
Abstract
Glioblastoma (GBM) is the most malignant brain cancer and one of the leading causes of cancer-related death globally. So, identifying potential molecular signatures and associated drug molecules are crucial for diagnosis and therapies of GBM. This study suggested GBM-causing ten key genes (ASPM, CCNB2, CDK1, AURKA, TOP2A, CHEK1, CDCA8, SMC4, MCM10, and RAD51AP1) from nine transcriptomics datasets by combining supervised and unsupervised learning results. Differential expression patterns of key genes (KGs) between GBM and control samples were verified by different independent databases. Gene regulatory network (GRN) detected some important transcriptional and post-transcriptional regulators for KGs. The KGs-set enrichment analysis unveiled some crucial GBM-causing molecular functions, biological processes, cellular components, and pathways. The DNA methylation analysis detected some hypo-methylated CpG sites that might stimulate the GBM development. From the immune infiltration analysis, we found that almost all KGs are associated with different immune cell infiltration levels. Finally, we recommended KGs-guided four repurposable drug molecules (Fluoxetine, Vatalanib, TGX221 and RO3306) against GBM through molecular docking, drug likeness, ADMET analyses and molecular dynamics simulation studies. Thus, the discoveries of this study could serve as valuable resources for wet-lab experiments in order to take a proper treatment plan against GBM.
Similar content being viewed by others

Introduction
Glioblastoma (GBM) is the grade-IV brain tumor in Gliomas according to the clinical and histopathological characteristics. Several genetic disorders like loss of heterozygosity, amplification, deletion, and mutation are associated with the initiation and development of GBM. Also, DNA methylation at CpG site in the promoter region of a gene is considered as a major cause of GBM. It affects the brain and central nervous system, accounting for approximately 14.3% of all tumors and comprising 49.1% of all malignant tumors1,2,3. Patients with GBM have only 3–5% survival rate for more than 5 years4. Despite the recent advancements in multimodality therapy like chemotherapy, radiotherapy, and supportive care, the overall prognosis for GBM patients remains unsatisfactory, and recurrence of the disease is frequently observed5,6. So, discovering new potential molecular biomarkers might play a crucial role in advancing GBM diagnosis, prognosis and therapies7. Because of the rapid expansion of high-throughput platforms, the vast amount of microarray gene expression data is generating rapidly associated with different diseases. Bioinformatics approaches are playing the significant role in identifying potential genomic biomarkers from those gene expression profiles for promptly diagnosis and therapies of diseases8,9,10,11.
There are some bioinformatics studies that have explored GBM-causing key genes (KGs) highlighting their pathogenetic processes through unsupervised WGCNA11,12,13,14 and LIMMA15,16,17,18,19,20,21,22,23,24,25 approaches. However, we observed that their KGs-sets are not so consistent and none of these studies provided KGs-guided therapeutic indications against GBM, though a therapeutic drug kills cancer cells by targeting the cancer-causing genes/proteins26,27. In this study, we attempted to explore more consistent GBM-causing KGs by analyzing multiple gene expression profile datasets generated from different countries using both supervised and unsupervised learning approaches for diagnostic and therapeutic indications,
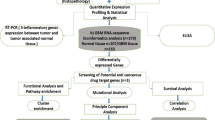
since supervised learning compared to the unsupervised learning gives more accurate results28,29. The workflow of this study is given in Fig. 1.

The workflow of the study.
Materials and methods
Source and description of data
In order to achieve our objectives, we considered both the raw data and metadata related to GBM as introduced below:
Collection of gene expression profiles from online databases
Total 10 microarray gene-expression profile datasets that contained GBM and control samples, were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database to explore GBM-causing key genes. The detail description of the datasets was given in Table 1.
Collection of meta-drug molecules from online sources
In order to repurpose potential drug molecules, we collected in total 139 drug molecules associated with GBM-causing genes from online databases DSigDB30 and GSCALite31, and published articles32,33,42,34,35,36,37,38,39,40,41 (Table S1).
Identification of differentially expressed genes (DEGs) by unsupervised approaches
To explore differentially expressed genes (DEGs) between GBM and control samples by the unsupervised approaches, we considered three microarray gene expression profile datasets (GSE104291, GSE86574 and GSE68848) from Table 1. To remove unimportant genes from each of these three datasets, at first, we considered variance-based gene filtering by CEMiTool43. For testing the significance of ith gene, this tool computes p-value as follows
where \(\:{\sigma\:}^{2}\) follows inverse gamma distribution and \(\:{s}_{i}^{2}\) is the variance for the expressions of ith gene. Obviously, \(\:{p}_{i}=1\) for \(\:{s}_{i}^{2}=0,\) while \(\:{p}_{i}=0\) for \(\:{s}_{i}^{2}=\infty\:,\) which indicates p-value decreases due to the increasing of \(\:{s}_{i}^{2}.\) Now \(\:{s}_{i}^{2}=0\) indicates all expressions in both case and control groups are equal. That is, ith gene is EE (equally expressed) between case and control group. It may be mentioned here that a gene is said to be equally expressed (EE) if its average expressions in case and control groups are equal; otehrwise, it is said to be differentially expressed (DE). It can also be shown that variance of differentially expression patterns is greater than variance of equally expression patterns for ith gene, that is \(\:{s}_{i,\:\:DE}^{2}>{s}_{i,\:\:EE}^{2}\) (see supplimentary section S1). Therefore, p-values computed by Eq. (1) can be used to select the differentially expressed genes (DEGs). This study considered p-values < 0.05 to select the DEGs. Then these DEGs sets were further filtered by using weighted correlation network analysis (WGCNA)44, which finds clusters (modules) of highly correlated genes. The WGCNA r-package was used to construct the co-expression network and gene modules for each of the three datasets. Module-trait relationships were determined by calculating the Pearson correlation coefficient between module eigengenes (MEs) and traits. Modules with significant correlations (>|0.6|, p-value < 0.005) were selected for further analysis. Further, signature genes were selected with module membership (MM) ≥ 0.8 and gene significance (GS) ≥0.7 as the cutoff value, also considered as DEGs for this study (Table S2).
Identification of DEGs) by supervised approaches
Random Forest (RF)45 and Support Vector Machine (SVM)46 are both popular supervised machine learning techniques for sample classifications. To explore differentially expressed genes (DEGs) between GBM and control samples by these two supervised approaches, we considered additional six datasets (GSE15824, GSE9171, GSE19728, GSE32374, GSE35493, GSE43289) from Table 1 with the previous three datasets (GSE104291, GSE86574 and GSE68848) that were analyzed by the unsupervised approaches as displayed in Fig. 1. It should be noted here that supervised approach requires more samples than unsupervised approaches to identify DEGs, since supervised approach requires more samples for partitioning dataset into training and test sets. We implemented both RF and SVM models to identify DEGs between GBM and control samples by the following steps.
Step 1
We combined nine preprocessing datasets from Table 1 to create a larger dataset of 444 with case and control.
Step 2
Then, for the ith gene (i = 1, 2, …, N), we trained both the RF and SVM prediction models by using randomly selected 60% of the total samples and leaving the remaining 40% as the test dataset. We implemented R-packages “randomForest” and “e1071” to train RF and SVM models, respectively.
Step 3
The trained model was used to classify the remaining 40% samples with the ith gene (i = 1, 2, …, N).
Step 4
After that, for the ith gene (i = 1, 2, 3,…,N), we computed the area under the ROC curve (AUC) and the classification accuracy (ACC) at a false positive rate (FPR) of 0.10 for both RF and SVM prediction models with the test samples.
Step 5
Finally, we detected the up- and down-regulated DEGs by satisfying the following criterion.
-
(i)
Up-regulated DEGs if AUCi ≥0.85, ACCi ≥0.85 for both prediction models & logFCi > 1.
-
(ii)
Down-regulated DEGs if AUCi ≥0.85, ACCi ≥0.85 for both prediction models & logFCi < -1.
Where logFC = log [(mean of controls) / (mean of cases)] indicates the log of fold change (logFC) value.
Selection of common DEGs (cDEGs) detected by both supervised and unsupervised approaches
From two DEGs lists computed by supervised and unsupervised methods respectively, we considered their common DEGs (cDEGs) as the most potential GBM-causing genes (cDEGs). Subsequently, these cDEGs were visualized by Venn diagram.
Protein-protein interaction (PPI) network analysis of cDEGs
To explore key genes (KGs), an online database and analysis tool (STRING v11.5) was used to create the PPI network of cDEGs. The network was visualized using the Cytoscape software47. The CytoHubba48 plugin in Cytoscape was used to select KGs based on six different topological measures including Closeness, Degree, Maximum Neighborhood Component (MNC), Edge Percolated Component (EPC), Maximal Clique Centrality (MCC) and Density of Maximum Neighborhood Component (DMNC). Further the “Molecular Complex Detection” (MCODE) plugin in Cytoscape was employed to detect the most prominent modules within the PPI network49.
In-silico verification of KGs using independent expression profiles
To verify the differential expression patterns of KGs, we used TCGA and GTEx databases from the GEPIA2 web tool50. We constructed Box plots to confirm the differential expression patterns of KGs between GBM and control groups. Also, to evaluate the predictive ability of KGs, we constructed prediction model based on Random Forest (RF) using independent expression profiles collected from NCBI database with accession ID GSE50161 (Table 1) and draw the ROC curves using the R-package “ROCR”51 to evaluate the prediction performance.
Enrichment analysis of KGs with GO terms and KEGG pathways
The Gene Ontology (GO) project is a bioinformatics tool that uses domain-specific ontologies to provide a complete source of functional data on gene products and descriptions of activities52. To investigate the Gene Ontology and KEGG pathway of KGs, we considered GeneCodis53, David54 and Enrichr55 database with P-value of 0.05 was chosen as threshold.
KGs regulatory network analysis
We investigated how transcription factors (TFs) and microRNAs (miRNAs) regulate KGs at both the transcriptional and post-transcriptional stages by analyzing their regulatory networks. The JASPAR database56 was used to identify the main TFs and the TarBase database57 was used to explore the main miRNAs. The NetworkAnalyst serve r58 was used to produce the networks. We used Cytoscape to visualize their interaction networks47.
DNA methylation analysis of KGs
MethSurv59 and ULCAN60 was used to investigate the methylation status of the KGs in GBM. Both of these web servers utilize TCGA methylation data. The level of DNA methylation was expressed by β -values (with a range from 0 to 1). M / (M + U + 100) is the formula used to calculate the β -values. The methylated and unmethylated intensities are denoted by M and U, respectively.
Immune infiltration level analysis of KGs
The Tumor Immune Estimation Resource (TIMER 2.0)61 is a comprehensive tool that estimates the quantity of tumor-infiltrating immune cell types from TCGA data. We utilized TIMER’s online tools to investigate the immune infiltration levels of CD8 + T cells, CD4 + T cells, neutrophils, B cells, macrophages, and dendritic cells with KGs in GBM.
Drug repurposing
To explore repurposable drug molecules, we performed molecular docking, Drug-Likeness and ADMET analysis, and MD simulation studies as discussed below.
Molecular docking
We considered 10 kg and associated top 2 TFs proteins as the target receptors. To explore potential ligands or drug molecules for treating GBM, molecular docking analysis between receptors and ligands was performed. Receptor proteins’ 3D structures were obtained from SWISS-MODEL62, Protein Data Bank63, and AlphaFold databases64. All 139 GBM-related meta-drug candidates’ 3D structures were taken from the PubChem database65. Following this, the binding affinity scores (in kcal/mol) between receptors and ligands (drug molecules) were determined through molecular docking using AutoDock Vina66. The arrangement of receptor proteins was based on the descending order of the average values in each row and drug-agents were arranged by the decreasing-order of column average in the score matrix to choose the top-ranked candidate drug molecules.
Evaluation of drug-likeness and ADMET properties of top-ranked drugs
We explored the structural features and chemical descriptors of the top ranked 25 drug molecules to understand their drug-like properties and assess their ADMET characteristics. We utilized SCFBio web tool to assess whether the compounds satisfied the Lipinski rule criteria67. Then, ADMET properties were computed by SwissADME68, amdetSAR69 and pkCSM70 for predicting the AMDET parameters. Further the interactions between these drugs and the top receptor protein were analyzed by PyMol71 and the Protein–Ligand Interaction Profiler (PLIP) web service72 by analyzing the docked complexes.
Molecular dynamic (MD) simulations studies
We carried out MD simulations by YASARA software73 and the AMBER1474 force field to investigate the dynamic properties of the top protein-ligand complexes. The hydrogen bonding network of the selected complexes was optimized and submerged using the TIP3P water model before the simulation was performed75. To maintain periodic boundary conditions, the solvent density was adjusted to 0.997 g/ml. Each simulation underwent a preliminary energy minimization process using steepest gradient algorithm with 5000 cycles. Each simulation was conducted under typical physiological conditions (298 K temperature, pH 7.4, 0.9% NaCl)76 and employed a multiple time-step algorithm77 which involved 2.50 femtoseconds (fs) time-step interval. A 100 ns molecular dynamics simulation was performed with a Berendsen thermostat78, and constant pressure. These conditions helped create a stable and realistic environment for the simulation. Trajectories of the simulation were captured at regular intervals of 250 picoseconds (ps), providing snapshots of the system’s behavior for subsequent in-depth analysis. The YASARA79 macro’s default script and the SciDAVis were used to conduct the primary analysis. Following that, using the YASARA software, all snapshots were accounted for MM-PBSA (MM-Poisson-Boltzmann surface area) binding free energy calculations. The following formula is used to calculate the binding free energy of the MM-PBSA80:
It is important to note that larger positive energies in the results indicated more favorable and stronger binding81.
Results
Identification of DEGs by unsupervised approaches
At first, we detected 5971, 6215 and 5776 DEGs from GSE68848, GSE86574 and GSE104292 respectively based on the variance property of genes expression with “CEMiTool”. For each filtered gene expression matrices, a soft threshold β value was chosen (11 for GSE68848, 18 for GSE86574 and 15 for GSE104291) based on a cutoff R2 value of 0.8 (Figure S1). Following that, some modules were found by hierarchical clustering with the minimal module size 30. To merge the modules, cut height of module eigengene was set to 0.1 for GSE68848, 0.15 for GSE86574 and, 0.30 for GSE104291 (Figure S2). All gene co-expression modules before and after merging were visualized in (Fig. 2). To uncover the relationship between modules and clinical traits (GBM and control), we selected six modules from GSE68848, six modules from GSE86574 and four modules from GSE104291 based on the module-trait relationship (>|0.6|, p-value < 0.005) (Figure S3). Further, a total of 699 signature genes (DEGs) were found from those significant modules with the cutoff at MM ≥ 0.8 and GS ≥ 0.7 (Figure S4). Combinedly, we got 502 DEGs between GBM and control samples (Table S4).

Cluster dendrogram of merged and unmerged modules for (A) GSE68848 (B) GSE86574 and (C) GSE104292.
Identification of differential expressed genes (DEGs) by machine learning (ML) approaches
We calculated AUC and ACC values for each gene in order to detect DEGs by using RF and SVM based prediction model as described in Sect. 2.2. Then we separated upregulated and down regulated DEGs by using logFC values. We obtained 1123 common DEGs, where 742 are downregulated, 381 upregulated, by using the criterion as given in step 5 of Sect. 2.2 (Fig. 3 and Table S3).

The volcano plot based on ACC and LogFC. (A) DEGs selection by RF-based prediction model (B) DEGs selection by SVM-based prediction model.
Selection of common DEGs (cDEGs) for ML and WGCNA approaches
Total 220 cDEGs were identified as GBM causing genes. These cDEGs were visualized using a Venn diagram in Fig. 4 (see also Table S5).

The cDEGs between the ML and WGCNA techniques are depicted in a Venn diagram.
Key gene (KG) identification from cDEGs by PPI network analysis
The PPI network was built using cDEGs, resulting in a network composed of 172 nodes and 977 edges. Top rank 10 kg (ASPM, CCNB2, CDK1, AURKA, TOP2A, CHEK1, CDCA8, SMC4, MCM10, and RAD51AP1) were selected from the PPI network by applying six topological measures (Fig. 5 and Table S6). Further we conducted module analysis with the cDEGs to locate key genes (KGs) in the clusters. Two modules were detected. Notably, all the KGs, detected by six topological measures were found in ‘module 1’ (Figure S5).

PPI network study of cDEGs. Green nodes indicate lower interactions, yellow nodes indicate medium interactions and red nodes indicate higher interactions (KGs).
Verification of differential expression patterns of KGs using independent datasets
At first, we verified the differential expression patterns of KGs in two independent databases (GTEx and TCGA) that combinedly contains 207 normal and 163 GBM samples through the box plot analysis (Figure S6.). We found that all KGs are upregulated that support our findings. To assess the prediction performance of KGs, we developed a Random Forest (RF) based prediction model using 60% samples as train data. The rest 40% data was used as test data. We also considered another independent test dataset from NCBI database with accession ID GSE50161. For both the test datasets, we constructed the ROC curves (Figure S7) and calculated some performance scores (AUC, TPR, TNR, and Accuracy) (Table S7). The performance of KGs in both prediction models was found to be strong with an AUC > 0.989 and ACC > 0.92.
Functional enrichment analysis of KGs with the GO-terms and KEGG pathways
For 10 kg, we carried out GO and KEGG pathway analysis. Here we took into account the most important GO terms from each cellular component (CC), biological process (BP), molecular function (MF), and KEGG pathways with P-value < 0.05 (Table S8).
KGs regulatory network analysis
The TFs and miRNAs networks were used to examine the regulators of KGs. We chose the top two TFs (GATA2, FOXC1) according to two topological measures, betweenness and degree with cutoff of 177 and 7 respectively as they play most prominent role in transcriptional level of the KGs (Figure S8-A). By employing the exact topological measures method, we chose the top five miRNAs (hsa-mir-16-5p, hsa-mir-34a-5p, hsa-mir-205-5p, hsa-mir-124-3p, and hsa-mir-147a) with betweenness and degree cutoff of 1225 and 10 respectively (Figure S8-B).
DNA methylation analysis of KGs in GBM
DNA methylation is an epigenetic process that controls the expression of genes82. DNA methylation allows researchers to uncover biomarkers for early detection, disease prognosis, and potential therapeutic targets. DNA methylation of essential genes helps researchers understand the regulatory mechanisms behind critical cellular processes and their disruption in disease, making it a vital aspect of genomic research83. Therefore, we examined DNA methylation status of KGs in GBM by MethSurv. We observed that except SMC4, the other nine KGs had significant CpG sites (p-value of ≤ 0.05) (Table S9). Additionally, ULCAN was also utilized to visualize the methylation status of the KGs in GBM. From Box whisker plot (Figure S9) it was found that almost all the KGs are hypomethylated in both GBM and Normal samples according to β-values ranging from 0 (completely unmethylated) to 1 (highly methylated). Some of KGs (TOP2A, CCNB2, CDK1, and, MCM10) showed almost no significant methylation differences between GBM and normal samples (β-values almost same). Rest of the KGs showed significant methylation differences between GBM and normal samples (lower β-values in GBM compared to normal samples).
Immune infiltration level analysis of KGs
The tumor microenvironment (TME) is a complex environment composed of different stromal components including immune cell along with the tumor cells84. To predict the infiltration of immune cells in GBM by the TIMER algorithm, we assessed the correlations between the expression levels of the KGs and the levels of infiltration of six immune cells (CD8 + T cell, B cell, CD4 + T cell, dendritic cell, neutrophil, and macrophage) (Figure S10). The findings indicate that the expression of KGs has a strong and positive relationship with the infiltration level of CD8 + T cell (0.12 ≤ Rho ≤ 0.35) and B cell (0.10 ≤ Rho ≤ 0.275) and weak and negative relationship with the infiltration level of CD4 + T cell (-0.15 ≥ Rho≥-0.39), Neutrophil (-0.19 ≥ Rho≥-0.298), Macrophage (0.11 ≥ Rho≥-0.016) and Dendritic cell (0.15 ≥ Rho≥-0.16). This result could help to discover potential immunotherapy for GBM.
KGs-guided drug repurposing by molecular docking
To explore KGs-guided repurposable drug molecules, we performed molecular docking between KGs mediated receptors and candidate drug molecules. The 3D structures of seven receptors (CDK1, AURKA, TOP2A, CHEK1, CDCA8, SMC4, and GATA2) were taken from the Protein Data Bank (PDB) using the following PDB codes: 6GU6, 2J4Z, 1ZXM, 1ZLT, 2KDD, 4U4P, 6ZFV. Four targets (CCNB2, MCM10, RAD51AP1, FOXC1) were obtained from the “AlphaFold Protein Structure Database” (AF-O95067-F1, AF-Q7L590-F1, AF-Q96B01-F1, AF-Q12948-F1) using their corresponding UniProt IDs, O95067, Q7L590, Q96B01, Q12948, respectively. The remaining one receptor (ASPM, uniport id Q8IZT6) was obtained from swiss model after homology modeling using template (P62295.1.A). Out of 139 drugs, the top-ranked 25 potential drugs were considered as potential drugs because all of them exhibited significant binding affinity (BA) < -7.0 (kcal/mol) after docking (Fig. 6). In terms of potential treatments for GBM, these 25 lead compounds appear promising.

Binding affinities between receptor proteins and drug agents.
Evaluation of drug-likeness and ADMET properties
Based on the Lipinski rule of five (ROF) we found that, out of top-ranked 25 drugs, 9 drugs (SNX2112, vatalanib, crenolanib, MLN4924, TGX221, fenbendazole, RO3306, fluoxetine and, vandetanib) violates no ROF (Table S10). Then, the ADME/T Properties of the 9 drugs were examined through various parameter. The water solubility (ESOL) score of all the 9 drugs were computed based on the LogP value. We found that the logP value off all the drugs were in range of -5.9 to -3.4 (poorly soluble < − 6 < moderately < − 4 < soluble) which indicates that they are water soluble68 (Table 2). A compound’s blood-brain barrier (BBB) permeability index indicates how likely it is to pass the BBB (a physiological barrier between the blood and the central nervous system). Compounds having a logBB > 0.3 can penetrate the BBB and logBB < -1 are poorly distributed to the brain. Though our study was based on GBM (one of the malignant brain cancers), it is very important that the drug molecules should cross BBB to exhibit its function inside the brain85,86. We found that 5 drugs (Fluoxetine, Vatalanib, TGX221, Fenbendazole and, RO3306) are more likely and 2 drugs (Crenolanib and Vandetanib) are less likely to possess the capability to penetrate the BBB. According to the Drug-Likeness and ADMET analysis of 25 drug molecules we concluded that, four compounds (Fluoxetine, Vatalanib, TGX221 and RO3306) could be the potential drugs for GBM. Table S11 displayed the interactions profile of the four drugs with the top ranked potential receptor AURKA.
Molecular dynamic (MD) simulations with the top-ranked drug-target complexes
After docking and ADME/T analysis, we selected four drugs - Fluoxetine, Vatalanib, TGX221 and RO3306 as four candidate drug molecules. Therefore, 100 ns MD-based MM-PBSA simulations were run on the top-ranked receptor (AURKA) and the four drug complexes (AURKA-RO3306, AURKA-Vatalanib, AURKA-TGX221, and AURKA-Fluoxetine) to evaluate their stability. All complexes showed a minor fluctuation in Cα backbone but remained stable rest of the simulation. The RMSD (root mean square deviation) related to the proposed receptor (AURKA) was displayed in Fig. 7(A). The estimated RMSDs ranged from 0.41 to 2.15. The AURKA complexes’ average RMSDs were 1.32, 1.35, 1.40, and 1.30, respectively. The RMSD of all the complexes raised slightly between 0 and 10 ns and remained stable till 100 ns. From the graph it can be clearly interpreted that all the complexes were structurally stable. The four complexes’ binding energies were calculated for the MM-PBSA shown in Fig. 7(B).

100ns MD simulation results for the top four complexes. (A) The RMSDs’ periodic progression. (B) MM-PBSA binding free energy (kJ/mol).
Discussion
Due to the heterogeneity of GBM, a high mortality and fatality rate still persists. So, it is essential to identify GBM causing key molecular signatures for diagnosis, prognosis and therapies. In this research, we considered nine microarray gene-expression datasets to discover GBM causing key genes (KGs). At first, we detected 220 overlapping DEGs between GBM and control samples by using machine learning (ML) and WGCNA approaches. Then the top-ranked 10 DEGs (ASPM, CCNB2, CDK1, AURKA, TOP2A, CHEK1, CDCA8, SMC4, MCM10, and RAD51AP1) were identified as the KGs via PPI network and module analysis. (Fig. 5, Table S6, Figure S5). Previous individual studies also reported some of these KGs as the GBM causing KGs. Among them, the key gene ‘Cyclin-dependent kinase 1 (CDK1)’ is a part of a group of cell cycle-regulating kinases. Its primary role involves overseeing the transition of the BP-term ‘G2/M phase’ (Table S7) of the cell cycle, and facilitating the initiation of mitosis through its interaction with cyclin B87. The G2/M phase has a potential role in the growth of GBM tumors88,89 Also, the inhibition of CDK1 through knockdown experiments resulted in a noteworthy reduction in the proliferation of GBM cells, specifically in U-87MG and U-251MG cell lines indicating that CDK1 is essential for the proliferation of GBM cells90. Aurora kinase A (AURKA) gene is a member of the MF-term “serine/threonine kinase”, and its activation plays a crucial role in governing cell division by controlling the process of mitosis91. It plays an important role in the development and spread of solid tumors, including glioblastomas92. A serine/threonine-specific protein kinase called checkpoint kinase 1 (CHEK1), also known as CHK1, controls the cell cycle checkpoint response and the DNA damage response93. It was also enriched in different KEGG pathways including cell cycle. Numerous CHK1 inhibitors were shown to interact with numerous MEK1/2 inhibitors to eradicate a variety of primary human glioblastoma isolates94. Moreover, other KGs (e.g., ASPM, CDCA8, MCM10 etc.) play an important role in developing GBM and associated with different biological process, molecular function and pathways 12,95,96,97. The expression analysis from the independent NCBI, TCGA and GTEx databases confirmed the differential expression patterns of KGs (Figure S6, S7). Some TFs and miRNAs were detected as the key transcriptional and post-transcriptional regulators of KGs by the gene regulatory network analysis (Figures S8) which might play a crucial role in the development of GBM. The DNA methylation study indicated that all the KGs (except SMC4) had CpG sites (Table S9) which might play an important role in GBM development.
DNA methylation analysis showed that most of the KGs are hypomethylated for which they become more active as oncogene98. From several studies, we found that these oncogenes are associated with the development of different cancers including GBM91,99,100,101,102,103,104. Tumor immunotherapy has emerged as a new area of study for tumors in recent years. In order to better understand the tumor microenvironment, more research has been concentrated on the immune cells’ penetration into tumor tissues105. We examined the association between the expressions of KGs and immune infiltrating cell types (CD8 + T and CD4 + T cell, B cell, neutrophil, dendritic cell and macrophage) in GBM and found their significant association in GBM progression and development (Figure S10). It has been found that CD8 + T cell infiltration positively correlates with the survival rate of patients with GBM106 and B cells were discovered to infiltrate in GBM107. However, it was found that almost all the KGs were poorly correlated with macrophage infiltration. It could indicate that higher gene expression is associated with lower immune cell infiltration, suggesting that these genes may contribute to immune evasion or suppression108,109. Some of these genes were also found to be weakly correlated with macrophage infiltration in previous studies12,110,111. Additionally, study showed that GBM located in the temporal lobe exhibited the highest levels of macrophage infiltration, while those in the frontal lobes had significantly lower levels of macrophage infiltration112.
We investigated potent drugs for the treatments against GBM and found four drugs (Fluoxetine, Vatalanib, TGX221 and RO3306) displayed favorable profiles. Among the identified candidate drugs, Fluoxetine has FDA approval for the therapy of major depressive disorder, as indicated by their Drug Bank (DB) database (DB accession number DB00472). It is one of the most prescribed selective serotonin reuptake inhibitor (SSRI), which increase the intracellular [Ca2+], thereby triggering apoptosis in gliomas113. Currently, research is being done on vatalanib to treat oral angiogenesis (accession numbers DB04879). The metastasis of GBM U87 cells after receiving CPI444 and vatalanib via a nanocarrier (GO-PEG) was significantly reduced114. It was discovered that TGX-221 prevented glioblastoma cells from migrating and invading, which allowed it to prevent cell growth and trigger apoptosis115. At dosages that inhibit CDK1, it was discovered that RO-3306 had no standalone cytotoxic impact but sensitized a number of GBM cells to Temozolomide (TMZ)87. Finally, the stability of the top-docked complexes (AURKA-RO3306, AURKA-Vatalanib, AURKA-TGX221 and AURKA-Fluoxetine) was assessed through molecular dynamics (MD)-based MM-PBSA simulation. The results indicated that these complexes exhibited consistent and stable behavior. Among the proposed drugs, TGX-222 and RO-3306 have not been approved yet and require further evaluation through wet-lab based experiments before clinical trial for the treatment of GBM.
Conclusion
This study identified GBM-causing 10 key genes (KGs) from nine transcriptomics datasets by using both supervised and unsupervised learning approaches. The association of KGs with GBM was also confirmed by some independent datasets/databases. The KGs-set enrichment analysis with GO-terms and KEGG pathways revealed some crucial biological process (DNA replication, G2/M transition of mitotic cell cycle), molecular functions (protein serine/threonine kinase activity, single-stranded DNA binding) and pathways (p53 signaling pathway, Cell cycle) associated with GBM. The KGs regulatory network analysis revealed two TFs (FOXC1 and GATA2) and five miRNAs (hsa-mir-16-5p, hsa-mir-34a-5p, hsa-mir-205-5p, hsa-mir-124-3p, hsa-mir-147a) as the transcriptional and post-transcriptional regulators. DNA methylation studies also showed that most of the KGs are hypomethylated which indicates their oncogenic activities. The infiltration level analysis of KGs revealed that, KGs are significantly associate with different tumor infiltrates immune cells such as, CD8 T cell, CD4 T cell, B cell, neutrophil, macrophage and dendritic cell (DC) of GBM. Four top-ranked potential drugs (Fluoxetine, Vatalanib, TGX221 and RO3306) were identified by molecular docking, drug-likeness and ADMET analysis. Therefore, the output of this study may play a vital role for diagnosis and therapies of GBM.
Data availability
The datasets analyzed in this study are freely available at the following links https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9171, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15824, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19728, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32374, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35493, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43289, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68848, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86574, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104291, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50161.
References
Louis, D. N. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 131, 803–820 (2016).
Ostrom, Q. T., Cioffi, G., Waite, K., Kruchko, C. & Barnholtz-Sloan, J. S. CBTRUS Statistical Report: primary brain and other Central Nervous System tumors diagnosed in the United States in 2014–2018. Neuro Oncol. 23, III1–III105 (2021).
Ohgaki, H. & Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 170, 1445–1453 (2007).
Szopa, W., Burley, T. A., Kramer-Marek, G. & Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. Biomed Res. Int. (2017). (2017).
Taphoorn, M. J. B., Sizoo, E. M. & Bottomley, A. Review on quality of Life issues in patients with primary brain tumors. Oncologist. 15, 618–626 (2010).
Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 22, e273–e281 (2015).
Sarhadi, V. K. & Armengol, G. Molecular biomarkers in Cancer. Biomolecules. 12, 1–39 (2022).
Islam, M. A. et al. Exploring Core genes by Comparative Transcriptomics Analysis for Early Diagnosis, prognosis, and therapies of Colorectal Cancer. Cancers (Basel) 15, (2023).
Hossain, M. T., Reza, M. S., Peng, Y., Feng, S. & Wei, Y. Identification of key genes as potential drug targets for gastric Cancer. Tsinghua Sci. Technol. 28, 649–664 (2023).
Islam, M. A. et al. Bioinformatics-based investigation on the genetic influence between SARS-CoV-2 infections and idiopathic pulmonary fibrosis (IPF) diseases, and drug repurposing. Sci. Rep. 13, 4685 (2023).
Li, C. et al. Identification of key modules and hub genes in glioblastoma multiforme based on co-expression network analysis. FEBS Open. Bio. 11, 833–850 (2021).
Zhou, J. et al. Construction of co-expression modules related to survival by WGCNA and identification of potential prognostic biomarkers in glioblastoma. J. Cell. Mol. Med. 25, 1633–1644 (2021).
Chen, X. et al. Identification of survival–associated key genes and long non–coding RNAs in glioblastoma multiforme by weighted gene co–expression network analysis. Int. J. Mol. Med. 43, 1709–1722 (2019).
Yang, Q. et al. Candidate Biomarkers and Molecular Mechanism Investigation for Glioblastoma Multiforme Utilizing WGCNA. Biomed Res. Int. (2018). (2018).
Yang, S., Gao, K. & Li, W. Identification of hub genes and pathways in glioblastoma by bioinformatics analysis. Oncol. Lett. 17, 1035–1041 (2019).
Arya, K. R. et al. Identification of hub genes and key pathways associated with anti-vegf resistant glioblastoma using gene expression data analysis. Biomolecules. 11, 1–20 (2021).
Cui, K. et al. Hub biomarkers for the diagnosis and treatment of glioblastoma based on microarray technology. Orig Artic https://doi.org/10.1177/1533033821990368
Lou, W., Ding, B., Xu, L. & Fan, W. Construction of potential glioblastoma multiforme-related miRNA-mRNA regulatory network. Front. Mol. Neurosci. 12, 66 (2019).
Luo, X. et al. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: a bioinformatics analysis. Oncol. Lett. 18, 6171–6179 (2019).
Li, L. et al. Identification of key candidate genes and pathways in glioblastoma by integrated bioinformatical analysis. Exp. Ther. Med. 18, 3439–3449 (2019).
Yang, L. et al. Bioinformatical Analysis of Gene Expression Omnibus Database Associates TAF7/CCNB1, TAF7/CCNA2, and GTF2E2/CDC20 pathways with Glioblastoma Development and Prognosis. World Neurosurg. 138, e492–e514 (2020).
Dong, C., Fan, W. & Fang, S. PBK as a potential Biomarker Associated with prognosis of Glioblastoma. J. Mol. Neurosci. 70, 56–64 (2020).
Geng, R. X. et al. Identification of core biomarkers associated with outcome in glioma: Evidence from bioinformatics analysis. Dis. Markers (2018). (2018).
Zou, Y. F. et al. Screening and authentication of molecular markers in malignant glioblastoma based on gene expression profiles. Oncol. Lett. 18, 4593–4604 (2019).
Guo, Y., Wang, X., Ning, W., Zhang, H. & Yu, C. Identification of two core genes in glioblastomas with different isocitrate dehydrogenase mutation status. Mol. Biol. Rep. 47, 7477–7488 (2020).
Wilkes, G. Targeted therapy: attacking Cancer with Molecular and Immunological targeted agents. Asia-Pacific J. Oncol. Nurs. 5, 137–155 (2018).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal. Transduct. Target. Ther. 6, 201 (2021).
Xia, H., Akay, Y. M. & Akay, M. Selecting relevant genes from microarray datasets using a Random Forest Model. IEEE Access. 9, 97813–97821 (2021).
Robust identification of Common genomic biomarkers from multiple gene expression profiles for the prognosis, diagnosis, and therapies of pancreatic cancer.
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 31, 3069–3071 (2015).
Liu, C. J. et al. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 34, 3771–3772 (2018).
Staedtke, V., Bai, R. Y. & Laterra, J. Investigational new drugs for brain cancer. Expert Opin. Investig Drugs. 25, 937–956 (2016).
Juillerat-Jeanneret, L. The targeted delivery of cancer drugs across the blood-brain barrier: chemical modifications of drugs or drug-nanoparticles? Drug Discov Today. 13, 1099–1106 (2008).
Zhuo, C. et al. Surprising anticancer activities of psychiatric medications: old drugs offer new hope for patients with brain cancer. Front. Pharmacol. 10, 1–7 (2019).
Kim, C., Shah, B. P., Subramaniam, P. & Lee, K. B. Synergistic induction of apoptosis in brain cancer cells by targeted codelivery of siRNA and anticancer drugs. Mol. Pharm. 8, 1955–1961 (2011).
Wainwright, D. A., Nigam, P., Thaci, B., Dey, M. & Lesniak, M. S. Recent developments on immunotherapy for brain cancer. Expert Opin. Emerg. Drugs. 17, 181–202 (2012).
Sontheimer, H. & Bridges, R. J. Sulfasalazine for brain cancer fits. Expert Opin. Investig Drugs. 21, 575–578 (2012).
Newton, H. B. Novel chemotherapeutic agents for the treatment of brain cancer. Expert Opin. Investig Drugs. 9, 2815–2829 (2000).
Laquintana, V. et al. New strategies to deliver anticancer drugs to brain tumors. Expert Opin. Drug Deliv. 6, 1017–1032 (2009).
Benjamin, W. No Titיליle. ペインクリニック学会治療指針3, 1–9 (2019).
Fung, N. H. et al. Understanding and exploiting cell signalling convergence nodes and pathway cross-talk in malignant brain cancer. Cell. Signal. 57, 2–9 (2019).
Bahmad, H. F. et al. Repurposing of Anticancer Stem Cell drugs in Brain tumors. J. Histochem. Cytochem. 69, 749–773 (2021).
Russo, P. S. T. et al. CEMiTool: a Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 19, 56 (2018).
Horvath, S. & Langfelder, P. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Cortes, Hohndel, U., Vladimir & Vapnik Corinna (AT&TBellLabs., (AT&TBellLabs., Hohndel, NJ07733, U. Support-Vector Networks. Mach. Learn. 297, 273–297 (1995).
Christmas, R. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Chin, C. H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, 11 (2014).
G.~D.~Bader. & C.~W.~Hogue. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2 (2003).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, 556–560 (2019).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics. 21, 3940–3941 (2005).
Dwight, J. T. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25 (2000).
Garcia-Moreno, A. et al. Funct. Enrich. Anal. Regul. Elem. Biomedicines 10, 590 (2022).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Khan, A. et al. JASPAR. : Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, (2018). (2018).
Sethupathy, P., Corda, B., Hatzigeorgiou, A. G. & TarBase A comprehensive database of experimentally supported animal microRNA targets. Rna. 12, 192–197 (2006).
Zhou, G. et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, 234–241 (2019).
Modhukur, V. et al. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 10, 277–288 (2018).
Chandrashekar, D. S. et al. An update to the integrated cancer data analysis platform. Neoplasia 25. UALCAN, 18–27 (2022).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, 296–303 (2018).
Berman, H. M. et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 58, 899–907 (2002).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, 439–444 (2022).
Kim, S. et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, 1102–1109 (2019).
Oleg, T., Arthur, J., AutoDock, O. & Vina Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and Multithreading. J. Comput. Chem. 31, (2010).
Lipinski, C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 1, 337–341 (2004).
Daina, A., Michielin, O., Zoete, V. & SwissADME A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Cheng, F. et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 52, 3099–3105 (2012).
Pires, Blundell, T. L. & Ascher, D. B. pkCSM: predicting small-molecule pharmacokinetic properties using graph-based signatures (Theory- How to Enterpret pkCSM Result). pKCSM 5 (2015).
Schrödinger, L. PyMOL the PyMOL Molecular Graphics System. CCP4 Newsl. Protein Crystallogr. 40, 82–92 (2010).
Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F. & Schroeder, M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 43, 443–447 (2015).
Krieger, E. & Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations | Bioinformatics | Oxford Academic. Bioinformatics. 30, 2981–2982 (2014).
Dickson, C. J. et al. Lipid14: the amber lipid force field. J. Chem. Theory Comput. 10, 865–879 (2014).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Krieger, E., Nielsen, J. E., Spronk, C. A. E. M. & Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph Model. 25, 481–486 (2006).
Krieger, E. & Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 36, 996–1007 (2015).
Schuler, L. D. et al. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3586–3616 (2001).
Krieger, E., Koraimann, G. & Vriend, G. Increasing the precision of comparative models with YASARA NOVA - A self-parameterizing force field. Proteins Struct. Funct. Genet. 47, 393–402 (2002).
Mitra, S. & Dash, R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J. Mol. Graph Model. 83, 42–52 (2018).
Srinivasan, E. & Rajasekaran, R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Adv. 6, 102744–102753 (2016).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology. 38, 23–38 (2013).
Jones, P. A. & Buckley, J. D. The role of dna methylation in cancer. Adv. Cancer Res. 54, 1–23 (1990).
Wei, R., Liu, S., Zhang, S., Min, L. & Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. (2020). (2020).
Wong, A. D. et al. The blood-brain barrier: an engineering perspective. Front. Neuroeng. https://doi.org/10.3389/fneng.2013.00007 (2013).
Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 37, 48–57 (2010).
Voce, D. J. et al. CDK1 is up-regulated by temozolomide in an NF-κB dependent manner in glioblastoma. Sci. Rep. 11, 5665 (2021).
Li, W. et al. 3-O-acetyl-11-keto-β-boswellic acid exerts anti-tumor effects in glioblastoma by arresting cell cycle at G2/M phase. J. Exp. Clin. Cancer Res. 37, 132 (2018).
Castro-Gamero, A. M., Pezuk, J. A., Brassesco, M. S. & Tone, L. G. G2/M inhibitors as pharmacotherapeutic opportunities for glioblastoma: the old, the new, and the future. Cancer Biol. Med. 15, 354–374 (2018).
Zhang, Y., Xia, Q. & Lin, J. Identification of the potential oncogenes in glioblastoma based on bioinformatic analysis and elucidation of the underlying mechanisms. Oncol. Rep. 40, 715–725 (2018).
Du, R., Huang, C., Liu, K., Li, X. & Dong, Z. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 20, 15 (2021).
Nguyen, T. T. T. et al. Aurora kinase a inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 12, 5203 (2021).
Zhang, Y. & Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 134, 1013–1023 (2014).
Tang, Y., Dai, Y., Grant, S. & Dent, P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. Ther. 13, 379–388 (2012).
Chen, X. et al. ASPM promotes glioblastoma growth by regulating G1 restriction point progression and Wnt-β-catenin signaling. Aging (Albany NY). 12, 224–241 (2020).
Horvath, S. et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl. Acad. Sci. U. S. A. 103, 17402–17407 (2006).
Kang, P. et al. Knockdown of MCM10 gene impairs Glioblastoma Cell Proliferation, Migration and Invasion and the implications for the regulation of Tumorigenesis. J. Mol. Neurosci. 70, 759–768 (2020).
Alshammari, E., Zhang, Y., Sobota, J. & Yang, Z. Aberrant DNA methylation of Tumor suppressor genes and oncogenes as Cancer biomarkers. Genomic Epigenomic Biomarkers Toxicol. Dis. 251–271. https://doi.org/10.1002/9781119807704.ch12 (2022).
Tsai, K. K., Bae, B. I., Hsu, C. C., Cheng, L. H. & Shaked, Y. Oncogenic ASPM is a regulatory hub of developmental and stemness signaling in cancers. Cancer Res. https://doi.org/10.1158/0008-5472.can-23-0158 (2023).
Cui, F., Hu, J., Ning, S., Tan, J. & Tang, H. Overexpression of MCM10 promotes cell proliferation and predicts poor prognosis in prostate cancer. Prostate. 78, 1299–1310 (2018).
Yan, Y. et al. SMC4 knockdown inhibits malignant biological behaviors of endometrial cancer cells by regulation of FoxO1 activity. Arch. Biochem. Biophys. 712, 109026 (2021).
Yi, Y. et al. Methylation-dependent and -independent roles of EZH2 synergize in CDCA8 activation in prostate cancer. Oncogene. 41, 1610–1621 (2022).
Zhuang, L., Zhang, Y., Meng, Z. & Yang, Z. Oncogenic roles of RAD51AP1 in Tumor tissues related to overall survival and disease-free survival in Hepatocellular Carcinoma. Cancer Control 27, (2020).
Sarmento, L. M. et al. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 34, 2978–2990 (2015).
Mikhail et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Wang, H. et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 496, 134–143 (2021).
Hussain, S. F. et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 8, 261–279 (2006).
Caner, A. Immune escape mechanism of Cancer. Curr. Mol. Biol. Rep. 10, 9–19 (2023).
Ge, H. et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int. J. Cancer. 141, 1434–1444 (2017).
Chinyama, H. A. et al. Identification of CDK1, PBK, and CHEK1 as an Oncogenic Signature in Glioblastoma: a Bioinformatics Approach to Repurpose Dapagliflozin as a therapeutic Agent. Int. J. Mol. Sci. 24, 16396 (2023).
Yang, S. et al. MCM4 is a novel prognostic biomarker and promotes cancer cell growth in glioma. Front. Oncol. 12, 1004324 (2022).
Zhou, Q. et al. Correlation of tumor-associated macrophage infiltration in glioblastoma with magnetic resonance imaging characteristics: a retrospective cross-sectional study. Quant. Imaging Med. Surg. 13, 5958–5973 (2023).
Liu, K. H. et al. Fluoxetine, an antidepressant, suppresses glioblastoma by evoking AMPAR-mediated calcium-dependent apoptosis. Oncotarget. 6, 5088–5101 (2015).
Mishra, V. S., Patil, S., Reddy, P. C. & Lochab, B. Combinatorial delivery of CPI444 and vatalanib loaded on PEGylated graphene oxide as an effective nanoformulation to target glioblastoma multiforme: in vitro evaluation. Front. Oncol. 12, 5088–5101 (2022).
Yang, X. et al. TGX-221 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol. Rep. 38, 2836–2842 (2017).
Acknowledgements
-.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.S., M.A.Z. & M.N.H.M. Data curation and processing: A.S., and A.A. Transcriptomic data analysis: M.B.H. and A.S. Molecular docking analysis: A.S., M.B.H., and A.A. Validation & Visualization: A.S. and M.B.H. Writing – original draft: A.S. Writing – review & editing: M.M.H.M., M.A.Z. and M.N.H.M. Project administration & Supervision: M.N.H.M. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sarker, A., Aziz, M.A., Hossen, M.B. et al. Discovery of key molecular signatures for diagnosis and therapies of glioblastoma by combining supervised and unsupervised learning approaches. Sci Rep 14, 27545 (2024). https://doi.org/10.1038/s41598-024-79391-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-79391-2
Keywords
This article is cited by
-
“First, do no harm” in the digital era: examining the practicality of the European Health Data Space proposal and ethical implications of artificial intelligence: A systematic literature review
BMC Medical Ethics (2026)
-
Deciphering RNA and protein expression discordance identifies TOP2A as a prognostic biomarker and potential therapeutic target in lung adenocarcinoma
Discover Oncology (2026)
-
Screening of common genomic biomarkers to explore common drugs for the treatment of pancreatic and kidney cancers with type-2 diabetes through bioinformatics analysis
Scientific Reports (2025)
