cells (Fig. 5f).

Effects of different RNA methyltransferases on dendritic cells (DCs). (a) Markers of different RNA methyltransferase-associated DCs clustered by negative matrix factorization. (b) Cell communication signaling patterns of different RNA methyltransferase-associated DCs. (c) Ligand-receptor linkages between CD8 + T cells and RNA methyltransferase-associated DCs. (d) Activated Kyoto Encyclopedia of Genes and Genomes pathways among different RNA methyltransferase-associated DCs. (e) Average expression of major histocompatibility complex (MHC) molecules genes in RNA methyltransferase-associated DCs. (f) DC subtype feature scores of different RNA methyltransferase-associated DCs.
Varied antitumor immunities in different RNA methyltransferase-associated CD8 + T cells and B cells
The pseudotime analysis revealed that various RNA methyltransferases exhibited differential expression patterns at distinct stages of the developmental trajectories of CD8 + T and B cells (Supplementary Figure S4 online). Using NMF clustering, we classified CD8 + T and B cells into six and five clusters, respectively (Fig. 6a, b). The network regulation analysis indicated significant variations in the RNA methyltransferase-mediated regulation of TFs between the CD8 + T cell and B cell clusters (Supplementary Figure S3 online). However, the cell communication analysis revealed no significant differences in the signal intensity of cell communication between RNA methyltransferase-mediated CD8 + T and B cells (Supplementary Figure S5 online). In the assessment of the average expression of co-stimulatory and co-inhibitory receptors on the surface of CD8 + T cells, m6A_WTAP + CD8 + T cells displayed relatively strong expression of multiple co-stimulatory receptors. In contrast, m7G_TRMT112 + CD8 + T cells exhibited relatively strong expression of co-inhibitory receptors and relatively weak expression of co-stimulatory receptors (Fig. 6c).

Effects of different RNA methyltransferases on CD8 + T cells and B cells. (a,b) Markers of different RNA methyltransferase-associated CD8 + T cells and B cells clustered via negative matrix factorization. (c) Average expression of immune checkpoints in RNA methyltransferase-associated CD8 + T cells. (d) Score of the exhaustion stage in different RNA methyltransferase-associated CD8 + T cells. (e) Average expression of immune checkpoints in RNA methyltransferase-associated B cells. (f) Average expression of major histocompatibility complex (MHC) molecules genes in RNA methyltransferase-associated B cells. (g) B cell subtype feature score of different RNA methyltransferase-associated B cells.
In addition, we used markers from previous studies to score different RNA methyltransferase-mediated CD8 + T cells, revealing that m6A_WTAP + CD8 + T cells were predominantly in the early stages of exhaustion, whereas m7G_TRMT112 + CD8 + T cells were mainly in the advanced stages of cell exhaustion (Fig. 6d). Similarly, an examination of the average expression of co-stimulatory and co-inhibitory receptors on the surface of B cells suggested that several co-stimulatory receptors were expressed at higher levels in m7G_RNMT + B cells (Fig. 6e). Further analysis of the expression of MHC receptors on the surface of B cells revealed higher MHC receptor expression on m7G_WBSCR22 + B cells (Fig. 6f). Finally, a comprehensive evaluation of the levels of different B cell subtype markers in different RNA methyltransferase-mediated B cells suggested that m7G_RNMT + B cells were associated with memory B cells, whereas m7G_WBSCR22 + B cells were associated with activated B cells (Fig. 6g). Notably, m6A_WTAP + CD8 + T cells and m7G_RNMT + B cells, as well as m7G_WBSCR22 + B cells, constituted relatively low percentages of their respective cell types within the tumor microenvironment (Supplementary Figure S2 online).
Effect of RNA methyltransferase-associated TME patterns on breast cancer prognosis and immunotherapy
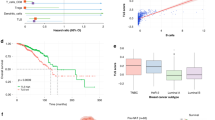
To characterize the RNA methyltransferase-associated TME cell types, we generated a signature matrix of different cell subtypes using CIBERSORTx, acquiring the cell abundance data from a number of different datasets. Initially, we examined the relationship between different cell types and prognosis in patients with TNBC using univariate Cox regression analyses (Fig. 7a). m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage were associated with poorer patient prognosis across multiple datasets. In contrast, m6A_WTAP + DC, m6A_WTAP + CD8 + T cells, m7G_WBSCR22 + B cells, and m7G_RNMT + B cells positively affected patient prognosis. We further compared the abundance of different RNA transferase-associated cell subpopulations in TNBC and normal breast samples (Supplementary Figure S6, online). We observed that in TNBC, m5C_NSUN5 + CAF and m5C_NSUN5 + macrophages exhibited increased infiltration, whereas m6A_WTAP + CD8 + T cells and m7G_RNMT + B cells showed decreased infiltration. This is consistent with the trend observed in our prognostic analysis.

Prognostic value and immunotherapy response prediction value of RNA methyltransferase-associated tumor microenvironment (TME) cells. (a) Overall survival analysis of RNA methyltransferase-associated TME cells. (b) Immunotherapy response analysis of RNA methyltransferase-associated TME cells. (c–e) Immunofluorescence staining for m6A_WTAP + CD8 + T cells in TNBC TMAs. Scale bars: left, 200 μm; right, 40 μm. (f) Comparison of m6A_WTAP + CD8 + T cell counts at different BC stages. For cell count calculation, three pictures were selected from each TNBC section. (g) Kaplan–Meier survival curves displaying the differences in DFS rates between patients classified based on the m6A_WTAP + CD8 + T cell count.
Furthermore, we used logistic regression to assess the significance of RNA methyltransferase-associated TME cell types in predicting the response to immunotherapy in patients with BC (Fig. 7b). m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage negatively affected immunotherapy response, whereas m6A_WTAP + DC, m6A_WTAP + CD8 + T cells, m7G_WBSCR22 + B cells, and m7G_RNMT + B cells were associated with a positive response.
Given the consistent trend in most of the datasets, we further investigated the abundance of m6A_WTAP + CD8 + T cells in different patients using immunofluorescence analysis of tissue microarrays; this revealed a decreasing trend of m6A_WTAP + CD8 + T cell infiltrates with a higher clinical stage (Fig. 7c-f). We categorized patients with TNBC into high and low infiltration groups based on the median number of m6A_WTAP + CD8 + T cells. Kaplan–Meier survival analyses and the log-rank test demonstrated that a higher infiltration of m6A_WTAP + CD8 + T cells was associated with extended disease-free survival (DFS) in patients (P = 0.019; Fig. 7g). Immunofluorescence experiments confirmed these RNA methyltransferase-associated cell subtypes in TNBC (Supplementary Figure S7, online).
Distribution features of RNA methyltransferase-mediated cellular subtypes in the ST
We utilized ST data to classify regions of RNA methyltransferase-associated TME cells in TNBC tissues. These classifications, namely “Invasive cancer + stroma + lymphocytes,” “Lymphocytes,” “Necrosis,” “Stroma,” and “TLS,” provided insights into their spatial distribution (Fig. 8a). The deconvolution algorithm yielded cell abundance data, revealing that m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage were predominantly localized in the tumor and mesenchymal zones. In contrast, m6A_WTAP + DC, m6A_WTAP + CD8 + T cells, m7G_WBSCR22 + B cells, and m7G_RNMT + B cells were primarily concentrated in the lymphocyte and tertiary lymphoid structure (TLS) regions (Fig. 8b).

Spatial transcriptome features of RNA methyltransferase-mediated cellular subtypes. (a) Spatial images of the tissue architecture of triple-negative breast cancer (TNBC). (b) The abundance of RNA-methyltransferase-mediated cellular subtypes in spatial organization. (c) Single-sample gene set enrichment analysis (ssGSEA) scores of tumor-associated pathways in the tumor zones.
We conducted a single-sample gene set enrichment analysis (ssGSEA) to assess tumor-associated pathways in the tumor zones, aiming to explore the spatial relationship between these prognostically relevant RNA methyltransferase-associated TME cells and tumor-associated pathways (Fig. 8c). This analysis indicated that tumor tissues surrounding highly infiltrated m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage regions exhibited increased hypoxia and glycolysis, the upregulation of several pathways linked to tumor proliferation (the PI3K-AKT and WNT pathways), and enhanced angiogenesis. In contrast, areas surrounding tumor tissues rich in m6A_WTAP + DC and m6A_WTAP + CD8 + T cells displayed relatively lower pathway scores.
Discussion
RNA methylation modifications play a crucial role in tumorigenesis6,19,38,39; however, their effects on the tumor immune microenvironment at the single-cell level are limited. The presnt study explored the expression patterns of RNA methylation regulators in major TNBC TME cell types, revealing their biological functions and cell-cell interactions. Our findings confirmed that different TME cell subtypes have different implications for TNBC prognosis and immunotherapy efficacy. This new perspective offers insights into how the modification of TME components by different RNA methyltransferases affects patient survival and treatment outcomes.
The TME, comprising various cell types such as stromal, vascular endothelial, and immune cells, significantly influences tumor development. This was also evident in our comparative analysis of stromal cells (CAF and PVL), endothelial cells, macrophages, and breast cancer epithelium in the TNBC TME. Research has highlighted the association of CAFs and macrophages with breast cancer invasion and metastasis25,40,41,42, whereas CD8 + T cells, B cells, and DCs contribute to antitumor immunity43,44,45. Notably, distinct subgroups exist within these TME cell clusters, playing unique roles in tumor immunity. Recent studies have emphasized the critical role of RNA methylation in orchestrating and reprogramming cells within the TME46,47,48,49. Similarly, the present study revealed diverse RNA methylation patterns in different TME cell types, with varying expression patterns of RNA methyltransferases along TME cell developmental trajectories, emphasizing the importance of investigating the effects of these modifications on TME cell function and antitumor immunity.
CAFs are crucial stromal cell components involved in tumor growth. They facilitate invasion by modulating ECM structures and engaging with tumor cells through the secretion of growth factors, cytokines, and chemokines41. However, few studies have explored the effects of diverse RNA methylation modifications on CAF phenotypes. The result of the present study underscore the tumor-promoting effect of m5C_NSUN5 + CAF. The cell communication analysis revealed the enhanced capacity of m5C_NSUN5 + CAF to export periostin signals, previously linked to the disruption of the lymphatic endothelial barrier and promotion of tumor lymphatic metastasis50. A gene set enrichment analysis revealed upregulated expression of MMPs and TGF-β in m5C_NSUN5 + CAF, indicative of ECM remodeling, a characteristic closely associated with the suppression of antitumor immune responses and promotion of tumor invasion51. Within the CAF subtype, myCAF is named owing to its high expression of α-smooth muscle actin and its contractile and secretory properties52. In the present study, a comparison of the fibroblast subtypes revealed that m5C_NSUN5 + CAF may be more similar to myCAF. Moreover, KEGG analysis revealed enrichment in the function of “regulation of actin cytoskeleton,” further suggesting that NSUN5 + CAF might resemble myCAFs. Notably, a higher abundance of m5C_NSUN5 + CAF correlates with a poorer prognosis in patients with TNBC who are non-responsive to immunotherapy. Therefore, high NSUN5 expression in CAFs could be a potential intervention target.
Tumor-associated macrophages (TAMs) represent a major component of immune cell infiltration in breast cancer, exhibiting substantial heterogeneity. These recruited macrophages can undergo reprogramming, shifting from anticancer to pro-cancer functionality in response to the TME42. Recent studies have revealed the critical role of RNA modifications in macrophage activation53. However, the modulation of RNA modification in TAMs within the TNBC microenvironment remains unexplored, as TAMs frequently undergo metabolic reprogramming in the TME54. The present study revealed that m5C_NSUN5 + Macrophages exhibit enhanced oxidative phosphorylation and glycolysis, which can induce a metabolic shift toward glycolysis. This shift leads to increased downstream lactate and TNF levels, ultimately promoting tumor progression55. Concurrently, TAMs reduce glucose consumption from tissues through oxidative phosphorylation, enhancing glucose availability in a hypoxic TME56. Enhanced fatty acid oxidation, methionine metabolism, and arginine metabolism in TAMs contribute to antitumor immunity suppression54. In the present study, these enhanced metabolic activities were also observed in m5C_NSUN5 + Macrophages. Notably, KEGG analysis revealed an enriched phagosome pathway in m5C_NSUN5 + Macrophages. Moreover, by reviewing previous studies, we found that phagosome maturation is closely related to antigen presentation. Immunosuppressed (M2) macrophages exhibit accelerated phagosome maturation and accelerated acidification of phagosomes, which inhibits phagosomal antigen cross-presentation57. This partly explains why m5C_NSUN5 + Macrophages might promote tumor development. In addition, enhanced glycolysis accelerates phagosomal acidification58, further suggesting that shifts in glucose metabolism in TAMs promote tumor function. In breast cancer, a subset of LAMs expressing immunosuppressive molecules, such as PD-L1 and PD-L2, is often present. LAMs promote breast cancer progression and are associated with a poor patient prognosis25,59. In the present study, we noted a high percentage of LAMs in m5C_NSUN5 + Macrophages, which are associated with a poor prognosis and immunotherapy resistance in patients with TNBC. Therefore, targeting these m5C_NSUN5 + Macrophages could improve the prognosis of patients with TNBC.
CD8 + T cells are potent effector cells in antitumor immune responses. However, intratumor CD8 + T cells often exhibit dysfunctional or exhausted phenotypes60. Beltra et al. delineated four phases of CD8 + T cell exhaustion29, where CD8 + T cell effector intensity progressively diminishes as depletion progresses, rendering them unresponsive to PD-L1/PD-1 blockade at the terminal exhaustion stage. The present study revealed that CD8 + T cells with high WTAP expression exhibit a pre-depletion stage, suggesting that these cells have not yet entered the depletion phase. In addition, immune checkpoint analysis demonstrated lower expression of immune co-inhibitory receptors and higher expression of immune co-stimulatory receptors in m6A_WTAP + CD8 + T cells, indicating robust antitumor immune activity. However, the lower proportions of m6A_WTAP + CD8 + T cells in the TME suggest that their antitumor effects may be diminished in TNBC. In addition, the bulk RNA-seq analysis revealed that greater m6A_WTAP + CD8 + T cell infiltration is associated with improved patient prognosis and a higher likelihood of responding to immunotherapy. Therefore, upregulating WTAP expression may inhibit depletion and enhance tumor-killing capacity.
The present study revealed that RNA modifications affect the antitumor immune functions of DCs and B cells in the TNBC TME. DCs play a crucial role in antitumor immunity, particularly in antigen presentation, which is essential in cancer therapies. DCs with high expression of m6A methyltransferase WTAP displayed upregulated MHC molecules and enhanced antigen presentation activity, and cell communication analysis revealed higher ligand-receptor interactions. Moreover, previous research underscores the necessity of DC-secreted chemokines, CXCL9 and CXCL10, in recruiting CXCR3-expressing CD8 + T cells for tumor tissue infiltration61. According to survival and immunotherapy response analyses, enhancing WTAP expression in DC may improve antitumor immunity. In addition to humoral immunity, B cells can also act as antigen-presenting cells to promote T-cell activation and thus play an anti-tumour role. In early-stage HER2 + breast cancer, the B cell signature exhibits superior prognostic and predictive values compared to TIL abundance62. The present study revealed that B cells with high expression of m7G methylation transferases RNMT and WBSCR22 have higher levels of MHC molecules and immune co-stimulatory molecules. These molecules are linked to improved patient prognosis and a higher response rate to immunotherapy, potentially enhancing antitumor immune responses.
In this study, we validated the clinical relevance of RNA methyltransferase-associated TME cell subtypes using spatial transcriptomic data. We found that the spatial spots with high m5C_NSUN5+CAF infiltration were surrounded by almost no infiltration of m6A_WTAP + DC, m6A_WTAP + CD8 + T cells, m7G_WBSCR22 + B cells and m7G_RNMT + B cells, further validating the inhibitory effect of m5C_NSUN5+CAF on antitumour immunity. Interestingly, cell subpopulations associated with favorable prognosis and immunotherapy response were noted near the TLS region. TLS is linked to improved prognosis across various cancers and holds substantial predictive value in immune checkpoint blockade therapy63. Moreover, spatial spots with high m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage infiltration are surrounded by regions with higher tumor-promoting biological functions, including EMT, angiogenesis, and tumor proliferation pathways64. These observations underscore the tumor-promoting role of m5C_NSUN5 + CAF and m5C_NSUN5 + Macrophage.
This study has limitations. The low depth of scRNA-Seq may have led to reduced expression of certain RNA methyltransferases and increased null observations, potentially introducing bias. In addition, due to the lack of a comprehensive treatment cohort, the role of RNA methyltransferase-associated cell subpopulations in immunotherapy responses requires further verification.
Conclusion
Using single-cell sequencing data, we identified distinct RNA methylation modification-mediated cellular subtypes within the TME of TNBC. We elucidated the biological functions of these RNA-methyltransferase-associated TME cellular subtypes and their potential contributions to tumor growth and antitumor immunomodulation. Our tissue microarray-based immunofluorescence analysis validated the importance of m6A_WTAP + CD8 + T cells, offering a novel perspective in regards to predicting the prognosis of patients with TNBC and enhancing the response to immunotherapy.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- CAF:
-
Cancer associated fibroblasts
- DFS:
-
Disease free survival
- ECM:
-
Extracellular matrix
- ICI:
-
Immune checkpoint inhibitors
- KEGG:
-
Kyoto Gene and Genome Encyclopedia
- LAM:
-
Lipid-associated macrophages
- MHC:
-
Major histocompatibility complex
- MMP:
-
Matrix metalloproteinases
- NMF:
-
Negative matrix factorization
- PBS:
-
Phosphate-buffered saline
- ST:
-
Spatial transcriptome
- TCGA:
-
The Cancer Genome Atlas
- TF:
-
Transcription factors
- TIDE:
-
Tumor Immune Dysfunction and Exclusion
- TIL:
-
Tumor Infiltrating Lymphocytes
- TLS:
-
Tertiary lymphoid structure
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- UMAP:
-
Uniform manifold approximation and projection
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. Cancer J. Clin. 72, (2022).
Bianchini, G., De Angelis, C., Licata, L. & Gianni, L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat. Rev. Clin. Oncol. 19, (2022).
Nolan, E., Lindeman, G. J. & Visvader, J. E. Deciphering breast cancer: from biology to the clinic. Cell. (2023).
Schmid, P. et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N Engl. J. Med. 386, 556–567 (2022).
Shi, H., Chai, P., Jia, R. & Fan, X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol. Cancer. 19, 78 (2020).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer. 20, 303–322 (2020).
Cui, L. et al. RNA modifications: importance in immune cell biology and related diseases. Signal. Transduct. Target. Ther. 7, 334 (2022).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell. 74, 640–650 (2019).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell. Biol. 20, 608–624 (2019).
Zhang, Y. et al. DirectRMDB: a database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Res. 51, D106–D116 (2023).
Chen, Y., Lin, Y., Shu, Y., He, J. & Gao, W. Interaction between N6-methyladenosine (m6A) modification and noncoding RNAs in cancer. Mol. Cancer. 19 (2020).
Huang, H., Weng, H. & Chen, J. m6A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 37, 270–288 (2020).
Bohnsack, K. E., Höbartner, C. & Bohnsack, M. T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes. 10, (2019).
Ma, J. et al. m5C-Atlas: a comprehensive database for decoding and annotating the 5-methylcytosine (m5C) epitranscriptome. Nucleic Acids Res. 50, D196–D203 (2022).
Trixl, L. & Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip Rev. RNA. 10, e1510 (2019).
Shinoda, S. et al. Mammalian NSUN2 introduces 5-methylcytidines into mitochondrial tRNAs. Nucleic Acids Res. 47, 8734–8745 (2019).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 22, 119–131 (2021).
Safra, M. et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 551, 251–255 (2017).
Luo, Y. et al. The potential role of N7-methylguanosine (m7G) in cancer. J. Hematol. Oncol. 15, (2022).
Adams, S. et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 5, 1205–1214 (2019).
Dong, L. et al. The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8 + T cell dysfunction and tumor growth. Cancer Cell. 39, (2021).
Wang, H. et al. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 10, 1898 (2019).
Galloway, A. et al. Upregulation of RNA cap methyltransferase RNMT drives ribosome biogenesis during T cell activation. Nucleic Acids Res. 49, 6722–6738 (2021).
Zheng, Z. et al. Control of early B cell development by the RNA N6-methyladenosine methylation. Cell. Rep. 31, 107819 (2020).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 14, 979–982 (2017).
Mahoney, K. M., Rennert, P. D. & Freeman, G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14, 561–584 (2015).
Gao, Y. et al. Single-cell N-methyladenosine regulator patterns guide intercellular communication of tumor microenvironment that contribute to colorectal cancer progression and immunotherapy. J. Translational Med. 20, 197 (2022).
Beltra, J-C. et al., Developmental relationships of four exhausted CD8 + T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 52, (2020).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell. Rep. 18, 248–262 (2017).
Zeng, D. et al. Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 12, 687975 (2021).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 160, 48–61 (2015).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14, 1083–1086 (2017).
Wu, Y. et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. 12, 134–153 (2022).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
An, Y. & Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 21, (2022).
Li, M. et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J. Translational Med. 20, 214 (2022).
Wu, S. Z. et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 39, e104063 (2020).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 16, 582–598 (2016).
Cassetta, L. et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 35, (2019).
Hu, Q. et al. Atlas of breast cancer infiltrated B-lymphocytes revealed by paired single-cell RNA-sequencing and antigen receptor profiling. Nat. Commun. 12, 2186 (2021).
Loi, S. et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann. Oncol. 25, 1544–1550 (2014).
Jhunjhunwala, S., Hammer, C. & Delamarre, L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer. 21, 298–312 (2021).
Yin, H. et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat. Commun. 12, 1394 (2021).
Song, H. et al. METTL3-mediated m6A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat. Commun. 12, 5522 (2021).
Han, D. et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 566, 270–274 (2019).
Tong, J. et al. m6A mRNA methylation sustains Treg suppressive functions. Cell. Res. 28, 253–256 (2018).
Wei, W-F. et al. Periostin + cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol. Oncol. 15, 210–227 (2021).
Chen, X. & Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov 18, (2019).
Kanzaki, R. & Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 111, 2708–2717 (2020).
Tong, J. et al. Pooled CRISPR screening identifies m6A as a positive regulator of macrophage activation. Sci. Adv. 7, (2021).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell. Metab. 30, 36–50 (2019).
Arts, R. J. W. et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology. 5, e1229725 (2016).
Wenes, M. et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell. Metab. 24, 701–715 (2016).
Pauwels, A. M., Trost, M., Beyaert, R. & Hoffmann, E. Patterns, receptors, and signals: regulation of phagosome maturation. Trends Immunol. 38, 407–422 (2017).
Gutiérrez, S. et al. Salmonella Typhimurium impairs glycolysis-mediated acidification of phagosomes to evade macrophage defense. PLoS Pathog. 17, e1009943 (2021).
Liu, Z. et al. Lipid-associated macrophages in the tumor-adipose microenvironment facilitate breast cancer progression. Oncoimmunology. 11, 2085432 (2022).
Philip, M. & Schietinger, A. CD8 + T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science. 322, 1097–1100 (2008).
Ding, S. et al. Single-cell atlas reveals a distinct immune profile fostered by T cell-B cell crosstalk in triple negative breast cancer. Cancer Commun. (Lond). 43, 661–684 (2023).
Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science. 375, eabf9419 (2022).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Acknowledgements
We express our gratitude to the funding agents and extend our sincere appreciation to all the individuals whose invaluable contributions played a major role in the successful completion of this project. We appreciate the support of the Shanghai Jiao Tong University Breast Cancer Database (SJTU-BCDB) for the collection of clinical data. We also thank the Medical Research Center of Quanzhou Medical College for maintaining the experimental conditions. We extend our heartfelt thanks to Li Shaoyang for assistance with image editing and Xu Xinyue for her support in experimental procedures. Each of these individuals contributed to the successful completion of this project, and we truly appreciate their efforts. The authors also thank www.figuredraw.com for providing the illustrations used to draw the flowcharts.
Funding
This work was supported by the Medical Innovation Project Foundation of Fujian Province, PRC [grant number 2021CX01010138]; Medical Innovation Project Foundation of Fujian Province, PRC [grant number 2021CXA045]; Science and Technology Innovation Joint Fund Project of Fujian Province [grant number 2019Y9049]; Science and Technology Project of Quanzhou [grant number 2020C047R], and the Special Fund for Clinical Research of the Wu Jieping Medical Foundation [grant number 320.6750.2021-10-60].
Author information
Authors and Affiliations
Contributions
DC, KC and TL developed the study design, with TL performing the data analysis and created the figures and tables. YH and SC edited and reviewed the manuscript, with YH collected patient samples. ZH performed the immunofluorescence analysis. XZ and ZL improved the language of the manuscript through reviews and revisions. All the authors made significant contributions to this article and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, T., Huang, Y., Cui, S. et al. RNA methylation patterns of tumor microenvironment cells regulate prognosis and immunotherapeutic responsiveness in patients with triple-negative breast cancer. Sci Rep 14, 26075 (2024). https://doi.org/10.1038/s41598-024-77941-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77941-2
