
Abstract
In this study, we analysed the mutation spectrum in subjects with suspected lipodystrophy using a targeted Next-generation sequencing (NGS) approach. Subjects with suspected lipodystrophy were for screened six genes (AGPAT2, BSCL2, LMNA, PPARG, ZMPSTE24, INSR) and the variants identified were confirmed through Sanger sequencing. The clinical and biochemical parameters were compared among the mutation positive and negative subjects. We identified eight individuals with pathogenic or likely pathogenic mutations, including both homozygous and heterozygous variants. Homozygous variants included AGPAT2(NM_006412.4):c.493-2A>G, AGPAT2(NM_006412.4):c.254_258dup, and BSCL2(NM_001122955.4):c.570del, while heterozygous variants encompassed LMNA(NM_170707.4):c.1444C>T, LMNA(NM_170707.4):c.1456A>G, LMNA(NM_170707.4):c.1445G>A, and PPARG(NM_015869.5):c.949T>C mutations. In this cohort, three subjects were diagnosed with congenital generalized lipodystrophy, while the remaining five had familial partial lipodystrophy. Majority (7/8) of the patients with lipodystrophy had hepatic involvement. Notably, more than half of the subjects (5/8) achieved optimal glycemic control through insulin sensitizers (PPARγ agonist and Metformin). Interestingly, even with a limited gene panel test, mutation-positive individuals exhibited a higher prevalence of typical clinical features and biochemical characteristics associated with lipodystrophy compared to their mutation-negative counterparts. In subjects with lipodystrophy, targeted NGS based screening may establish a genetic diagnosis and aid in family screening and genetic counselling. Knowing the clinical and biochemical features typical to lipodystrophy may help in diagnosis especially in resource limited setting.
Similar content being viewed by others
Introduction
Inherited lipodystrophy syndromes (LS) are a group of genetically heterogenous disorders characterised by the deficiency of adipose tissue in the absence of nutritional deprivation and a catabolic state. The loss of adipose tissue leads to ectopic fat accumulation in organs such as the liver, muscle and pancreas thereby leading to metabolic dysregulation and its subsequent clinical manifestations1. Although, it is considered a rare genetic disorder with the estimated prevalence of congenital generalised lipodystrophy (CGL) to be 1 in 10 million and that of familial partial lipodystrophy (FPLD) to be 1 in a million1, a recent study from a large clinical care cohort has estimated the prevalence of FPLD to be 1 in 70002. Inherited LS are broadly classified based on the extent of adipose tissue loss into CGL and FPLD3.
Lipodystrophy predisposes to metabolic and cardiovascular comorbidities which include insulin resistance related diabetes mellitus (DM), non-alcoholic fatty liver disease (NAFLD), polycystic ovary syndrome (PCOS), lipid abnormalities, and premature atherosclerosis. The major causes of mortality in LS include coronary artery disease (CAD), chronic liver disease (CLD), chronic kidney disease (CKD), acute pancreatitis, and sepsis4.
To date, there are around 400 cases of genetically proven CGL and more than 500 cases of FPLD that have been described. However, the most commonly described clinical phenotype of LD is based almost entirely on the topography of adipose tissue, and this feature alone lacks the specificity to identify this rare disorder. Therefore, there is a need to identify specific clinical features that point more precisely towards the diagnosis of LS5. The Endocrine Society guidelines have suggested that the diagnosis be based on history, physical examination, body composition, and metabolic status, and eventually genetic testing helps to establish the diagnosis of suspected inherited LS6. The data from the Indian subcontinent on inherited LS is scarce, and is limited to a few case reports7,8,9,10,11. In this article, we describe the clinical features, biochemical parameters, and the pattern of fat distribution among eight genetically proven subjects with inherited LS. We also compared these parameters among the mutation positive subjects and mutation negative subjects with a clinical suspicion of LS.
Methods
In this case series, we present eight subjects with inherited LS from seven kindreds, identified through targeted next-generation sequencing (NGS) of six genes that are implicated in the pathogenesis of insulin resistance forms of diabetes - including two cases already published as individual case reports7,12. The six genes that are included involve AGPAT2, BSCL2, LMNA, PPARG, ZMPSTE24, and INSR. Subjects with diabetes mellitus under care in our department with the any of the following features were enrolled and underwent genetic screening over a 10-year duration (2012–2022).
-
1.
A clinical suspicion of lipodystrophic diabetes (LipD) bearing features of young onset diabetes mellitus (< 35 years of age) and with complete or partial loss of adipose tissue with any of the following features - signs of insulin resistance, consanguineous marriage, family history of a similar illness, hypertriglyceridemia, premature CAD, CKD, CLD, NAFLD, PCOS.
-
2.
High insulin requirements > 2 units/kg/day or > 200 units/day.
Subjects whose autoimmune antibodies (GAD and IA2) were positive and/or those with a history of diabetic ketoacidosis were excluded. This study was approved by the Institutional Review Board (IRB) and Ethics Committee of Christian Medical College, Vellore (IRB Min. No.16154). The study was conducted in accordance with the ethical principles for medical research stipulated by the declaration of Helsinki, 2013. A written informed consent was obtained from all study participants. The clinical and laboratory investigations of these subjects were retrieved from the electronic medical records.
Clinical history and evaluation
A detailed clinical assessment which included the family history, consanguinity, age of onset of diabetes and history of co-morbidities (CKD, CAD, CLD, PCOS) were obtained from the electronic medical records. In addition, the data collected included anthropometry (height, weight, BMI), presence of signs of insulin resistance, pattern of fat loss and fat distribution, presence of signs of lipodystrophy such as acromegaloid or cushingoid appearance, phlebomegaly, hirsutism and features of micro and macrovascular complications of diabetes.
Laboratory evaluation
The laboratory data included fasting blood glucose, HbA1C, C-peptide levels, serum triglycerides and cholesterol, liver function tests (LFT), creatinine and ultrasound of the abdomen to assess the liver morphology (fatty liver). Severe hypertriglyceridemia was defined as a serum triglyceride > 5.6 mmol/l (with or without treatment) or > 2.8 mmol/l (on treatment)6. Deranged LFT was defined as presence of transaminitis (SGOT/SGPT > 3 times the ULN or elevated bilirubin). Metabolic complications were defined as the presence of either of the following – diabetes mellitus, hepatic dysfunction (CLD, deranged LFT, fatty liver grade ≥ 2 on ultrasound), CKD (GFR < 60 ml/1.73m2/min or macroalbuminuria), cardiomyopathy or premature CAD and severe hypertriglyceridemia. Body composition was assessed using a Hologic Discovery A-QDR 4500 DXA (dual-energy X-ray absorptiometry) scanner. Thus, objective data on the pattern of fat distribution was available from the DXA images. The total body fat percentage in centiles was compared with the normative data of body fat in the Asian Indian population for age and gender from the DXA images, published by Marwaha et al13..
Genetic analysis
DNA extraction
A 2 ml whole blood EDTA sample was collected from the subjects, and the automated DNA extraction was carried out using Maxwell® RSC Genomic DNA Kit on Maxwell® 16 MDx Instrument.
NGS workflow
-
a.
Target enrichment: We have designed and utilized a multiplex PCR approach covering the exons & splice site junctions for the panel of six genes (AGPAT2, BSCL2, LMNA, PPARG, ZMPSTE24, INSR).
-
b.
Library preparation: Pooled amplicons were fragmented using Bioruptor® (Diagnenode) followed by end repair, adaptor ligation, and amplification and size-selection using E -gel size select in 2% precast gels.
-
c.
Template preparation: Clonal amplification was carried on OT2 Emulsion PCR (Ion Torrent, Life Technologies) followed by streptavidin dynabead based enrichment in the Ion one-touch enrichment system.
-
d.
Sequencing and Bioinformatic analysis: Sequencing was performed on Ion Torrent PGM using Ion PGM™ 200 Sequencing Kit (Ion Torrent, Life Technologies) using 316 chips. The sequencing data were mapped to the reference hg19 human genome using Torrent Mapping Alignment Program (TMAP). Following Primary analysis on the overall quality of the sequencing run, coverage analysis of the targeted genes was carried out on the Ion torrent server with bed files designed for the targeted genes. The FASTQ files were analyzed using DNASTAR software. The identification of potentially significant variants was based on prevalence as compared to publicly accessible genomic databases (1000Genomes, ExAc, GnomAD, Clinvar& HGMD) in-silico predictions (mutation taster, SIFT, Poly Phen, Varsome based on ACMG guidelines)14and literature review. All variants identified by NGS were confirmed through Sanger sequencing and found no false positives, demonstrating a high level of specificity. Additionally, all target regions were sequenced with a mean coverage of 200x, and 99% of the regions (coding regions and splice site regions) achieved a minimum coverage of 20x, ensuring robust variant detection with high sensitivity. Over the years, we have employed Multiplex PCR coupled with NGS for targeted sequencing, and this strategy has consistently shown high sensitivity and specificity across various gene panels15.
Comparison of mutation positive and mutation negative subjects
The clinical, biochemical features, metabolic complications and body composition data were compared among the eight-mutation positive subjects and 16 mutation negative subjects with a clinical suspicion of LipD (5 subjects with inadequate clinical data were excluded).
Statistical analysis
The data was analyzed using SPSS version 21.0 (IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp). Continuous variables such as age, body fat percentage and serum triglyceride levels were reported using Mean ± SD/ median (IQR); the categorical variables were reported as frequencies and percentages. The association between clinical features, metabolic status and body composition and lipodystrophy diagnosis was studied using chi-square test/Fisher’s exact test and independent t test (whichever being contextually appropriate).
Results
In this study, 29 subjects with young onset diabetes and a clinical suspicion of the presence of insulin-resistant syndromes, underwent comprehensive genetic screening using a 6 gene panel, which have been previously implicated in the etiology of insulin resistant forms of diabetes. The 6 genes included AGPAT2, BSCL2, LMNA, PPARG, ZMPSTE24 and INSR. The mean age at diagnosis of diabetes among the subjects who underwent genetic testing was 24 ± 8.1 years and majority (72%) were females (21/29). The mean BMI of the subjects was 20.4 ± 4.7 kg/m2. Among these subjects, 8/29 (27%) tested positive for a gene mutation in LS genes. Majority of them (7/8) were females.
Subjects with inherited lipodystrophy (mutation positive)
Clinical characteristics
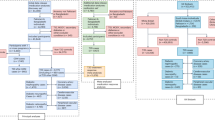
Eight subjects tested positive for gene mutations associated with LS. Three subjects were positive for gene mutations that were associated with CGL and the other five for FPLD. The clinical characteristics of the mutation positive subjects are described in Table 1. LD1 and LD2 were born of consanguineous marriage, however, none of the subjects with CGL had affected family members. All subjects with FPLD had a family history of young onset diabetes and relatives with a similar phenotype. LD4 and LD5 belonged to the same family. The pedigree chart of the eight mutation positive subjects is depicted in Fig. 1.

Pedigree of subjects with inherited lipodystrophy.
Complications of diabetes mellitus
Four out of eight subjects had micro/macroalbuminuria (LD1, LD5, LD6, LD7). LD6 had macroalbuminuria (447 mg/day), LD7 had nephrotic range proteinuria (4610 mg/day) and others had microalbuminuria. One subject (LD7) had proliferative diabetic retinopathy in both eyes and required laser photocoagulation. The same subject had chronic kidney disease in addition. None of the subject had clinical neuropathy (using monofilaments and biosthesiometer testing) or macrovascular complications.
Laboratory parameters
All subjects with LS had hypertriglyceridemia and 7/8 had hepatic steatosis (one subject with FPLD (LD7) had normal liver echotexture). Although all subjects had hypertriglyceridemia only two subjects had severe hypertriglyceridemia Table 1. Six (6/8) patients achieved optimal glycemic control (HbA1c < 7%) with treatment. Seven (7/8) patients with LS had hepatic involvement in the form of hepatic steatosis or chronic liver disease (Table 1).
Treatment
Two subjects with CGL required a high dose of insulin in addition to insulin sensitizers for glycaemic control. Among those with FPLD, only LD5 required high dose insulin therapy. The other subjects with FPLD were managed with oral insulin sensitisers alone. Among the three subjects on insulin, LD2 and LD5 had poor glycaemic control despite higher insulin doses (Supplementary data table S1).
Body composition
All subjects with LS had their total body fat percentage below the 50th centile for the age, gender, and BMI (Supplementary data table S1). Subjects with CGL had a severe reduction in body fat when compared to those with FPLD. However, contrary to the fact that CGL are expected to have a generalised reduction in body fat, all three subjects with CGL had relative sparing of fat in the head and neck. Among subjects with FPLD, all subjects with the LMNA mutation (LD4, LD5, LD6, LD8) had an increased deposition of fat in the neck, and loss from the limbs and trunk, and had associated calf pseudohypertrophy (Fig. 2). One subject with PPARG mutation(LD6) did not demonstrate increased fat deposition in the neck, and pseudohypertrophy of the calf was less prominent than expected (Supplementary data table S2 and Fig. 2).

Comparison of body composition of subjects with LMNA and PPARG muta.
Genetic mutation in our cohort
The mutation types of these subjects are presented in Table 1. LD1 was positive for a homozygous AGPAT2 splice mutation AGPAT2(NM_006412.4):c.493-2A>G, which has been previously reported as likely pathogenic3,16. LD3 was found to carry a homozygous AGPAT2 mutation AGPAT2(NM_006412.4):c.254_258dup, which results in a frameshift at codon 87, followed by a premature termination: p.(Gln87GlyfsTer20). This variant has one submission in ClinVar and was classified as likely pathogenic. LD2 was positive for a novel homozygous BSCL2 deletion, BSCL2(NM_001122955.4):c.570del, leading to a frameshift and premature termination at p.(Leu191TrpfsTer21). While CGL is an autosomal recessive disorder, we did not find any family history of the condition. However, both LD1 and LD2 were born to consanguineous parents. Although CGL is an autosomal recessive disorder, no family history of the condition (suggesting a skipped generation) was identified. However, both LD1 and LD2 were born to consanguineous parents.
All subjects with familial partial lipodystrophy (FPLD), which follows an autosomal dominant inheritance pattern, had a family history of early-onset diabetes and relatives with similar phenotypic traits. LD4 and LD5 were both positive for the previously reported LMNA mutation LMNA(NM_170707.4):c.1444C>T p.(Arg482Trp), which has over 10 submissions in ClinVar and is classified as pathogenic. LD8 tested positive for LMNA mutation LMNA(NM_170707.4):c.1445G>A p.(Arg482Gln), which also has over 10 ClinVar submissions and is classified as pathogenic.
LD6 carried a novel LMNA variant, LMNA(NM_170707.4):c.1456A>G p.(Lys486Glu). The meta-analysis score from multiple in silico tools is 9, with a score > 8 considered strong evidence of pathogenicity. Additionally, pathogenic variants have been reported at the same codon (p.Lys486Asn). Our findings, along with reports of other mutations between codons 482–486, suggest this region may be a mutation hotspot within the LMNA gene. Based on the available evidence, the p.(Lys486Glu) variant is classified as likely pathogenic.
LD7 was found to carry a heterozygous PPARG mutation, PPARG(NM_015869.5):c.949T>C p.(Ser317Pro). This variant has not been reported in gnomAD or ExAC databases. However, there is a single submission in ClinVar, where it is classified as a variant of uncertain significance. Further family screening and additional investigations are needed to better classify this variant.
Family screening
All subjects were given genetic counselling about the mode of inheritance of the suspected disorder and risk of transmission to the next generation. After testing, those with mutation were counselled and given the option for family screening. In those who gave consent, family screening has been done. Three subjects underwent family screening (LD1,4,5 and 8). Both parents and sister of LD1 were found to have heterozygous mutation of the same mutation as LD1. In the second family (LD4,5), mother also carried the same mutation as the subjects and manifested the similar clinical phenotype. Daughter of LD8 also carried the same mutation and had a similar phenotype as the proband, however was euglycemic.
Comparison of characteristics between mutation positive and mutation negative subjects
The clinical and laboratory parameters were compared among the mutation positive subjects and mutation negative subjects with a clinical suspicion of LipD. It was found that the typical clinical features of LS (Grade ≥ 3 acanthosis nigricans, phlebomegaly and muscular appearance) and hepatic dysfunction was more prevalent among the mutation positive subjects compared to the mutation negative subjects (p < 0.05) (Table 2).
Discussion
The study describes the clinical and biochemical profile, the pattern of fat distribution of eight subjects with genetically proven inherited LS.
Although inherited LS, does not have any gender based predisposition, a female predisposition has been noted in literature17. Seven out of eight subjects were females (5/5 FPLD being females). It has been found that women with FPLD are more severely affected with metabolic complications of insulin resistance than men18. This differential manifestation of FPLD in females has been attributed to be due to the glucocorticoid receptor GRβ overexpression or increased proinflammatory cytokines in females19.
Lipodystrophy has been associated with a pre-receptor defect in insulin action20, and subjects generally have relatively higher insulin requirements5. The same pattern has been has been noted in a majority of reported cases4with a few exceptions21,22. In our study group, one subject with CGL and four subjects with FPLD achieved glycaemic control with oral insulin sensitizers alone. Although this may be expected in cases of FPLD, as per the literature, it seldom occurs in cases of CGL21. Whether the benefit of thiazolidinediones extends beyond glycaemic control in FPLD, appears to be a point for discussion, with some studies supporting the improvement in body fat pattern and metabolic profile of patients23,24,25and some studies not showing any impact on these parameters26.
Due to the underlying insulin resistance in LipD, the c-peptide levels are commonly expected to be very high in subjects with LS. Serum c-peptide levels were available for four subjects in our study group and all four had significantly elevated levels (Table 1). However, the level in one subject (LD5) was comparable to the level that may be seen in type 2 diabetes mellitus27, and may have been impacted by glucotoxicity, that was secondary to poor glycaemic control (HbA1C 12%). Seven out of eight patients with inherited lipodystrophy had hepatic involvement. Therefore, when a patient with young onset diabetes presents with hepatic involvement (hepatic steatosis grade ≥ 2/chronic liver disease/ elevated liver enzymes > 5 times ULN) especially with low/normal BMI, LipD should be considered as a differential diagnosis.
All mutation positive subjects had a total body fat percentage of less than the 50th centile for age, gender, and BMI. All subjects with CGL had a generalised loss of body fat. As described before, in our cohort, subjects with LMNA mutations had an accumulation of fat on the face, neck and supraclavicular areas, thereby giving patients a “pseudo-Cushingoid appearance” and calf muscle pseudohypertrophy28,29. This is evident from the body composition data (LD4, LD6, LD8) in Supplementary data table S2 and the DXA image (Fig. 2). Also, LD7 with PPARG mutations have depicted a loss of subcutaneous fat in the extremities and gluteo-femoral region with sparing of facial and nuchal regions28 (Supplementary data table S2 and Fig. 2). The fat distribution in PPARG runs contrary to the description of fat distribution in FPLD2, which is characterised by redistribution of fat from over the trunk and gluteofemoral region to the face and neck. This feature has also been described in the study by Gosseaume et al., wherein they focused on the metabolic and reproductive features of 26 women with LD due to a heterozygous mutation in the PPARG gene. All women had limb lipoatrophy, muscular hypertrophy, android fat distribution, and acanthosis nigricans in this cohort which bears similarity to the phenotype in LD7 and without a pseudocushingoid appearance30.
This study, to the best of our knowledge, is the first case series from India on inherited LS and LipD. Moreover, it comprehensively describes the clinical phenotype with concurrent body composition data. The current study is limited by the fact that the genetic panels did not cover all the known genes involved in the LS spectrum of diseases. However, the existing panel does have the potential to derive the diagnosis in at least 90% of subjects with LS as has been viewed in current published literature31. CGL 1,2 and FPLD 1,2,3 constitute a majority of the reported cases and all these variants, except for FPLD 1 are covered in the current genetic panel. Moreover, the genetic mutations which are associated with FPLD 1 (Kobberling type) is unknown till date. Although some data was not comprehensive in the mutation negative group, patients with a paucity of data have been excluded from the analysis.
Conclusion
In our cohort, all subjects with genetically proven LS had hepatic involvement. Two thirds of the subjects in our cohort with LS, which includes both FLPD and CGL, responded to oral insulin sensitizers. In subjects with LS, targeted NGS based screening may establish a genetic diagnosis and may aid in further family screening and genetic counselling. As the diagnosis of LS is based largely on clinical and body composition data, knowing the clinical and biochemical features typical to LS may help in diagnosis especially in resource limited setting where genetic testing is not widely available.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Agarwal, A. K. & Garg, A. Genetic disorders of adipose tissue development, differentiation, and death. Annu. Rev. Genom Hum. Genet. 7 (1), 175–199 (2006).
Gonzaga-Jauregui, C. et al. Clinical and molecular prevalence of Lipodystrophy in an unascertained large clinical care cohort. Diabetes. 69 (2), 249–258 (2019).
Agarwal, A. K. et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab. 88 (10), 4840–4847 (2003).
Fourman, L. T. & Grinspoon, S. K. Approach to the patient with Lipodystrophy. J. Clin. Endocrinol. Metabolism. 107 (6), 1714–1726 (2022).
Angelidi, A. M., Filippaios, A. & Mantzoros, C. S. Severe insulin resistance syndromes. J. Clin. Invest. 131 (4), e142245 (2021).
Brown, R. J. et al. The diagnosis and management of Lipodystrophy syndromes: a Multi-society Practice Guideline. J. Clin. Endocrinol. Metabolism. 101 (12), 4500–4511 (2016).
Asha, H. S., Chapla, A., Shetty, S. & Thomas, N. Next-generation sequencing-based genetic testing for familial partial lipodystrophy. AACE Clin. Case Rep. 1 (1), e28–31 (2015).
Joshi, R. & Sharma, S. Berardinelli Seip congenital lipodystrophy syndrome: 10 year follow-up. Indian Pediatr. 56 (10), 877–878 (2019).
Mandal, K., Aneja, S., Seth, A. & Khan, A. Berardinelli-Seip congenital lipodystrophy. Indian Pediatr. 43 (5), 440–445 (2006).
Kannan, S., Khanna, I., Kaur, M. & Radin, M. Visual vignette. Type 2 familial partial lipodystrophy syndrome of the Dunnigan variety. Endocr. Pract. 17 (4), 665 (2011).
Chakraborty, P. P., Datta, S., Mukhopadhyay, S. & Chowdhury, S. Pseudoacromegaly in congenital generalised lipodystrophy (Berardinelli-Seip syndrome). BMJ Case Rep. 2016, bcr2016214493 (2016).
Shetty, S., Chapla, A., Kapoor, N., Thomas, N. & Paul, T. V. A novel variant of the AGPAT2 mutation in generalized congenital lipodystrophy, detected by next generation sequencing. Australas Med. J. (Online). 9 (6), 164 (2016).
Marwaha, R. K. et al. Normative data of body fat mass and its distribution as assessed by DXA in Indian adult population. J. Clin. Densitom. 17 (1), 136–142 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424 (2015).
Chapla, A. et al. Maturity onset diabetes of the young in India–a distinctive mutation pattern identified through targeted next-generation sequencing. Clin. Endocrinol. 82 (4), 533–542 (2015).
Ceccarini, G. et al. Congenital generalized lipoatrophy (Berardinelli-Seip syndrome) type 1: description of novel AGPAT2 homozygous variants showing the highly heterogeneous presentation of the disease. Front. Endocrinol. 11, 39 (2020).
Costa-Riquetto, A. D. et al. Targeted massively parallel sequencing for congenital generalized lipodystrophy. Arch. Endocrinol. Metab. 64 (5), 559–566 (2021).
Garg, A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 85 (5), 1776–1782 (2000).
Resende, A. T. P. et al. Phenotypic diversity and glucocorticoid sensitivity in patients with familial partial lipodystrophy type 2. Clin. Endocrinol. (Oxf). 91 (1), 94–103 (2019).
Golden, M. P. et al. Insulin resistance in total lipodystrophy: evidence for a pre-receptor defect in insulin action. Metabolism. 34 (4), 330–335 (1985).
Chaves, C., Chaves, M., Anselmo, J. & César, R. Successful long-term use of pioglitazone in Berardinelli–Seip lipodystrophy-associated diabetes. Endocrinol. Diabetes Metab. Case Rep. 2021, 20–0183 (2021).
Moreau, F. et al. Efficacy of pioglitazone in familial partial lipodystrophy of the Dunnigan type: a case report. Diabetes Metab. 33 (5), 385–389 (2007).
Iizaka, T. et al. Clinical characteristics and efficacy of pioglitazone in a Japanese patient with familial partial lipodystrophy due to peroxisome proliferator-activated receptor γ gene mutation. Endocr. J. 70 (1), 69–76 (2023).
Sleilati, G. G., Leff, T., Bonnett, J. W. & Hegele, R. A. Efficacy and safety of pioglitazone in treatment of a patient with an atypical partial lipodystrophy syndrome. Endocr. Pract. 13 (6), 656–661 (2007).
Agostini, M. et al. A Pharmacogenetic Approach to the treatment of patients with PPARG mutations. Diabetes. 67 (6), 1086–1092 (2018).
Simha, V., Rao, S. & Garg, A. Prolonged thiazolidinedione therapy does not reverse fat loss in patients with familial partial lipodystrophy, Dunnigan variety: Letter to the Editor. Diabetes, Obesity and Metabolism. ;10(12):1275–6. (2008).
Deep, H. S., Singh, B. P. & Singh, S. P. Evaluation of serum c-peptide levels in type 2 diabetics in Punjabi population. Int. J. Adv. Med. 4 (4), 1026–1030 (2017).
Bagias, C., Xiarchou, A., Bargiota, A. & Tigas, S. Familial partial lipodystrophy (FPLD): recent insights. Diabetes Metab. Syndr. Obes. 13, 1531–1544 (2020).
Meral, R. et al. Fat shadows from DXA for the qualitative Assessment of Lipodystrophy: when a picture is Worth a Thousand numbers. Diabetes Care. 41 (10), 2255–2258 (2018).
Gosseaume, C. et al. Perinatal, metabolic, and reproductive features in PPARG-related lipodystrophy. Eur. J. Endocrinol. 188 (3), 273–281 (2023).
Akinci, B., Meral, R. & Oral, E. A. Phenotypic and genetic characteristics of Lipodystrophy: pathophysiology, metabolic abnormalities, and comorbidities. Curr. Diab Rep. 18 (12), 143 (2018).
Funding
The study has been supported by a grant from Department of Biotechnology, Government of India (ref. no: BT/PR26420/MED/12/774/2017).
Author information
Authors and Affiliations
Contributions
Conceptualization: N.T, A.C, R.R.,H.S.A.; Data curation: R.R.; Formal analysis: R.R., N.T., A.C., H.S.A., F.J., T.P., N.K., ; Investigation: J.J., D.V., ; Methodology: R.R., N.T., A.C., H.S.A., F.J., T.P., N.K.; Project administration and resources: A.C., J.J., D.V., N.T., R.R.; Supervision: N.T., A.C.; Writing original draft: R.R.; Writing review and editing: N.T., A.C., H.S.A., F.J., T.P., N.K.; Funding acquisition: N.T., A.C.; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rajan, R., Chapla, A., Johnson, J. et al. A series of genetically confirmed congenital lipodystrophy and diabetes in adult southern Indian patients. Sci Rep 14, 28277 (2024). https://doi.org/10.1038/s41598-024-79516-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-79516-7
