
Abstract
Liver cancer is globally the most frequent fatal malignancy, and its identification is critical for making clinical decisions about treatment options. Pathological diagnostics and contemporary imaging technologies provide insufficient information for tumor identification. Hydrogen peroxide (H2O2), an emerging biomarker is a powerful oxidant found in the tumor microenvironment, and stimulates the invasion, proliferation, and metastasis of liver cancer cells. This study describes a medically effective and sensitive electrochemical sensor based on ascorbic acid immobilized zinc selenide nanoparticles (AsA@Zn-Se NPs) decorated on a glassy carbon electrode (GCE) for determining H2O2 in PBS and human serum samples of liver cancer patients. The morphological and structural characterization of fabricated sensor is done by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), ultraviolet-visible (UV-Vis) spectroscopy, energy dispersive X-ray (EDX), and scanning electron microscopy (SEM). Ascorbic acid (AsA), an antioxidant for H2O2 redox behavior, is immobilized on Zn-Se NPs to aid H2O2 detection through cyclic voltammetry (CV). The sensor exhibits a low detection limit, and board linear range of 0.49 µM and 0–70 µM, respectively. The low-cost electrochemical sensor is robust for up to 100 cycles. Elecys AFP assay results validate that increasing alpha-fetoprotein (AFP) concentration, a biomarker for liver cancer, can increase the H2O2 levels in serum samples. Therefore, the proposed sensor can be used to diagnose liver cancer in clinical settings.
Similar content being viewed by others

Introduction
Malignant tumors are health risk for human beings worldwide. About 15.2 million people are diagnosed with malignant tumors annually. Various challenges hinder their earlier diagnoses and thus lead to surgical procedures1. Magnetic resonance imaging (MRI), biofluid-based analysis, and mass spectrometry (MS) are the available techniques however they are expensive options. Rapid pathology and frozen sectioning are fast techniques but require trained pathologists. These limitations raise the need for developing cheaper, more accurate, and faster techniques for cancer diagnosis1, resulting in lower mortality rates and less morbidity.
The alterations of nucleic acid and small biomolecules in normal and malignant tissues provide information regarding disease and physiology. The elevated concentrations of free radicals are reported in metabolically active cells to change cellular metabolism and cause metastasis and malignancy. H2O2 is a stable reactive oxygen species (ROS) involved in oxidative damage, intracellular signaling, cellular physiology, host defense, protein folding, and disease progression. The cancer progression is associated with abnormal H2O2 levels. Malignant cells exhibit increased H2O2 levels compared to healthy cells, and lead to cancer cells metastasis and proliferation. H2O2 dissociation leads to formation of hydroxyl free radicals (.OH−), which are extremely destructive towards lipids, nucleic acids, amino acids, and carbohydrates. It is an effective biomarker for earlier onco-treatment1,2,3,4,5. The exact H2O2 concentration remains unsettled, but likely the amount in healthy individuals is < 10 µM in plasma and blood, however levels elevate to 50 µM in unhealthy individuals5,6.
Hydrogen peroxide (H2O2) is involved in the pathogenesis of liver cancer, prostate cancer, and smoking associated lung cancer7. Hepatocarcinogenesis involves variety of inducers, having different modes of cytotoxic effect and cellular targets, but formation of ROS is their common denominator. Liver cancer is one of the most malignant human cancers globally, and studies report that imbalance in antioxidant defense system leads to alleviation in its property to scavenge ROS i.e., H2O2 and O2.−8. Overproduction of free radicals or ROS, lead to initiation, and progression of liver damage, because of the severe oxidative stress these species induce. H2O2 lower concentration, and reactivity make its detection difficult in normal physiological conditions, and diseased state9. H2O2 levels abnormally elevate in tumor microenvironment or tumor cells10.
Flow cytometry, titrimetric analysis, high-pressure liquid chromatography, colorimetry, chemiluminescence, and fluorometry are employed for in vitro H2O2 determination, however complex sample preparations, matrix interferences, short-shelf-time, and low sensitivities hamper their routine use. Commercial kits are used in clinical immunoassays but with limitations. Electrochemical sensors have a number of merits i.e., miniaturization, fast detection time, cost-efficacy, direct measurability, and high selectivity. Sensors provide sensing interface between electrodes and cells for enhancing suitability of screening H2O2 secreted from tumors1,4,6,11,12. H2O2 sensors are in demand, and are being studied by a number of researchers. Thus, it is necessary to fabricate sensors having good reproducibility, high stability, rapid response rate, and high sensitivity13.
H2O2 electrochemical sensors employ enzymes like glucose oxidase and horseradish peroxidase (HRP), however they have strict environmental requirements, complicated preparation procedures, get deactivated easily, have poor chemical stability, are expensive1,2,3,4,14, and due to indirect electron transfer have lower sensitivity5. Non-enzymatic sensors, rely on nanostructures, conducting polymer composite, carbon-based nanoparticles, metal nanoparticles, metal oxides, and metal organic frameworks can overcome the defects of enzyme-based sensors due to their remarkable properties i.e., low-cost electrodes, fast response time, high stability, longer shelf-life, enhanced surface renewability, and low detection limits. Thus, are on-demand in modern research5,15,16.
Antioxidants are classed as enzymatic including superoxide dismutase (SOD), glutathione reductase (GRD), glutathione peroxidase (GSHPx), catalase (CAT), and non-enzymatic i.e., selenium, vitamin E, ascorbic acid, etc. The former ones function by scavenging and decomposing free radicals, whereas the latter through chain reactions of free radicals17. Ascorbic acid (AsA) being an antioxidant scavenges toxic free radicals from cellular metabolism, and prevents mental illness, cold, scurvy, acquired immunodeficiency syndrome etc18. It protects the organisms from deterioration, and harmful effects of oxidative stress. It is an effective ROS scavenging agent used in biological processes19.
Selenium (Se) as a semiconductor with photoelectric characteristics is employed in number of fields i.e., photographic exposure meters, solar cells, rectifiers, xerography, and sensing devices15. Se is a free radical scavenger with high catalytic activity20. Various studies have discussed the Se role in anticarcinogenic activities15. Nanomaterials are ideal electrode materials because of applied potential window, large surface area, flexibility, and low charge transfer resistance12. Particularly, metal nanoparticles are reported in literature for H2O2 detection because they not only favor the phenomenon of charge transfer but also enhance the catalytic sites, due to their excellent electrocatalytic activity, high surface to volume ratio, and outstanding electrical conductivity. According to recent literature, Ag-Cu alloys21, Fe3O4/3DGr NCs13, AuNPs-UiO-6622, Co3O4-ATNTs23, MOF-Au@Pt nanflowers, NdFeO324, core shell Fe3O4@CuAlNSs25, have been employed for H2O2 detection in different matrices considering its versatile applications. The inorganic materials utilized for H2O2 detection include Pd/PEDOT, carbon-based nanocomposite, Pd NPs on graphene oxide nanosheets, Se/Pt, TiO2, polyethyleneimine-capped Ag nanoclusters, and CuO nanoflowers11.
In different real samples i.e., urine, cancer cells and human serum, Asif et al.,25 evaluated the electrocatalytic activity of H2O2, via core shell Fe3O4@CuAl nanospheres. The enhanced electrocatalytic reduction of H2O2 was result of LDHs p-type semiconductive channels, and Fe3O4@CuAl NSs abundant active sites. Similarly, another nanosphere based sensing probe comprising of CuO@MnAl LDHs, had been fabricated for analyzing H2O2 reduction behavior26. Through corrosion engineering strategy, iron substrates were converted into Cu-Fe (OH)2-FeS/PCF electrode for the in vitro and in situ determination of H2O2 in human brain normal and tumor cells27. ß-Mo2C/C/GCE was fabricated and employed by Li et al.,28 for the non-enzymatic electrochemical quantification of H2O2 in pretreated human serum samples.
Herein, we prepare enzyme-free sensor based on Asa@Zn-Se NPs for H2O2 detection in synthetic standard buffer solutions and real serum samples of liver cancer patients. The determination of H2O2 via fabricated nanoparticles modified on glassy carbon electrode could assist in earlier prognosis of liver cancer. The electrochemical analysis via cyclic voltammetry was done to attain comprehensive data regarding H2O2 detection. The selectivity, sensitivity and limit of detection were evaluated to access the potential of fabricated sensor. The higher surface area of ZnSeNPs, along with immobilized ascorbic acid proved to be effective combination for H2O2 sensing. The immobilized AsA has electrocatalytic activity towards H2O2 reduction. The relationship of oxidative stress marker i.e., H2O2, and tumor marker i.e., AFP, is thus explored to monitor the liver cancer. The AFP level in liver cancer patients is determined by employing Elecsys Total Alpha-feto Protein (AFP) Assay. Our research study, provides possibility to synthesize enzyme-free electrochemical sensor to detect analytes present in trace amount in the living organisms.
Experimental
Materials used in synthesis
Zinc acetate dihydrate (Zn(CH3COO)2.2H2O, ≥ 99%), selenium oxide (SeO2, ≥ 98.9%), trisodium citrate (Na3C6H5O7, ≥ 99.9%), sodium borohydride (NaBH4, ≥ 99.6%), ethanol (C2H5OH, ≥ 99%), and sodium hydroxide (NaOH, 97%) were procured from Sigma Aldrich (USA). The solutions for nanocomposites’ fabrication were prepared in distilled and deionized water. The mentioned precursors were of laboratory grade, and employed without further purification.
Preparation of zinc oxide (ZnO) NPs
The chemical reduction method was used to synthesize ZnO NPs from Zn (CH3COO)2.2H2O by following a recent study with small modifications29. Zn (CH3COO)2.2H2O (1.5 g) was added in ultrapure water (50 mL), followed by heating at 50 °C under constant stirring for 30 min. The obtained homogeneous solution was reduced by Na3C6H5O7 (10 mL). The solution was further stirred for 2 h until milky composite was obtained. The obtained white product was centrifuged for 30 min at 5000 rpm followed by washing with EtOH and vacuum dried at 60 °C for 3 h. ZnO NPs were then annealed at 480 °C for 4 h.
Preparation of selenium (Se) NPs
Se NPs were also prepared by the chemical reduction method. Aqueous SeO2 (2 g, 50 mL) was heated for 2 h, at 60 °C. NaBH4 (10 mL) was used for reduction and was added slowly under continuous stirring in cold environmental condition. SeO2 solution was dropwise added into NaBH4 solution. The resultant solution was centrifuged and obtained Se crystals were washed with EtOH and water to eliminate unreacted precursors, and oven dried at 60 °C.
Preparation of zinc selenide (Zn-Se) NPs
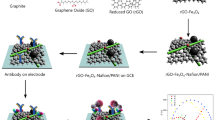
The chemical precipitation method was employed to fabricate bimetallic NPs (Fig. 1). The reaction mixture of Se NPs, and ZnO NPs in 3:2 was dissolved in 30 mL distilled water, under vigorous stirring for 2–3 h. NaOH (10 mL) was added dropwise for precipitation, and to maintain pH of solution. The ZnSeNPs were precipitated and collected by centrifugation. They were washed with distilled H2O and EtOH several times to remove impurities and unreacted products and dried for several hours at specific temperature (around 60–80 °C).

Representative diagram for the fabrication of ZnSeNPs (the green color corresponds to pristine SeNPs, and spherical purple-red network depicts pristine ZnONPs. Whereas, the resulting ZnSeNPs show both green and purple-red network, indicating the formation of ZnSeNPs from chemical precipitation method).
Fabrication of working electrode
To activate the electrode surface, and to remove contaminants, the bare working electrode was polished using alumina powder suspension, and polishing cloth. It was then rinsed by ultrapure water by employing sonicator. Zn-Se NPs (5 mg), and ascorbic acid (5 mg) were mixed in deionized water in 1:1 (w/w) to produce homogeneous paste for bare GCE, and was deposited via micropipette and dried. H2O2 solutions of varying concentrations and pH were prepared. The prepared electrode had a shelf life of six weeks.
Electrochemical investigation of hydrogen peroxide (H2O2) by AsA@Zn-Se NPs
Potentiostat (PG-STAT) PGSTAT302N electrochemical system was comprised of three electrodes i.e., working (AsA@Zn-Se NPs/GCE), reference (Ag/AgCl), and auxiliary (Pt wire) in a glass cell (volume ~ 100 mL). The redox behavior of AsA@Zn-Se NPs for H2O2 detection was analyzed by CV.
Serum sample collection and processing
Blood samples of liver cancer patients were collected after obtaining written informed consent from all individuals. Blood was centrifuged for 10 min at 4000 rpm to obtain serum in serum collection tubes. The protocols were approved by the Ethical Committee of MINAR Cancer Hospital Multan, Pakistan. In addition, all methods were performed in accordance with the relevant guidelines and regulations of the Ethical Committee of MINAR Cancer Hospital Multan, Pakistan.
Elecsys total alpha-feto protein (AFP) assay
The total Elecsys AFP assay was used to evaluate AFP levels in biofluids. It was based on two monoclonal antibodies i.e., AFP-ACT, and unbound AFP from Fujirebio Diagnostics. Blood of five liver cancer patients was collected in sample collection tubes. Detection of AFP antigens were made via sandwich ELISA, and procedure was carried for 18 min. The first incubation used 6 µL sample. Biotinylated monoclonal AFP specific antibody (mAb-AFP-1) (1 mg/mL) preserved in phosphate buffer ruthenium complex (100 mmol/L, pH 7.4) formed the sandwich complex with monoclonal AFP-specific antibody. In second incubation, 0.72 mg/mL streptavidin coated microparticles were attached to the solid phase via streptavidin and biotin interaction. This bound mixture was aspirated to measuring cell, followed by magnetic capturing of microparticles on electrode surface. The Procell/Procell M was employed to eliminate unbound substrate. Chemiluminescence was produced by providing voltage to the electrode. The emission intensity was measured by photomultiplier detector, and an automatic two-point calibration curve was created.
Results and discussion
Characterization of synthesized NPs
IR spectra (Fig. 2A) are obtained on INVENIO FTIR Spectrometer from Bruker Germany, to identify chemical structure of synthesized material. The peak at 2918 cm− 1 represents -NH2. Peak at 615 cm− 1 is attributed to Zn-Se bend30. Peaks at 1712 cm− 1, and 1250 cm− 1 correspond to C=O, and C-N, respectively31.
X-ray diffraction (XRD) study is conducted on Bruker D8 Advance Powder Diffractometer. XRD spectra are shown in Fig. 2B. Peaks at 47.5°, 56.1°, 61.6°, 65.1°, and 67.9° have hkl planes of (102), (110), (103), (200), and (112), respectively. These patterns align with the known patterns of ZnO 36-1451. Zn-Se NPs exhibit additional peaks at 23.3°, 29.4°, 41.3°, 43.5°, 45.2° and 51.7°, corresponding to hkl values of (100), (101), (110), (102), (111), and (201), respectively. These peaks coincide with standard pattern of Se 01-0848. The low intensity peak of Se is found at 23.3° which arises from untreated Se in Zn-Se NPs.
The crystallite size is calculated by Sherrer formula:
λ indicates X-rays wavelength, θ the incident angle, and β depicts the peak width at half maximum. The average crystallite size of Zn-Se NPs is 8.7 nm. Sharp peaks indicate NPs crystallinity. The value is comparable with those reported in literature32.
AQ7100APAC Thermo Fischer Scientific UK Spectrophotometer records the absorptions of Se NPs, ZnO NPs, and Zn-Se NPs in 200–800 nm range (Fig. 2C). Sharp absorption at 255 nm shows formation of Se NPs. ZnO NPs absorb at 207 nm. A shoulder peak at 267 nm is related to the electron transfer from the valence to conduction band. Zn-Se NPs show peak at 213 nm, and shoulder peak at 266 nm. The addition of Se slightly shifts the peak towards right. Band gap is measured by the following equation:
The observed wavelengths are 332 nm, 287 nm, and 280 nm for Se NPs, ZnO NPs, and Zn-Se NPs, respectively. The calculated band gaps are 3.7 eV, 4.3 eV, and 4.4 eV for Se NPs, ZnO NPs, and Zn-Se NPs, respectively. There is no notable change in the band gaps. According to a reported study, the band gap of 2.99 eV33, is observed for Zn-Se NPs, which is smaller than our calculated one, indicating the successful formation of nanoparticles having higher conductivity rate, enhancing its applicability to be employed as sensor. Energy dispersive X-ray spectroscopy (EDX) analysis exhibit the elemental make-up of Zn-Se NPs (Fig. 2C). The material exhibits six peaks, three for Zn at 1, 8.7, and 9.6 keV, and three for Se at 1.5, 11.3, and 12.5 keV.

(A) FTIR, (B) XRD, and (C) UV of Se NPs, ZnO NPs, and Zn-Se NPs, and (D) EDX of Zn-Se NPs.
Scanning electron microscopic (SEM) images (JSM-7200 F JEOL Japan) are shown in Fig. 3. Se NPs have uniform and homogeneous hexagonal morphology, similar to a reported study32. Its analysis by Image J software shows 50–110 nm diameter with average particle size of 80.5 nm. SEM image of ZnO NPs shows clusters from 50 to 110 nm with average particle size of 75.6 nm. SEM image of Zn-Se NPs depicts uniform geometry with good distribution. The average particle size is 39.8 nm, while literature suggests the range from 2 to 50 nm, depending on the synthetic technique. The obtained size distribution range is from 30 to 60 nm, which is in accordance to reported range of ZnSeNPs (~ 40–60 nm)33.

(A), (C), and (E) SEM images of Se NPs, ZnO NPs, and ZnSe NPs. (B), (D), and (E) Particle size distributions of Se NPs, ZnO NPs, and ZnSe NPs, respectively.
Electrochemical evaluation of AsA@Zn-Se/GCE
The immobilization and electrochemical behaviors of all fabricated electrodes i.e., Se NPs, ZnO NPs, Zn-Se NPs, and AsA@Zn-Se NPs are assessed by computer-controlled in 0.1 M KCl, and 0.1 mM K4[Fe(CN)6] solutions. The cyclic voltammograms in Fig. 4A show redox behavior of modified electrodes. AsA@Zn-Se NPs modified GCE displayed prominent current response, whereas other modifiers generate negligible current response. A broad oxidation peak is found at + 0.3 V, and reduction peak at + 0.15 V. The maximum conductivity is yielded by AsA@Zn-Se/GCE. Varying scan speeds i.e., 10–70 mV/s are tested regarding the redox action of fabricated electrode. The peak potential shifts to more positive upon increasing the scan rate (Fig. 4B). 30 mV/s is the optimum value to provide reasonable time and avoid mass transport limitations.
Stability of AsA@Zn-Se/GCE
Stability is assessed by setting 100 cycles of CV in 0.1 M KCl, and 0.1 mM K4[Fe(CN)6] solutions. Voltammograms are shown in Fig. 4C where reduction and oxidation peaks after 100 cycles on AsA@Zn-Se/GCE are obtained at the same potential as of first cycle. The steady activity is evaluated by monitoring amperometric response at 0.01 V for 9 h. The chronoamperometric curve is shown in Fig. 4D to confirm the stability.
Electrochemical active surface area (ECSA), and roughness factor (rf) of AsA@Zn-Se/GCE
A 1:1 ratio of 0.1 mM K4[Fe(CN)6], and 0.1 M KCl is used to evaluate AsA@Zn-Se/GCE regarding surface area. CV analysis is carried out at scan rates from 10 to 60 mV/s (Fig. 4E). Randles-Sevcik equation is used to compute ECSA of AsA@Zn-Se modified GCE, and bare GCE. The calculated values are 0.44 cm2 and 0.073 cm2, respectively. The ECSA values are acquired by the line graph illustrated in Fig. 4F. ECSA of AsA@Zn-Se/GCE shows moderate surface area compared to bare GCE.
Roughness factor (Rf) is measured by computing peak current (Ip.a.) of [Fe(CN)6]−3/−4 via redox probe corresponding to blank electrode. The amount of reduction and oxidation cores specified by electrochemical process determine the factor, and electrode dimension. Surface determine the cores34. The oxidation peaks, and surface areas of modified and bare GCE determine the Rf factor. The alteration in electrode´s surface area is depicted by current ratio for bare and modified electrodes. Here is the equation:
Peak currents for bare and AsA@Zn-Se modified GCE are presented by Ip2, and Ip1, respectively. The bare GCE surface area (A1), and AsA@Zn-Se modified GCE (A2) are 0.073 and 0.44 cm2, respectively. The measured Rf is 6.02.

(A) Cyclic voltammograms representing electrical conductivities of bare GCE, Se NPs/GCE, ZnO NPs/GCE, Zn-Se NPs/GCE, and AsA@Zn-Se/GCE, (B) effect of varying scan rates on AsA@Zn-Se/GCE redox activity, (C) cyclic voltammograms representing stability of AsA@Zn-Se/GCE up to 100 cycles, (D) chronoamperometry of AsA@Zn-Se/GCE in the presence of redox probe, (E) cyclic voltammograms representing ECSA of AsA@Zn-Se/GCE at varying scan rates in 0.1 mM K4[Fe(CN)6] containing 0.1 M KCl, and (F) corresponding calibration plot of ECSA.
Electrochemical performance of AsA@Zn-Se/GCE for H2O2 sensing
We looked into the electrochemical capabilities of AsA@Zn-Se/GCE for H2O2 detection. The effect of varying H2O2 concentrations including 10, 20, 30, 40, 50, 60, and 70 µM in PBS (0.1 M, pH 7) is evaluated via CV (Fig. 5A). The voltammogram depict increase in oxidation peak current with increase in H2O2 concentration. The peak current is observed at + 0.4 V. The least and maximum oxidation peak currents are observed at 10 µM and 70 µM, respectively. The current responses are due to electrocatalytic properties of AsA@Zn-Se. The calibration plot for linearity check between the concentration of H2O2, and peak current revealed that this enhanced current response was linear, and have linear dependency of R2 = 0.957 (Fig. 5B). The redox activity of AsA@Zn-Se modified GCE for H2O2 detection is also evaluated in pH solutions i.e., 6.6, 7.0, 7.2, 7.4, 7.6, 7.8, and 8.0 (Fig. 5C). AsA@Zn-Se/GCE shows good detection at pH 7.4 which is appropriate for variety of analytes. Figure 5D represents the calibration plot between pH and current.

(A) Cyclic voltammograms illustrating the effect of varying H2O2 concentrations i.e., 10–70 µM in PBS solution (pH 7.4), (B) line graph at varying H2O2 concentrations versus peak current, (C) influence of pH 6.8-8.0 on electrochemical behavior of AsA@Zn-Se for H2O2 in PBS (0.1 M), and (D) calibration plot at different pH versus peak current.
Mechanism of H2O2 electrochemical sensing
AsA@ZnSe assist the redox reaction of H2O2 at GCE surface. It favors the electrochemical determination due to its catalytic properties, and elevation in the number of active sites for hydrogen peroxide detection. The enhanced electrocatalytic activity is due to nanostructured morphology of ZnSe NPs, increasing efficiency of electron transfer between electrode surface and synthesized material. Several processes occur at electrodes to sense H2O2. Equations A, B, and C show Zn-Se reacting with K4[Fe(CN)6] to achieve electrical conductivity via oxidation. Zn-Se breaks into Zn2+ and Se2+, and Fe3+ converts to Fe2+. Zn-Se is immobilized by ascorbic acid to achieve [C6H8O6]Zn−Se+ (D). Ascorbic acid (C6H8O6) undergoes self-oxidation in K4[Fe(CN)6] presence and converts to dehydroascorbic acid (C6H6O6) (E). Equation (F) depicts the oxidation reaction of H2O2 occurring at electrode surface yielding oxygen. The increase in concentration of H2O2, lead to enhanced oxidation current. In electrochemical H2O2 detection, 2 e− reduce H to generate two H2O molecules, (G) and (H). The overall mechanism indicates complete H2O2 detection (G).
Oxidation at Zn-Se electrode:
Reduction by ferrocyanide ions:
Overall reaction facilitated by K4[Fe(CN)6] as catalyst:
Immobilization of ascorbic acid on Zn-Se:
Self-oxidation of ascorbic acid:
Hydrogen peroxide (H2O2) oxidation at electrode:
Hydrogen peroxide (H2O2) reduction at electrode:
Overall reaction at electrode surface:
Electrochemical impedance spectroscopic (EIS) analysis
EIS assesses electrode response at varying pH, and concentration. Charge transfer resistance (Rct) at electrode surface is determined by the probe kinetics. Nyquist curves is plotted in the presence of redox probe vs. AgCl/AgCl/3 M KCl to examine Rct parameter. Nyquist diagrams for bare GCE, Se/GCE, ZnO/GCE, and AsA@Zn-Se/GCE in the presence of 0.1 M KCl and 0.1 M K4[Fe(CN)6] are shown in Fig. 6A. Randles equivalent circuit uses Nyquist plot to extract quantitative data from impedance measurements. We observe a wide semi-circle for bare GCE, thus large charge-transfer kinetics is expected. Whereas, AsA@Zn-Se/GCE displayed extremely low semi-circle as compared to SE/GCE, and ZnO/GCE, thus indicate smaller charge transfer resistance. Rct for bare GCE, SE/GCE, ZnO/GCE, and AsA@Zn-Se/GCE are 23.4 kΩ, 3.4 kΩ, 2.4 kΩ, and 1.2 kΩ, respectively. These outcomes show that AsA@Zn-Se modified GCE has the maximum transfer of electrons indicating maximum conductivity with minimum resistivity.
Figure 6B illustrates how concentration affects impedance. H2O2 concentration improves electron transfer resistance on electrode due to electrostatic interactions which lead to better analyte detection. Rct values in maximum (70 µM), and minimum (10 µM) concentrations are 13.4 kΩ and 4.9 kΩ. Figure 6C depicts the impedance measured at varying pH including 6.8, 7.0, 7.2, 7.4, 7.6, 7.8, and 8.0. The impedance is lowest at pH 7.4 with Rct value of 0.24 kΩ, while Rct fluctuates with pH. The highest resistance is recorded at pH 8.0 with 8.6 kΩ.

Electrochemical impedance of (A) bare GCE, Se/GCE, ZnO/GCE, and AsA@Zn-Se/GCE, (B) varying H2O2 concentrations, and (C) varying pH at 70 µM H2O2 concentration.
Heterogeneous electron transfer rate constant (ko)
EIS investigates the charge transfer at surface of electrode. Electrochemical impedance behavior is measured across many electrodes in the same circuit. The semi-circle diameter at electrode interface detects Rct by measuring electron transfer kinetics from redox probe. Rct of AsA@Zn-Se modified GCE and bare GCE are 1.2 kΩ and 23.4 kΩ, respectively. AsA@Zn-Se works by increasing the rate of electron transport. Electrochemical characteristics show that AsA@Zn-Se/GCE outperforms bare electrode for the electron transport kinetics due to increased surface area.
EIS is used to calculate standard heterogeneous rate constant (ko):
R represents gas constant (8.314 J K− 1 mol− 1), T depicts the thermodynamic temperature (298.15 K), F the Faraday constant (96,458 C mol− 1), Rct the electron transfer resistance (1.2 kΩ and 23.4 kΩ for AsA@Zn-Se/GCE and bare GCE, respectively), and A the electrode surface area (0.073 and 0.44 cm2 for bare GCE and AsA@Zn-Se/GCE, respectively), and C the concentration of [Fe(CN)6]3−/4− solution. The calculated ko values for AsA@Zn-Se/GCE and bare GCE are 4.84 × 10− 5, and 1.49 × 10− 5 respectively. Higher ko values suggest faster equilibrium and more electron transfer.
Test of analytical parameters i.e., reproducibility and interference studies of AsA@Zn-Se/GCE for H2O2 sensing
Reproducibility is studied by fabricating three independent electrodes for cyclic voltametric detection of H2O2 at optimum scan rates, and in selected potential window (Fig. 7A). The studied parameter depicts excellent reproducibility. The selectivity of fabricated sensor for H2O2 detection is assessed by analyzing various compounds i.e., cysteine, lysine, tyrosine, glucose, ascorbic acid, uric acid, and arginine (Fig. 7B). The interference studies depict that no interferent produce noticeable current change, therefore current response to H2O2 remain unaffected, indicating exceptional selectivity of fabricated sensor.

(A) Reproducibility of AsA@Zn-Se/GCE for H2O2 analyzed via cyclic voltammetry. (B) Interference plot for H2O2 detection with interfering agents analyzed via cyclic voltammetry.
Limit of detection and sensitivity of fabricated sensor
The limit of detection, and sensitivity of AsA@Zn-Se/GCE for H2O2 was calculated by employing the equations mentioned below:
where S indicates slope, and σ indicates standard error or standard deviation.
The LOD, and sensitivity of AsA@Zn-Se/GCE for H2O2 determination via electrochemical approach was recorded as 0.49 µM, and 1.23 × 10− 3 A/(mol/L/cm2), respectively.
H2O2 detection in blood serum of liver cancer patients
AsA@Zn-Se modified GCE is used for the electrochemical H2O2 detection in human serum. The serum is diluted with 0.1 M PBS of pH 7.4 and H2O2 is detected (Fig. 8). Prior to electrochemical analysis, five serum samples are analyzed for AFP levels using Elecsys AFP assay to confirm liver cancer. All samples contain varied amounts of AFP (Table 1). After examining AFP levels in serum, H2O2 concentration is determined using CV (Table 1), with AsA@Zn-Se/GCE under optimized conditions in respective potential windows. The altered redox balance of H2O2 in liver cancer can lead to cancer progression and growth. The current values are used to compute H2O2 concentration in each sample. H2O2 concentration increases with increasing AFP levels in serum samples.

H2O2 detection by cyclic voltammogram in human serum of liver cancer patients by AsA@Zn-Se/GCE.
The voltammograms of sensing modalities show prominent H2O2 peaks. Change in behavior during the analysis depict that H2O2 concentration varies from individual to individual depending on cancer severity, and alteration in metabolic rates.
The comparison of fabricated sensor with reported ones (Table 2).
Conclusion
A novel non-enzymatic electrochemical sensor, AsA@Zn-Se, with a wide sensing range, low detection limit, anti-interference characteristics, and high durability is reported for H2O2 detection in human serum of liver cancer patients, pre-diagnosed by Elecsys total AFP assay. The sensor fabrication is facile with excellent detection outcomes. The fabricated sensing materials are characterized via FTIR, UV, XRD, EDX, and SEM. The obtained structural, and morphological results ensure the formation of SeNPS, ZnONPs, and Zn-SeNPs. Enhanced surface area of fabricated sensor and immobilized ascorbic acid is an effective combination for H2O2 determination. Ascorbic acid is an antioxidant with electrocatalytic activity towards H2O2. H2O2 redox approach is straightforward and does not complicate the fabrication procedure. The electrochemical sensor shows lower detection limits of 0.49 µM with good linear range i.e., 0–70 µM. The simplicity of preparation, availability in wide range of prospective applications, the flexible and freestanding AsA@Zn-Se/GCE is projected to provide a modular strategy for future sensing material manufacturing.
Data availability
All data is provided in the manuscript file.
References
Yu, Y. et al. A nonenzymatic hydrogen peroxide electrochemical sensing and application in cancer diagnosis. Small Methods. 5 (5), 2001212 (2021).
Wang, H. et al. Metal-organic framework (MOF)-Au@ pt nanoflowers composite material for electrochemical sensing of H2O2 in living cells. J. Electroanal. Chem. 897, 115603 (2021).
Dang, W. et al. AuNPs-NH2/Cu-MOF modified glassy carbon electrode as enzyme-free electrochemical sensor detecting H2O2. J. Electroanal. Chem. 856, 113592 (2020).
Zhu, Y. et al. Graphene frameworks-confined synthesis of 2D-layered NiCoP for the electrochemical sensing of H2O2 at lower overpotential. Microchim. Acta. 189 (9), 345 (2022).
Luo, Y. et al. Tailored fabrication of defect-rich Ion implanted CeO2-x nanoflakes for Electrochemical sensing of H2O2. J. Electrochem. Soc. 170 (5), 057519 (2023).
Murugan, P. et al. Electrochemical detection of H2O2 using an activated glassy carbon electrode. ECS Sens. Plus. 1 (3), 034401 (2022).
Feig, D. I., Reid, T. M. & Loeb, L. A. Reactive oxygen species in tumorigenesis. Cancer Res. 54 (7_Supplement), 1890s–1894s (1994).
Kung, M. L. et al. Enhanced reactive oxygen species overexpression by CuO nanoparticles in poorly differentiated hepatocellular carcinoma cells. Nanoscale 7 (5), 1820–1829 (2015).
Ahmad, T. et al. Recent advances in electrochemical sensing of hydrogen peroxide (H2O2) released from cancer cells. Nanomaterials 12 (9), 1475 (2022).
An, H. et al. Hydrogen peroxide-activatable nanoparticles for luminescence imaging and in situ triggerable photodynamic therapy of cancer. ACS Appl. Mater. Interfaces. 12 (15), 17230–17243 (2020).
Fatima, B. et al. Catalase immobilized antimonene quantum dots used as an electrochemical biosensor for quantitative determination of H2O2 from CA-125 diagnosed ovarian cancer samples. Mater. Sci. Engineering: C. 117, 111296 (2020).
Thatikayala, D. et al. Progress of advanced nanomaterials in the non-enzymatic electrochemical sensing of glucose and H2O2. Biosensors 10 (11), 151 (2020).
Sobahi, N. et al. Electrochemical sensing of H2O2 by employing a flexible Fe3O4/graphene/carbon cloth as working electrode. Materials 16 (7), 2770 (2023).
Şen, F. B. et al. Redox-based colorimetric sensing of H2O2 after removal of antioxidants with ABTS radical oxidation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 248, 119266 (2021).
Dumore, N. S. & Mukhopadhyay, M. Sensitivity enhanced SeNPs-FTO electrochemical sensor for hydrogen peroxide detection. J. Electroanal. Chem. 878, 114544 (2020).
Sudhakara, S. M. et al. Phthalocyanine pendented polyaniline via amide linkage for an electrochemical sensing of H2O2. Microchem. J. 161, 105781 (2021).
Munteanu, I. G. & Apetrei, C. A review on electrochemical sensors and biosensors used in assessing antioxidant activity. Antioxidants 11 (3), 584 (2022).
Yang, Y. et al. Fabrication of nonenzymatic electrochemical interface for ratiometric and simultaneous detection of hydrogen peroxide, dopamine, and ascorbic acid. Microchem. J. 178, 107344 (2022).
Han, F. et al. Oxidized titanium carbide MXene-enabled photoelectrochemical sensor for quantifying synergistic interaction of ascorbic acid based antioxidants system. Biosens. Bioelectron. 177, 112978 (2021).
Dumore, N. S. & Mukhopadhyay, M. Development of novel electrochemical sensor based on PtNPs-SeNPs-FTO nanocomposites via electrochemical deposition for detection of hydrogen peroxide. J. Environ. Chem. Eng. 10 (2), 107058 (2022).
Shafa, M. et al. Ag-Cu nanoalloys: an electrochemical sensor for H2O2 detection. Surf. Interfaces. 36, 102616 (2023).
Wang, Q. et al. An electrochemical sensor for H2O2 based on au nanoparticles embedded in UiO-66 metal–organic framework films. ACS Appl. Nano Mater. 4 (6), 6103–6110 (2021).
Ullah, R. et al. Electrochemical sensing of H2O2 using cobalt oxide modified TiO2 nanotubes. Curr. Appl. Phys. 38, 40–48 (2022).
Rajaitha, P. M. et al. An electrochemical sensor based on multiferroic NdFeO3 particles modified electrode for the detection of H2O2. J. Alloys Compd. 915, 165402 (2022).
Asif, M. et al. Core-shell iron oxide-layered double hydroxide: high electrochemical sensing performance of H2O2 biomarker in live cancer cells with plasma therapeutics. Biosens. Bioelectron. 97, 352–359 (2017).
Asif, M. et al. Metal oxide intercalated layered double hydroxide nanosphere: with enhanced electrocatalyic activity towards H2O2 for biological applications. Sens. Actuators B. 239, 243–252 (2017).
Asif, M. et al. Unveiling microbiologically influenced corrosion engineering to transfigure damages into benefits: a textile sensor for H2O2 detection in clinical cancer tissues. Chem. Eng. J. 427, 131398 (2022).
Li, B. et al. Carbon-doping mesoporous β-Mo2C aggregates for nanomolar electrochemical detection of hydrogen peroxide. ACS Appl. Nano Mater. 3 (8), 7499–7507 (2020).
Mustafa, M., Ahmed, A. M. & Mohammed, R. Synthesis And Characterization. (2018).
Sridevi, D. et al. A facile synthesis of Mn-doped ZnSe nanoparticles for an enhanced photocatalytic activity and biological applications. Ceram. Int. 48 (19), 29394–29402 (2022).
Salem, A. et al. Simple synthesis of ZnSe nanoparticles by thermal treatment and their characterization. Results Phys. 7, 1175–1180 (2017).
Sanchez-Martinez, A. et al. ZnSe nanoparticles prepared by coprecipitation method for photocatalytic applications. Mater. Lett. 282, 128702 (2021).
Shakir, M. et al. Characterization of ZnSe nanoparticles synthesized by microwave heating process. Solid State Commun. 149 (45–46), 2047–2049 (2009).
Krzyczmonik, P. et al. Honeycomb-structured porous poly (3, 4-ethylenedioxythiophene) composite layers on a gold electrode. Thin Solid Films. 565, 54–61 (2014).
Ye, M. et al. ZnFe2O4/graphitic carbon nitride nano/microcomposites for the enhanced electrochemical sensing of H2O2. ACS Appl. Nano Mater. 5 (8), 10922–10932 (2022).
Ma, Y. et al. Trimetallic metal–organic framework nanosheets as nanozymes for the electrochemical sensing of H2O2. J. Electroanal. Chem. 940, 117490 (2023).
Acknowledgements
The authors acknowledge the Higher Education Commission (HEC) of Pakistan for this research.
Author information
Authors and Affiliations
Contributions
S.Z.J. performed the experiment and wrote the initial draftS.A. supervised the project and wrote the final draft.D.H. designed the methodology and experimentsJ.N. provided resources and testing facilitiesA.A. performed the experimentsM.N.H. validated and results and reviewed the final draftB.F. provided the concept and validated the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jawad, S.E.Z., Ahmed, S., Hussain, D. et al. Ascorbic acid-immobilized zinc selenide for electrochemical monitoring of hydrogen peroxide in liver cancer samples. Sci Rep 15, 237 (2025). https://doi.org/10.1038/s41598-024-81411-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-81411-0
