
Abstract
Human gut microbiota plays a crucial role in health and disease. Infancy is a critical period for gut microbiota maturation and immune system development and has the potential to affect long-term health. Understanding the development of gut microbiota in Japanese children is essential because of regional differences and the long-term health effects of the early gut microbiota. However, while several longitudinal studies in Japan have explored the development of the gut microbiota after birth, more extended follow-up periods are still needed. In this study, we aimed to analyze the gut microbiota of 106 Japanese mother–child pairs from the Chiba Study of Mother and Child Health, Japan, over 3.5 years. The results showed that the alpha diversity of the gut microbiota in children increased with age, and its composition began to resemble that of adults. We identified four distinct clusters of gut microbiota that reflected different maturation stages. The similarity between the maternal and child gut microbiota appeared to follow a bimodal-like distribution, suggesting that the presence of older siblings may enhance this similarity. This study highlights the dynamic nature of gut microbiota development in Japanese children and deepens our understanding of the similarities between maternal and child gut microbiota.
Similar content being viewed by others

Introduction
The human gut microbiota comprises a complex ecosystem of microorganisms and plays a vital role in health and disease1,2. The relationship between the gut microbiota during infancy and health is linked to gastrointestinal diseases, such as inflammatory bowel disease3 and irritable bowel syndrome4, as well as to the development of allergy-related diseases5,6, obesity7, diabetes mellitus8,9, autoimmune disorders10, and autism spectrum disorders11. For instance, changes in the gut microbiota have been observed in infants with allergies, showing a correlation between serum immunoglobulin E levels and specific gut bacteria12. These associations suggest that infancy is a critical period for the maturation of the gut microbiota and the development of the immune system. The gut microbiota develops dynamically during infancy and usually stabilizes at approximately 2–3 years of age9,13,14. Although early colonization by the gut microbiota is suggested to have lasting health effects throughout life, as supported by theories such as the “developmental origin of health and disease”15,16, the actual mechanisms and extent of these impacts remain unclear17.
Factors contributing to the formation of early gut microbiota include mother-to-child transmission18, duration of pregnancy9, mode of delivery9,19, and nutritional methods (breast milk or formula) 9,20. Moreover, environmental factors such as the presence of siblings and other household conditions play a significant role in shaping the gut microbiota9,21. In the context of mother to child transmission, infants have been seen to possess the same Bifidobacterium strains as their mothers during the first 90 days post-birth22,23. Furthermore, within the same household, family members have been observed to share common bacterial strains that are not limited to the mother–child relationship24. However, the link between mother–child relationship and changes in the child’s gut microbiota throughout growth is not yet fully understood.
Cross-sectional studies have investigated the dynamics of early gut microbiota in various countries. The most recent studies, however, have been conducted on large cohorts in Western countries and China9,15,20,21,25,26. The gut microbiota differs by region and population27; therefore, the development of the gut microbiota in Japanese children, particularly in terms of its composition, is expected to differ from that observed in children from other countries. Despite this, there are only a few longitudinal studies in Japan that have tracked the development of gut microbiota for several years post-birth28.
In this study, we aimed to analyze the gut microbiota of Japanese mother–child pairs, investigating its diversity, composition, and development over several years post-birth by utilizing an ongoing prospective birth cohort study—the Chiba Study of Mother and Child Health (C-MACH) 29. Furthermore, we aimed to understand the developmental patterns of the gut microbiota in Japanese children, and explore the similarities between maternal and child gut microbiota as well as the effects of factors contributing to the vertical transmission of the gut microbiota between mothers and infants.
Results
Cohort overview
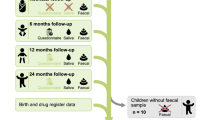
This study was based on the C-MACH. The study design is illustrated in Fig. 1. Participants were recruited from three hospitals in the Chiba and Saitama prefectures in Japan. A total of 434 pregnant women participated in the study between February 2014 and June 2015. Of these, 106 women consented to provide fecal samples during the first and third trimesters of their pregnancy, as well as from their children6,30. The characteristics of the participating mother–child pairs are summarized in Table 1. A total of 106 mothers from the C-MACH cohort were recruited and followed longitudinally for 3.5 years postpartum. Fecal samples were collected from 95 children (41 boys and 54 girls), 12 of whom were delivered via cesarean section. The median birth weight was 3034 g, and the median gestational age at birth was 39 weeks. Regarding nutrition during the first month of life, 68% of children received exclusive breastfeeding, and 30% received mixed feeding, based on the available data. However, there was a significant amount of missing data regarding infant feeding practices at 1 month. Additionally, 40 individuals (37%) received antibiotic therapy during the initial 3.5 years of life. Comparisons of the microbiota study participants (106 pairs) with the entire cohort (434 pairs) are presented in Table 1. The characteristics of the target maternal-child cohort showed no differences when compared with various statistical surveys in Japan31,32,33.

Study overview. (a) Participant flowchart. (b) Overview of the timing and number of fecal samples. The cohort consisted of pregnant women who lived in Chiba, Japan and were enrolled in the Chiba Study of Mother and Child Health. Children were included in the prospective cohort after obtaining parental consent.
Fecal samples were obtained from mothers at approximately 12 (n = 59) and 32 (n = 58) weeks of gestation and from children at 1 month (n = 66), 4 months (n = 65), 10 months (n = 64), 18 months (n = 52), and 3.5 years (n = 38) of age (Fig. 1). On average, four fecal samples were obtained from each child. The study included 285 infant and 117 maternal samples.
Changes in the diversity of gut microbiota in children
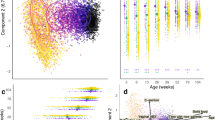
We performed 16S rRNA gene sequencing using DNA extracted from fecal samples. A median of 51.0 k high-quality processed reads was obtained per sample (median of 44.1 k and 65.3 k reads for infant and maternal sample, respectively). Based on rarefaction analysis, the sequencing depth was rarefied to 12,000 reads. The Shannon index, which indicates gut microbial α-diversity, showed a statistically significant increase with advancing age in the children (Fig. 2a; 4 vs 10 months, 2.46 vs 3.14, p < 0.001; 10 vs 18 months, 3.14 vs 3.60, p < 0.001; 18 months vs 3.5 years, 3.60 vs 4.11, p = 0.009; Wilcoxon rank-sum test with Bonferroni correction) (Fig. 2a). Figure 2b illustrates the overall compositional structure of the gut microbiota in the children and their mothers at various time points. Permutational multivariate analysis of variance (PERMANOVA) and pairwise adonis with Bray–Curtis dissimilarity metrics revealed significant differences among age groups (Fig. 2b). The microbial community converged toward an adult-like configuration.

Alpha diversity and compositional structure of gut microbiota in children and their mothers. (a) Shannon diversity index indicating an increase in gut microbial alpha diversity in children with age. Each symbol represents an individual sample, with the median value shown. The x-axis represents age points, while the y-axis represents the Shannon diversity index. The asterisks denote statistically significant differences determined by the Wilcoxon rank-sum test with Bonferroni correction for multiple comparisons, where p < 0.001. (b) PCoA plot illustrating the beta diversity based on Bray–Curtis dissimilarity, demonstrating the differences in gut microbiota composition at various time points for both children and their mothers. The x-axis represents PCo1 (6.4%), while the y-axis represents PCo2 (3.9%). Each point represents an individual sample, with different shapes and colors indicating different ages and stages. PCoA: Principal Coordinate Analysis; PCo1: Principal Coordinate 1; PCo2: Principal Coordinate 2.
At the phylum level, the following observations were noted: an association between age and increased relative abundance of Firmicutes, a decrease in Proteobacteria, an inverse U-shaped trend with peak prevalence at 4 months for Actinobacteria, and a nonlinear association between Bacteroidetes and age (Fig. 3a). At 3.5 years, the phylum distribution in the children was similar to that of their mothers. The main bacterial taxonomic groups (top 10 genera with the highest average relative abundance across all fecal samples) were examined across all ages tested (1, 4, 10, 18 months, and 3.5 years; Fig. 3b). Bifidobacterium was consistently the predominant genus throughout the age range of 1–3.5 years. Bacteroides was detected in the selected children at as early as 1 month of age, with its abundance notably increasing from 18 months onward. The presence of Blautia became evident at 18 months of age.

Phylum-level and genus-level distribution of gut microbiota in children and their mothers. (a) Boxplots depicting the relative abundance of major phyla in the gut microbiota of children at 3.5 years of age and their mothers. (b) Relative abundance of the top 10 genera at different ages and during the mother’s pregnancy. Mo_E: mothers during the early trimester; Mo_L: mothers during the late trimester; 1M: 1-month-old child; 4M: 4-months-old child; 10M: 10- months-old child; 18M: 18-months-old child; 42M: 3.5-year-old child.
Cluster analysis of intestinal microbiota composition among participants
We used the Dirichlet-multinomial mixture (DMM) model to conduct a cluster analysis based on the structure of the gut microbial community and to characterize the gut microbiota of the entire participant population. Four DMM clusters were identified based on their lowest Laplace approximation scores (see Supplementary Fig. S1 online). Cluster Type 1 primarily included children aged 1–4 months; while 64 (47%) were 1-month-old, 58 (43%) were 4-month-old. Cluster Type 2 mainly included children aged 10–18 months; while 49 (58%) were 10-month-old, 25 (30%) were 18-month-old. Cluster Type 3 mainly included children aged 18 months and 3.5 years; while 25 (29%) were 18-month-old, 27 (32%) were 3.5-year-old. Cluster Type 4 mainly included mothers in their first or third trimester of pregnancy; while 43 (44%) were in their first trimester, 43 (44%) were in their third trimester (Fig. 4a). Cluster Type 3 included 31 out of 117 adults, whereas Cluster Type 4 included 11 out of 285 children (Fig. 4a). The alpha diversity (Shannon index) was substantially different among the clusters (Fig. 4b). The major bacterial taxa (top 20 genera) also differed among the clusters; Bifidobacterium was predominant in Cluster Types 1, 2, and 3, whereas Blautia and Bacteroides started gaining dominance in Cluster Type 3 and became predominant in Cluster Type 4 (Fig. 4c).

Cluster analysis of gut microbiota composition of participants. (a) The distribution of fecal samples across four clusters identified using the Dirichlet-multinomial mixtures (DMM) model at each age and the mothers’ gestational period. The bubble size represents the number of fecal samples in each cluster. (b) Boxplot depicting the alpha diversity (Shannon index) of different clusters. The y-axis represents the Shannon index. The asterisks denote statistically significant differences determined by the Wilcoxon rank-sum test with Bonferroni correction for multiple comparisons, where p < 0.001. (c) Heatmap showing the relative abundance of the top 20 genera in the gut microbiota across the four DMM clusters. Mo_E: mothers during the early trimester, Mo_L: mothers during the late trimester, 1M: 1-month-old child, 4M: 4-months-old child, 10M: 10-months-old child, 18M: 18-months-old child, 42M: 3.5-year-old child.
Similarity in intestinal microbiota between mother and child and its associated factors
We evaluated the similarity in gut microbiota between mothers and their infants using the mother-infant cohort. We analyzed the Bray–Curtis dissimilarity indices among the 3.5-year-old infants, whose gut microbiota composition is commonly understood to be transitioning toward that of adults, and their mothers. The Bray–Curtis dissimilarity indices were significantly lower between mother–child pairs compared to unrelated pairs (Fig. 5a). The data appeared to show a distribution like bimodality for the gut microbiota similarity of biological mother–offspring pairs.

Similarity in gut microbiota between mothers and children. (a) Violin plot showing the Bray–Curtis dissimilarity indices between 3.5-year-old children and their biological mothers compared to unrelated mother–child pairs (Wilcoxon rank-sum test). (b) Violin plot illustrating the Bray–Curtis dissimilarity indices for mother–child pairs with or without older siblings (Wilcoxon rank-sum test).
We performed exploratory analyses to investigate factors associated with mother-infant microbiota similarity. The placement of 3.5-year-old children in various cluster types from the DMM cluster analysis was examined based on the hypothesis that the gut microbial composition of children is similar to that of their mothers. Mother–child pairs were divided into two groups based on the median of the Bray–Curtis dissimilarity index. The results showed that children who resembled their mothers did not necessarily belong to Cluster Type 4 (the predominantly adult cluster) (see Supplementary Table S1 online). Subsequent exploratory analysis of various factors within the cohort revealed that, when older siblings were present, there was a tendency for the similarity in gut microbiota between mothers and their children to increase (Fig. 5b; see Supplementary Table S2 online). No significant relationships were found for other variables, such as the method of delivery, nutrition methods, or maternal pre-pregnancy nutrition (see Supplementary Table S2 online).
Discussion
In this study, the alpha diversity of the children’s gut microbiota increased from 1 month to 3.5 years of age, whereas its beta diversity resembled that of adult microbiota as the children grew older. At the phylum level, by 3.5 years of age, the composition of children’s gut microbiota closely resembled that of adults. Four clusters based on the DMM model appear to reflect the growth and development of the children. The similarity between the gut microbiota of mothers and that of their children exhibited a bimodal-like distribution, suggesting the potential influence of the presence of older siblings on this increased similarity.
The compositional complexity (diversity) of the gut microbiota in children changes with age. In this study, the Shannon index, which is indicative of α-diversity, increased with age. This is consistent with the results of previous reports9,34. Our longitudinal analyses of the gut microbiota of participants, show that although the α-diversity of children’s gut microbiota increases up to 3.5 years of age, it does not reach the levels observed in their mothers. This result is consistent with those of previous cross-sectional studies15,35, suggesting that in Japan, the diversity of the gut microbiota of children does not fully attain adult levels during early childhood.
The composition of gut microbiota in children undergoes significant changes as they grow older9,13,15. In the present study, the Bray–Curtis dissimilarity index revealed that the gut microbiota of children gradually became more similar to that of adults as they aged. More pronounced changes were observed between the ages of 10–18 months and 18 months–3.5 years (see Supplementary Table S3 online). The dietary transition period, defined as complementary feeding, typically spans from 6 to 24 months of age36. Furthermore, according to the weaning food guidelines provided by the Japanese government, weaning is generally completed between the ages of 12 and 18 months37. During the exclusive breastfeeding period, the proliferation of Bifidobacterium is facilitated by the presence of human milk oligosaccharides, leading to an increased abundance of these bacteria38. In the present study, many infants were mixed-fed; however, a study using qPCR to examine Bifidobacterium in newborns up to three months old reported no significant difference in the total amount of Bifidobacterium between the exclusively breastfed and mixed-fed groups39. The shift from milk-based feeding, including mixed feeding, to complementary foods plays a key role in shaping gut microbiota diversity and composition in early life.
In the present study, Bifidobacterium was found to be the most abundant among the gut microbiota of children between the ages of 1 month and 3.5 years, peaking at approximately 40% at 4 months and remaining at > 10% (median value) till 3.5 years (Fig. 3b). Japanese individuals harbor higher levels of Bifidobacterium in their intestines compared to people from other countries; for example, the prevalence of Bifidobacterium was 10–20% in Japanese adults as compared to < 2% in those in Western countries and some parts of Asia27,40,41. In addition, the prevalence of Bifidobacterium was 5–20% in school-aged children (7–11 years) in 10 Asian countries, with 20% in those in Japan, which is higher compared to the < 5% in those in Western countries13,42,43. Although the proportion of Bifidobacterium in infants is generally high, the results of the present study showed that the prevalence of Bifidobacterium in infants and young children in this Japanese cohort was higher than that reported in cohort studies conducted in the United States, Denmark, and Sweden9,20,21. The high prevalence of Bifidobacterium in Japanese individuals is influenced by dietary habits, genetic factors, and cultural practices. The levels of Bifidobacterium, which possesses glycoside hydrolases for starch break down and enzymes for dietary fiber digestion, decrease on a low-carbohydrate, high-fat diet and increase on a high-carbohydrate, high-fiber diet44,45. Japanese individuals consume a traditional diet rich in carbohydrates such as rice and fermented products, which promote the growth of Bifidobacterium27. Furthermore, approximately 65–75% of the global population exhibits lactase nonpersistence (LNP)46. In individuals with LNP, lactose is not digested in the small intestine but instead reaches the colon. A study involving 1214 Japanese adults reported that all participants had a genotype associated with low lactase expression47. Lactose is a primary carbohydrate that promotes the growth of Bifidobacterium, and LNP is a contributing factor to the high levels of Bifidobacterium in the Japanese population. The LNP phenotype becomes evident in individuals aged 3–5 years48. Therefore, the high abundance of Bifidobacterium in Japanese infants and young children is likely due to genetic factors, as well as due to transmission from parents and family members. In addition, unique Japanese household bathing practices, which are not prevalent in Western cultures, might facilitate the transmission of Bifidobacterium not only from mother to child, but also from father to child and among other family members49. The observed richness of Bifidobacterium in Japanese infants in this study corroborates these findings and those of previous studies. The fluctuations in the major bacterial taxa were consistent with the changes observed in previous cohort studies that investigated the detailed longitudinal development of gut microbiota in children up to 1 year of age in Belgium and in those up to 5 years of age in Sweden14,50.
In the present study, four distinct clusters of the gut microbiota were identified among all participants. The proportional composition of bacteria constituting these clusters differed from those identified in similar cluster analyses conducted in other countries9,14. A cross-sectional study involving 367 healthy Japanese individuals aged 0–104 years revealed the composition of the gut microbiota across various age groups in the Japanese population35; post-weaning changes included an increase in Bacteroides, Ruminococcus, Blautia, and Faecalibacterium and a decrease in Bifidobacterium, Veillonella, Streptococcus, and Enterococcus. These characteristics are consistent with the changes observed in Cluster Types 1–4 in the present study. Therefore, the clusters identified in this study reflect changes (maturation) associated with aging. Each cluster included children who did not fit into the expected cluster. For example, a few infants aged 1 to 4 months were assigned to a cluster composed primarily of children aged 10 to 18 months. However, these early maturing children did not consistently transition to higher clusters (levels of maturation) during subsequent growth stages. This phenomenon may be influenced by factors such as mode of delivery, nutritional intake, and post-weaning dietary patterns. Children born via cesarean section have lower microbial diversity in their gut microbiota than those born vaginally9,20. Furthermore, infants receiving mixed nutrition have a higher microbial diversity in their gut microbiota than those exclusively breastfed9,21. However, the impact of such differences decreases over time with infant growth25. Correspondingly, we observed that differences in birth method and nutritional status affected microbial diversity and composition in the early infantile period; however, the impact of these variables decreased by 18 months of age. Overall, these findings highlight the dynamic nature of gut microbiota development during the early postnatal period and its modulation by birth method, nutrition, and weaning, emphasizing the convergence toward a more mature microbiota composition in children by the age of 18 months.
The data appeared to suggest a bimodal-like distribution in the similarity of gut microbiota between 3.5-year-old children and their mothers. This suggests that while some children have a gut microbial composition that closely resembles that of their mothers’, others do not. Factors contributing to the vertical transmission of the gut microbiota from mothers to infants include the mode of delivery, breastfeeding, physical contact, and the household environment9,14,21. Owing to these factors, the neonatal gut microbiota often resembles the maternal microbial community22,25. However, this similarity in microbial communities typically diminishes after the first year of life21,24,25. Conversely, some studies have reported that even children older than three years exhibit a gut microbiota composition more similar to that of their biological mothers than to that of unrelated children’s mothers18,24,25. To date, there have been no reports identifying the presence of children with high or low similarity to their mothers, indicating a bimodal distribution, as observed in the present study. Furthermore, our study suggests that the presence of older siblings may enhance the similarity of gut microbiota between mothers and children. Older siblings contribute to the diversity and maturation of a child’s gut microbiota9,18,51. The influence of siblings on gut microbiota, similar to the hygiene hypothesis52, is thought to be mediated through physical contact, which facilitates bacterial transmission among siblings51.
In addition, the impact of siblings on a child’s gut microbiota has been observed to be more pronounced when the age difference between siblings is smaller19. However, our study did not detect an effect of the age difference between siblings. The limited sample size in this study might have constrained our ability to detect significant differences, particularly in subgroup analyses, where smaller sample sizes can obscure meaningful trends. Further research with a larger sample size is needed to fully understand the impact of sibling relationships on gut microbiota similarity. Nonetheless, this study is the first to report the possibility that older siblings act as mediating factors, enhancing the similarity of gut microbiota between mothers and children.
The present study has some limitations. First, our cohort was based on a specific region and the results need to be validated in multiple cohorts from diverse populations. Second, while we collected early infant nutrition data, detailed post-weaning dietary information for both mothers and children was not gathered, which is a significant limitation given the strong influence of diet on gut microbiota composition. Third, we were unable to explore the relationship between genotypes affecting LNP and gut microbiota due to the absence of genetic data. Fourth, the lack of metabolome analysis limits our understanding of the microbiota’s functional potential. Lastly, the small sample size hindered the possibility of adjusting for co-variables such as delivery mode, feeding regimen, and other relevant factors.
In conclusion, this study provides insights into changes in the gut microbiota of Japanese children over a period of 3.5 years after birth. In addition, the results show that the similarity in the gut microbiota between mothers and their offspring may be influenced by the presence of older siblings. The findings of this study enhance our understanding of the changes in the gut microbiota of Japanese children and the similarities between maternal and child microbiota.
Methods
Ethics statement
This study was conducted in accordance with the Declaration of Helsinki and the Ethical Guidelines for Medical and Health Research Involving Human Subjects of the Japanese Ministry of Health, Labor, and Welfare. The study protocol was approved by the Biomedical Research Ethics Committee of the Graduate School of Medicine at Chiba University, Japan (Approval ID 989; application submitted on September 20, 2019). Written informed consent was obtained from all study participants.
Cohort design
The C-MACH is an ongoing prospective birth cohort study in Japan. The study protocol has been reported elsewhere29. For the current study, pregnant women were recruited from three hospitals in Chiba and Saitama prefectures in Japan between February 2014 and June 2015 for the first phase of the study. A total of 434 pregnant women participated in this study. Information on birth characteristics (sex, mode of delivery, gestational age at birth, birth length, and weight) and maternal weight at delivery was obtained from medical records. Maternal and infant information, including maternal age, height, and pre-pregnancy weight; feeding practices; and child weight after birth was obtained from self-administered questionnaires completed at 12 (first trimester) and 32 (third trimester) weeks of gestation; then at 1, 4, 10, and 18 months; and finally at 3.5 years postpartum.
Gut microbiota analysis
Participants at the Onodera Ladies Clinic in Chiba, Japan, were informed about the microbiota study, and 66 women were consented to participate in it. Fecal samples were obtained from mothers during early and late pregnancy, and from their children at age 1, 4, 10, and 18 months, and then at 3.5 years. The fecal samples were collected at home and stored at − 18 °C. Thereafter, the samples were transferred to the Chiba University Center for Preventive Medical Sciences Biobank and stored at − 80 °C until DNA extraction. The numbers of fecal samples from mothers and their children included in this study are shown in Fig. 1.
DNA was extracted from fecal samples and 16S rRNA gene sequences were amplified as described previously6,30,53. The fecal samples were initially processed with methanol, followed by filtration and centrifugation. The pellet was used for fecal DNA extraction using a procedure adapted from previously reported methods54, beginning with resuspension of the pellet and incubation with lysozyme (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) and achromopeptidase (Fujifilm Wako Pure Chemical Corporation), followed by treatment with sodium dodecyl sulfate and proteinase K (Merck, Darmstadt, Germany). The subsequent DNA purification process involved phenol/chloroform/isoamyl alcohol extraction, ethanol precipitation, RNase treatment, and polyethylene glycol precipitation.
Variable regions V1–V2 of the 16S rRNA gene were sequenced using Illumina MiSeq (Illumina, San Diego, CA, USA)55. Briefly, the 16S rRNA V1–V2 amplicon was amplified using KAPA HiFi Hot Start Ready Mix (2 ×) (TaKaRa Bio Inc., Shiga, Japan) with the following universal bacterial 16S rRNA gene primers: forward primer (27Fmod) 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGRGTTTGATYMTGGCTCAG-3′ and reverse primer (338R) 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTGCTGCCTCCCGTAGGAGT-3′. After 20 PCR cycles and purification with AMPure XP, the product was confirmed using 3% agarose gel electrophoresis.
Indices and sequencing adapters were added to the PCR products using the Nextera XT Index Kit (Illumina). The amplicons were purified, quantified, and pooled, following which the pooled libraries were analyzed and quantified. After denaturation, the library spiked with 20% PhiX, was sequenced on an Illumina MiSeq using a 500-cycle kit.
The sequence reads obtained were analyzed using the Quantitative Insights into Microbial Ecology2 (QIIME2) software (version 2021.04, https://qiime2.org). The sequences were subjected to quality control using the DADA2 plugin in QIIME2. This process includes denoising, paired-end read merging, chimera checking, and generating a feature table of amplicon sequence variants (ASVs). Trimming and truncation parameters were optimized for maximum sequence retention while maintaining data quality. Specifically, the forward reads were truncated at 220 bp and the reverse reads at 160 bp. ASVs were subjected to taxonomic classification using a pre-trained Naïve Bayes classifier, a probabilistic model based on the Silva 138 database (downloaded on June 2021 from https://docs.qiime2.org/2021.4/data-resources/).
Statistical analyses
We considered five different aspects of the variation in the infant gut microbiota as outcomes: (1) alpha diversity (taxonomic diversity of the microbiota in the sample) assessed by the Shannon index, (2) beta diversity (overall community differences between samples) assessed by the Bray–Curtis dissimilarity, (3) relative abundance at both phylum and genus levels, (4) DMM model cluster analysis to identify distinct microbial community structures, and (5) assessment of similarity of the mother-infant gut microbiota and related factors using the Bray–Curtis dissimilarity.
All analyses were conducted using the statistical software R, version 4.2.2 (R Foundation, Vienna, Austria)55. Both alpha-diversity (Shannon index) and beta-diversity (Bray–Curtis dissimilarity) metrics were calculated using the R package “microeco”56. Beta diversity was visually represented using Principal Coordinate Analysis plots. Comparison between groups were performed using PERMANOVA, the R package ‘pairwise.adonis2′57 and the unpaired Wilcoxon test, with P values < 0.05 considered statistically significant. The P values were adjusted using the default Bonferroni method to account for multiple comparisons.
DMM models, which can be used to describe variability in microbial metagenomic data, were run using the R package “Dirichlet-multinomial”58. DMM model clustering was used to determine the gut microbiota community types at the genus level for the entire sample. The number of community types was selected based on the minimal Laplace approximation of the negative log model evidence. Samples were assigned to community types based on their maximum posterior probabilities.
The Bray–Curtis dissimilarity is a compositional distance measure that accounts for both the presence and abundance of taxa in the communities (BC = 1 − (2 Cij/ (Si + Sj)), where Cij is the sum of the lowest abundance of each taxon in the two communities and Si and Sj are the total numbers of taxa present in the two communities. Exploratory analyses were conducted to examine the factors influencing the similarity of the gut microbiota between mothers and children using data from the cohort study. This was exploratory analyses and no multiplicity adjustments were made.
Data availability
The raw sequence files are registered in the DDBJ under DRA accession number PRJDB19560.
References
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. https://doi.org/10.1038/nri2515 (2009).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267. https://doi.org/10.1126/science.1223813 (2012).
Conrad, M. A. et al. The intestinal microbiome of inflammatory bowel disease across the pediatric age range. Gut Microbes 16, 2317932. https://doi.org/10.1080/19490976.2024.2317932 (2024).
Saulnier, D. M. et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology 141, 121. https://doi.org/10.1053/j.gastro (2011).
Subbarao, P. et al. The Canadian healthy infant longitudinal development (CHILD) study: Examining developmental origins of allergy and asthma. Thorax 70, 998–1000. https://doi.org/10.1136/thoraxjnl-2015-207246 (2015).
Tanabe, H. et al. Association of the maternal microbiome in Japanese pregnant women with the cumulative prevalence of dermatitis in early infancy: A pilot study from the Chiba study of mother and child health birth cohort (vol 12, 100065, 2019). World Allergy Organ. J. 14, 100507. https://doi.org/10.1016/j.waojou.2021.100507 (2021).
Indiani, C. et al. Childhood obesity and firmicutes/bacteroidetes ratio in the gut microbiota: A systematic review. Child. Obes. 14, 501–509. https://doi.org/10.1089/chi.2018.0040 (2018).
Wen, L. et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109-U1110. https://doi.org/10.1038/nature07336 (2008).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583. https://doi.org/10.1038/s41586-018-0617-x (2018).
Tremlett, H. et al. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 23, 1308–1321. https://doi.org/10.1111/ene.13026 (2016).
Sharon, G., Sampson, T. R., Geschwind, D. H. & Mazmanian, S. K. The central nervous system and the gut microbiome. Cell 167, 915–932. https://doi.org/10.1016/j.cell.2016.10.027 (2016).
Ling, Z. X. et al. Altered fecal microbiota composition associated with food allergy in infants. Appl. Environ. Microbiol. 80, 2546–2554. https://doi.org/10.1128/aem.00003-14 (2014).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222. https://doi.org/10.1038/nature11053 (2012).
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765. https://doi.org/10.1016/j.chom.2021.02.021 (2021).
Barker, D., Winter, P., Osmond, C., Margetts, B. & Simmonds, S. Weight in infancy and death from ischemic heart-disease. Lancet 2, 577–580 (1989).
Hoffman, D., Reynolds, R. & Hardy, D. Developmental origins of health and disease: Current knowledge and potential mechanisms. Nutr. Rev. 75, 951–970. https://doi.org/10.1093/nutrit/nux053 (2017).
Michel, C. & Blottière, H. M. Neonatal programming of microbiota composition: A plausible idea that is not supported by the evidence. Front. Microbiol. 13, 2022. https://doi.org/10.3389/fmicb.2022.825942 (2022).
Van Daele, E., Knol, J. & Belzer, C. Microbial transmission from mother to child: Improving infant intestinal microbiota development by identifying the obstacles. Crit. Rev. Microbiol. 45, 613–648. https://doi.org/10.1080/1040841x.2019.1680601 (2019).
Wernroth, M. L. et al. Development of gut microbiota during the first 2 years of life. Sci. Rep. 12(1), 9080. https://doi.org/10.1038/s41598-022-13009-3 (2022).
Fehr, K. et al. Breastmilk feeding practices are associated with the co-occurrence of bacteria in mothers’ milk and the infant gut: The child cohort study. Cell Host Microbe 28, 285. https://doi.org/10.1016/j.chom.2020.06.009 (2020).
Christensen, E. D. et al. The developing airway and gut microbiota in early life is influenced by age of older siblings. Microbiome 10, 106. https://doi.org/10.1186/s40168-022-01305-z (2022).
Makino, H. et al. Transmission of intestinal Bifidobacterium longum subsp. longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl. Environ. Microbiol. 77, 6788–6793. https://doi.org/10.1128/aem.05346-11 (2011).
Makino, H. et al. Mother-to-infant transmission of intestinal bifidobacterial strains has an impact on the early development of vaginally delivered infant’s microbiota. PLoS ONE 8, e78331. https://doi.org/10.1371/journal.pone.0078331 (2013).
Lax, S. et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052. https://doi.org/10.1126/science.1254529 (2014).
Jokela, R. et al. Sources of gut microbiota variation in a large longitudinal Finnish infant cohort. Ebiomedicine 94, 104695. https://doi.org/10.1016/j.ebiom.2023.104695 (2023).
Ouyang, R. Z. et al. Maturation of the gut metabolome during the first year of life in humans. Gut Microbes 15, 2231596. https://doi.org/10.1080/19490976.2023.2231596 (2023).
Nishijima, S. et al. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 23, 125–133. https://doi.org/10.1093/dnares/dsw002 (2016).
Tsukuda, N. et al. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. 15, 2574–2590. https://doi.org/10.1038/s41396-021-00937-7 (2021).
Sakurai, K. et al. Chiba study of mother and children’s health (C-MACH): Cohort study with omics analyses. Bmj Open 6, e010531. https://doi.org/10.1136/bmjopen-2015-010531 (2016).
Sato, Y. et al. Maternal gut microbiota is associated with newborn anthropometrics in a sex-specific manner. J. Dev. Origins Health Dis. 10, 659–666. https://doi.org/10.1017/s2040174419000138 (2019).
Government of Japan. Ministry of Internal Affairs and Communications. Population Estimates. e-Stat, Portal Site of Official Statistics of Japan., https://www.e-stat.go.jp/stat-search/files?page=1&toukei=00200524 (Accessed 19 August 2024)
Government of Japan. Ministry of Health, Labour and Welfare. Medical Facilities Survey. e-Stat, Portal Site of Official Statistics of Japan., https://www.e-stat.go.jp/statsearch/files?page=1&toukei=00450021&tstat=000001030908 (Accessed 19 August 2024)
Government of Japan. Ministry of Health, Labour and Welfare. Infant Nutrition Survey. e-Stat, Portal Site of Official Statistics of Japan., https://www.e-stat.go.jp/surveyplan/p00450271001 (Accessed 19 August 2024)
Bokulich, N. A. et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8, 343ra82. https://doi.org/10.1126/scitranslmed.aad7121 (2016).
Odamaki, T. et al. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. Bmc Microbiol. 16, 1–12. https://doi.org/10.1186/s12866-016-0708-5 (2016).
World Health Organization. WHO Guideline for complementary feeding of infants and young children 6–23 months of age (2023).
Study group on the revision of the Support Guide to Breastfeeding and Weaning. Support Guide to Breastfeeding and Weaning in Japanese (revised 2019); https://www.mhlw.go.jp/content/11908000/000496257.pdf (Accessed 25 June 2024)
Laursen, M. F. Gut microbiota development: Influence of diet from infancy to toddlerhood. Ann. Nutr. Metab. 77, 21–34. https://doi.org/10.1159/000517912 (2021).
Saturio, S. et al. Early-life development of the bifidobacterial community in the infant gut. Int. J. Mol. Sci. 22, 3382. https://doi.org/10.3390/ijms22073382 (2021).
Nam, Y. D., Jung, M. J., Roh, S. W., Kim, M. S. & Bae, J. W. Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS ONE 6, e22109. https://doi.org/10.1371/journal.pone.0022109 (2011).
Escobar, J. S., Klotz, B., Valdes, B. E. & Agudelo, G. M. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 14(1–14), 2014. https://doi.org/10.1186/s12866-014-0311-6 (2014).
Nakayama, J. et al. Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 5, 8397. https://doi.org/10.1038/srep08397 (2015).
Lin, A. et al. Distinct distal gut microbiome diversity and composition in healthy children from Bangladesh and the United States. PLoS ONE 8, e53838. https://doi.org/10.1371/journal.pone.0053838 (2013).
Brinkworth, G. D., Noakes, M., Clifton, P. M. & Bird, A. R. Comparative effects of very low-carbohydrate, high-fat and high-carbohydrate, low-fat weight-loss diets on bowel habit and faecal short-chain fatty acids and bacterial populations. Br. J. Nutr. 101, 1493–1502. https://doi.org/10.1017/s0007114508094658 (2009).
Yasukawa, Z. et al. Effect of repeated consumption of partially hydrolyzed guar gum on fecal characteristics and gut microbiota: A randomized, double-blind, placebo-controlled, and parallel-group clinical trial. Nutrients 11, 2170. https://doi.org/10.3390/nu11092170 (2019).
Mattar, R., Mazo, D. F. D. & Carrilho, F. J. Lactose intolerance: Diagnosis, genetic, and clinical factors. Clin. Exp. Gastroenterol. 5, 113–121. https://doi.org/10.2147/ceg.s32368 (2012).
Kato, K. et al. Association between functional lactase variants and a high abundance of Bifidobacterium in the gut of healthy Japanese people. PLoS ONE 13, 0206189. https://doi.org/10.1371/journal.pone.0206189 (2018).
Troelsen, J. T. Adult-type hypolactasia and regulation of lactase expression. Biochim. Et Biophys. Acta-Gen. Subj. 1723, 19–32. https://doi.org/10.1016/j.bbagen.2005.02.003 (2005).
Odamaki, T. et al. Impact of a bathing tradition on shared gut microbes among Japanese families (vol 9, 4380, 2019). Sci. Rep. https://doi.org/10.1038/s41598-020-59023-1 (2020).
Beller, L. et al. Successional stages in infant gut microbiota maturation. Mbio https://doi.org/10.1128/mBio.01857-21 (2021).
Laursen, M. F. et al. Having older siblings is associated with gut microbiota development during early childhood. BMC Microbiol. https://doi.org/10.1186/s12866-015-0477-6 (2015).
Strachan, D. P. Hay fever, hygiene, and household size. BMJ 18, 1259–1260. https://doi.org/10.1136/bmj.299.6710.1259 (1989).
Sakurai, K. et al. Association between gut microbiota composition and glycoalbumin level during pregnancy in Japanese women: Pilot study from Chiba study of mother and child health. J. Diabetes Investig. 11, 699–706. https://doi.org/10.1111/jdi.13177 (2020).
Kim, S. W. et al. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253. https://doi.org/10.1093/dnares/dst006 (2013).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2022). https://www.R-project.org/ (Accessed 19 August 2024).
Liu, C., Cui, Y. M., Li, X. Z. & Yao, M. J. microeco: An R package for data mining in microbial community ecology. Fems Microbiol. Ecol. https://doi.org/10.1093/femsec/fiaa255 (2021).
Martinez Arbizu, P. pairwiseAdo0nis: Pairwise multilevel comparison using adonis; https://github.com/pmartinezarbizu/pairwiseAdonis (2020).
Holmes, I., Harris, K. & Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE https://doi.org/10.1371/journal.pone.0030126 (2012).
Acknowledgements
We would like to express our gratitude to the study participants and members of the C-MACH for their cooperation. We thank Dr. Hiromi Tanabe for the technical support. This research was supported by grants from the Japan Society for the Promotion of Science (16H01781, 17K00577, 22H03347), Chiba Foundation for Health Promotion & Disease Prevention, and the Collaborative Research Program of the Science and Technology Platform Program for Industry-Academia Co-Creation (JPMJOP1831) from the Japan Science and Technology Agency. This study was also supported by the Japan Agency for Medical Research and Development (JP18gm0710009) and the Yakult Bio-Science Foundation. We also appreciate the assistance provided by Editage (www.editage.jp) for the English language editing of this manuscript.
Author information
Authors and Affiliations
Contributions
The authors are justifiably credited with authorship according to the authorship criteria. C.M. was the primary investigator in this study. C.M., K.S., and Y.T. conceived the study idea and designed the study. K.S. supervised the study design. M.W. handled the organization and storage of the research data, including maintaining the database and ensuring data integrity. O.H., A.N., and T.K. processed the samples using MiSeq. K.S., T.K., and Y.T. performed data management and statistical analyses. Y.T. drafted and edited the manuscript. All authors participated in discussions on protocol development and revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tsuruoka, Y., Kato, T., Watanabe, M. et al. Changes in the intestinal microbiota of Japanese children during the first 3.5 years of life. Sci Rep 14, 29302 (2024). https://doi.org/10.1038/s41598-024-78844-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-78844-y
Keywords
This article is cited by
-
Understanding the bacteriome, phageome and phage-associated bacteriome in healthy Vietnamese children under two years of age
Archives of Microbiology (2026)
-
Immunomodulatory Role of Milk on Gut Microbiota: Implications for Enhancing Oral Vaccine Efficacy
Probiotics and Antimicrobial Proteins (2025)
