
Abstract
Five metal dithiocarbamate complexes [M(PTHIQDTC)2] [where PTHIQDTC is (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate anion and M is Ni(II) (1), Sn(II) (2), Hg(II) (3), Pb(II) (4) and Zn(II) (5)] were synthesized from the reaction of MX2 (X is Cl– for 1–3 and OAc– for 4–5) with ligand of triethylammonium (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate [Et3NH][PTHIQDTC] in methanolic solution at room temperature. The five complexes were characterized by IR, 1H and13C NMR, mass spectrometry, elemental analysis and TGA analysis. Recrystallization of [Zn(PTHIQDTC)2] (5) in dimethylsulfoxide (DMSO) converts 5 to [Zn(PTHIQDTC)2(DMSO)] (6). The structure of complex 6 has been determined by X-ray crystallography. The X-ray structural analysis of 6 indicated that the zinc(II) is five-coordinated in a distorted trigonal-bipyramidal by four S atoms from two (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate anion ligands (PTHIQDTC) and one O atom from DMSO.
Similar content being viewed by others

Introduction
The rich chemistry of dithiocarbamates as mono- and bidentate ligands in coordination chemistry spreads their applications in various fields of science including chemistry, biochemistry, material science, medicine, and environmental science. Beside their applications as herbicides, fungicides and pesticides in agriculture1,2,3, they have found extensive applications in medicinal chemistry for detection and treatment of cancers4,5,6, as inhibitors of enzymes7, positron emission tomography (PET) scanning8–9 and as antibacterial agents10. Their application for detection of gaseous nitric oxide is noteworthy11. These complexes were also applied as efficient precursors for the synthesis of nanoscale and quantum dots of metal sulfides12,13,14. Some complexes of dithiocarbamates such as MoDTC/ZDDP have found applications as engine oil additive and are commercialized15. Dithiocarbamates are efficient ligands for stabilization of transition metals in unusual oxidation states16. Recently, the dithiocarbamate complexes were applied as catalyst for organic transformations17–18. Furthermore, these complexes were applied extensively as reverse addition-fragmentation chain transfer (RAFT) polymerization agents19, rubber vulcanization agents20, and for the determination and removal of metals and dyes from aqueous solutions21,22.
There are many reports in recent years on the synthesis and applications of metal dithiocarbamate complexes based on secondary amines. In this context, in 2024, Singh and coworkers reported the synthesis of three new Ni(II), Cu(II), and Zn(II) dithiocarbamate complexes based on potassium N-(4-fluorobenzyl) N-(naphthalen-1-ylmethyl) dithiocarbamate ligand and investigated their anticonvulsant and antianxiety activities23. In addition, Prasad et al., reported the synthesis and physiochemical properties of four new homoleptic dithiocarbamate complexes of Co(II), Ni(II), Cu(II), and Zn(II) with N-(4-methoxybenzyl) N-(phenylethyl) dithiocarbamate ligand bearing a chiral center24. In 2022, Hrubaru and coworkers have investigated the synthesis and antimicrobial activities of Ni(II), Pd(II) and Pt(II) complexes of N,N-bis(3,3-dimethyl-allyl)-dithiocarbamate25. Among various secondary amines, (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline (PTHIQ) is an efficient substrate for the synthesis of chiral active pharmaceutical ingredients and some of them such as solifenacin and solifenacin succinate (Fig. 1) are available in market, drugs for the treatment of overactive bladder and neurogenic detrusor overactivity (NDO)26. Due to the aforementioned applications of dithiocarbamates and PTHIQ in drug discovery, in this paper, we are investigating the synthesis and characterization of novel chiral dithiocarbamate complexes based on PTHIQ (PTHIQDTC), the thia-analogue of PTHIQ-carbamate.

PTHIQ-based commercially available drugs.
Experimental
Materials and physical methods
All reagents and solvents were purchased from Merck and used without further purification. All NMR spectra were recorded on Bruker DRX Avance B300 spectrometer for protons at 300.13 MHz and for carbon at 75.47 MHz. Melting points were measured on a Büchi type B-545 instrument. Elemental analysis performed by a Perkin–Elmer 2004 (II) CHNS/O analyzer. Infrared spectra were recorded in KBr pellets on a Perkin Elmer FT spectrum RX1 over the range of 400–4000 cm−1. TGA measurement has been performed by a NETZSCH TG 209 F1 Iris instrument. Mass spectra were determined at an ionizing voltage of 70 eV by 5973 Network Mass Selective Detector.
Synthesis of triethylammonium 1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate [Et3NH][PTHIQDTC]
(1S)-1-Phenyl-1,2,3,4-tetrahydroisoquinoline (2.09 g, 10 mmol) and triethylamine (1.40 mL, 10 mmol) were added to a round bottom flask containing THF (15 mL). The mixture was stirred at room temperature for 15 min, then CS2 (1.69 g, 1.40 mL, 20 mmol) was added dropwise, and stirring was continued for 14 h at room temperature. After evaporation of the resulting yellow solution in a rotary evaporator, an oily product was obtained. By adding diethyl ether to the crude product, a white precipitate was formed. The white precipitate was collected by filtration and washed several times with diethyl ether to remove any unreacted starting materials (yield 3.16 g, 82.0%, m. p. 97–99 °C). FT-IR (KBr, ν cm−1): 3100 − 2300 (broad peak), 1598, 1492, 1451, 1381, 1282, 1272, 1218, 971, 770, 744, 704, 625;1H NMR (300 MHz, CDCl3), δ (ppm) 8.39 (s, 1 H, N-H), 7.51–7.02 (m, 10 H, Ar–H and CH), 5.45–5.30 (m, 1 H, CH2–CH2N), 3.52 (m, 1 H, CH2–CH2N), 3.27 (q, J = 7.3 Hz, 6 H, 3CH2), 3.19–3.06 (m, 1 H, CH2–CH2N), 2.72 (m, 1 H, CH2–CH2N), 1.39 (t, J = 7.3 Hz, 9 H, 3CH3);13C NMR (75 MHz, CDCl3) δ (ppm) 210.7 (C=S), 142.0 (Ar), 136.1 (Ar), 136.0 (Ar), 128.8 (Ar), 128.6 (Ar), 128.5 (Ar), 128.0 (Ar), 127.8 (Ar), 126.7 (Ar), 125.5 (Ar), 63.1 (CH), 45.7 (3CH2N), 44.8 (CH2–CH2N), 28.3 (CH2–CH2N), 8.4 (3 CH3); Anal. Calc. for C22H30N2S2: C, 68.35; H, 7.82; N, 7.25. Found: C, 68.33; H, 8.16; N, 7.34%.
Synthesis of [Ni(PTHIQDTC)2] (1)
A solution of [Et3NH][PTHIQDTC] (0.39 g, 1 mmol) in methanol (5 mL) was added to a solution of NiCl2·6H2O (0.12 g, 0.5 mmol) in methanol (5 mL). The mixture was stirred for 30 min at room temperature to give a green precipitate. The green precipitate was collected by filtration and then washed several times with methanol to remove any unreacted starting materials (yield 0.26 g, 83%, m. p. 160–162 °C); FT-IR (KBr, ν cm−1) 3024, 2924, 1599, 1486, 1445, 1231, 1148, 928, 768, 749, 708, 626;1H NMR (300 MHz, CDCl3) δ (ppm) 7.20–7.06 (m, 9 H, Ar–H), 6.88 (s, 1 H, CH), 4.41 (brs, 1 H, CH2–CH2N), 3.43 (brs, 1 H, CH2–CH2N), 3.10 (brs, 1 H, CH2–CH2N), 2.88 (s, 1 H, CH2–CH2N);13C NMR (75 MHz, CDCl3), δ (ppm): 205.9 (C=S), 139.1 (Ar), 133.9 (Ar), 133.1 (Ar), 128.9 (Ar), 128.8 (Ar), 128.6 (Ar), 128.5 (Ar), 128.1 (Ar), 127.8 (Ar), 126.6 (Ar), 60.1 (CH), 41.2 (CH2–CH2N), 27.7 (CH2–CH2N); MS (EI) m/z (%): 411.3, 324.3, 252.2, 206.2 (100), 178.2, 132.2, 76.1, 51.2; Anal. Calc. for C32H28N2S4Ni: C, 61.25; H, 4.50; N, 4.46. Found: C, 62.19; H, 4.21; N, 4.64%.
Synthesis of [Sn(PTHIQDTC)2] (2)
A solution of [Et3NH][PTHIQDTC] (0.39 g, 1 mmol) in methanol (5 mL) was added to a solution of SnCl2·2H2O (0.11 g, 0.5 mmol) in methanol (5 mL). The mixture was stirred for 30 min at room temperature to give a yellow-orange precipitate. The yellow-orange precipitate was collected by filtration and then washed several times with methanol to remove any unreacted starting materials (yield 0.29 g, 84%, m. p. 157–159 °C). FT-IR (KBr, ν cm−1): 3058, 3024, 2927, 1599, 1492, 1480, 1435, 1292, 1230, 1143, 930, 752, 700, 622;1H NMR (300 MHz, CDCl3), δ (ppm) 8.39–6.29 (m, 10 H, Ar–H and CH), 4.46 (brs, 1 H, CH2–CH2N), 3.53 (brs, 1 H, CH2–CH2N), 3.10 (brs, 1 H, CH2–CH2N), 2.80 (brs, 1 H, CH2–CH2N);13C NMR (75 MHz, CDCl3) δ (ppm) 212.2 (C=S), 139.4 (Ar), 134.5 (Ar), 134.2 (Ar), 128.6 (Ar), 128.5 (Ar), 128.5 (Ar), 128.4 (Ar), 128.2 (Ar), 127.6 (Ar), 126.5 (Ar), 66.1 (CH), 47.0 (CH2–CH2N), 28.2 (CH2–CH2N); MS (EI) m/z(%): 594.2, 310.0, 284.1, 252.1, 206.2 (100), 178.1, 132.2, 102.2, 76.1, 51.1; Anal. Calc. for C32H28N2S4Sn: C, 55.90; H, 4.10; N, 4.07. Found: C, 55.02; H, 3.84; N, 3.94%.
Synthesis of [Hg(PTHIQDTC)2] (3)
A solution of [Et3NH][PTHIQDTC] (0.39 g, 1 mmol) in methanol (5 mL) was added to a solution of HgCl2 (0.14 g, 0.5 mmol) in methanol (5 mL). The mixture was stirred for 30 min at room temperature to give a white precipitate. The white precipitate was collected by filtration and then washed several times with methanol to remove any unreacted starting materials (yield 0.32 g, 83%, m. p. 184–186 °C). FT-IR (KBr, ν cm−1) 3024, 2925, 2605, 1598, 1492, 1470, 1445, 1425, 1292, 1230, 1143, 965, 929, 753, 732, 697, 621;1H NMR (300 MHz, DMSO-d6) δ (ppm) 7.42–7.14 (m, 10 H, Ar–H and CH), 4.46–4.29 (m, 1 H, CH2–CH2N), 3.78–3.70 (m, 1 H, CH2–CH2N), 3.14–2.93 (m, 1 H, CH2–CH2N), 2.83–2.48 (m, 1 H, CH2–CH2N);13C NMR (75 MHz, DMSO-d6), δ (ppm): δ 201.7(C=S), 139.4 (Ar), 134.3 (Ar), 134.2 (Ar), 128.6 (Ar), 128.5 (Ar), 128.1 (Ar), 127.9 (Ar), 127.7 (Ar), 127.2 (Ar), 126.5 (Ar), 66.5 (Ar), 48.4 (CH2–CH2N), 27.5 (CH2–CH2N); MS (EI) m/z(%) : 492.2, 285.1, 252.2, 206.2 (100), 178.1, 152.1, 132.1, 103.1, 76.0, 51.1; Anal. Calc. for C32H28N2S4Hg: C, 49.95; H, 3.67; N, 3.64. Found: C, 48.90; H, 3.36; N, 3.71%.
Synthesis of [Pb(PTHIQDTC)2] (4)
A solution of [Et3NH][PTHIQDTC] (0.39 g, 1 mmol) in methanol (5 mL) was added to a solution of Pb(OAc)2·3H2O (0.19 g, 0.5 mmol) in methanol (5 mL). The mixture was stirred for 30 min at room temperature to give a yellow precipitate. The yellow precipitate was collected by filtration and then washed several times with methanol to remove any unreacted starting materials (yield 0.33 g, 85%, m. p. 165–167 °C); FT-IR (KBr, ν cm−1) 3021, 2916, 1597, 1491, 1459, 1415, 1394, 1290, 1224, 1142, 1108, 1038, 961, 929, 750, 732, 698, 615;1H NMR (300 MHz, CDCl3); δ (ppm) 7.56 (s, 1 H, CH), 7.47–7.09 (m, 9 H, Ar–H), 4.87–4.79 (m, 1 H, CH2–CH2N), 3.71–3.41 (m, 1 H, CH2–CH2N), 3.20–3.12 (m, 1 H, CH2–CH2N), 2.89–2.81 (m, 1 H, CH2–CH2N);13C NMR (75 MHz, CDCl3) δ (ppm) 202.8 (C=S), 139.9 (Ar), 135.4 (Ar), 134.3 (Ar), 128.7 (Ar), 128.6 (Ar), 128.3 (Ar), 128.3 (Ar), 127.8 (Ar), 127.5 (Ar), 126.3 (Ar), 62.6, 44.1 (CH2–CH2N), 28.0 (CH2–CH2N); MS (EI) m/z(%): 492.2, 411.3, 310.1, 284.1, 252.2, 206.2, 178.2, 132.2 (100), 103.1, 76.1, 51.1; Anal. Calc. for C32H28N2S4Pb: C, 49.53; H, 3.64; N, 3.61. Found: C, 50.20; H, 3.29; N, 3.89%.
Synthesis of [Zn(PTHIQDTC)2] (5)
A solution of [Et3NH][PTHIQDTC] (0.39 g, 1 mmol) in methanol (5 mL) was added to a solution of Zn(OAc)2·2H2O (0.11 g, 0.5 mmol) in methanol (5 mL). The mixture was stirred for 30 min at room temperature to give a white precipitate. The white precipitate was collected by filtration and then washed several times with methanol to remove any unreacted starting materials (yield 0.27 g, 85.4%, m. p. 225–227 °C). FT-IR (KBr, ν cm−1) 3055, 3023, 2924, 1598, 1491, 1478, 1436, 1282, 1234, 1155, 969, 930, 754, 732, 693;1H NMR (300 MHz, CDCl3), δ (ppm) 7.39 (s, 1 H, CH), 7.36–7.14 (m, 9 H, Ar–H), 4.91–4.59 (m, 1 H, CH2–CH2N), 3.77–3.51 (m, 1 H, CH2–CH2N), 3.18–3.10 (m, 1 H, CH2–CH2N), 2.98–2.74 (m, 1 H, CH2–CH2N);13C NMR (75 MHz, CDCl3) δ (ppm) 202.6 (C=S), 139.5 (Ar), 134.3 (Ar), 134.0 (Ar), 128.8 (Ar), 128.7 (Ar), 128.5 (Ar), 128.1 (2 C, Ar), 127.7 (Ar), 126.5 (Ar), 64.9 (CH), 45.8 (CH2–CH2N), 28.1 (CH2–CH2N); MS (EI) m/z(%): 594.2, 358.1, 310.0, 281.1, 253.1, 206.1, 178.1,139.1, 115.1, 85.2, 57.2 (100); Anal. Calc. for C32H28N2S4Zn: C, 60.60; H, 4.45; N, 4.42. Found: C, 60.36; H, 3.94; N, 4.72%.
X-ray structure analysis
The X-ray diffraction measurement for [Zn(PTHIQDTC)2(DMSO)] (6) was carried out on a STOE IPDS-II diffractometer with graphite monochromated Mo-Kα radiation at 298 K. Data were collected in a series of ω scans in 1° oscillations and integrated using the Stöe X-AREA software package27. A numerical absorption correction was applied using the X-RED228 and X-SHAPE29 softwares. The structure was solved by direct methods using SHELXS-9730 and refined with full-matrix least-squares on F2 using the SHELXL-2019/331 program package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added at ideal positions and constrained to ride on their parent atoms, with Uiso(H) = 1.2Ueq. All refinements were performed using the OLEX2 crystallographic software package32. The ORTEP and crystal packing diagram for 6 were drawn with the Mercury 2.4 program33.
Results and discussion
Synthesis of chiral ligand and complexes 1–6
In this research work, the chiral ligand namely triethylammonium 1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate (PTHIQDTC) was prepared via the reaction of an equimolar amount of (1S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline and triethylamine in the presence of two equivalents of CS2 in THF at room temperature. Although an equivalent of CS2 is needed for completion of the reaction according to the stoichiometry, but we used two equivalents of CS2 to get higher yield. Then, we investigated the complexation behavior of this chiral ligand with different metals, such as Ni(II) (1), Sn(II) (2), Hg(II) (3), Pb(II) (4) and Zn(II) (5) in methanol solution at room temperature, which led to the formation of [M(PTHIQDTC)2] complexes. It is notable that no racemization was observed during the synthesis. To investigate the exact structure of these complexes, suitable crystals of the complex [Zn(PTHIQDTC)2(DMSO)] (6) for X-ray diffraction measurement were obtained by methanol diffusion into a DMSO solution of 5 for 1 week. By this method, the complex [Zn(PTHIQDTC)2] (5) was converted to complex [Zn(PTHIQDTC)2(DMSO)] (6) (Fig. 2).

Preparation of [Et3NH][PTHIQDTC] and complexes 1–6.
Spectroscopic characterization of chiral ligand and 1–6
The infrared absorption bands for free chiral ligand and title complexes are listed in the “Experimental” section. The infrared spectrum of the free chiral ligand [Et3NH][PTHIQDTC] shows a broad absorption band at 3100–2300 cm−1 for the N–H stretching vibrations in ammonium dithiocarbamate group (Et3NH+) and several bands at 3100–2900 cm−1 for the C–H vibrations of phenyl rings and methylene groups. This broad absorption band disappears after coordination of this ligand to metals. Several absorption bands were observed in the range of 1600–1381 cm−1 for complexes which were assigned to C–N, C=C and C=S stretching vibrations34,35,36,37. The ν(C–N) stretching band for the dithiocarbamate moiety in free ligand appeared at 1451 cm−1, and this peak experienced a blue shift to around 1480 cm−1 in the complexes, suggesting the coordination of dithiocarbamate group to metals in bidentate mode38. Also, the single absorption band in 1000–940 cm−1 region, without any splitting, was assigned to the completely symmetrical bonding of the dithiocarbamate ligand in these complexes, acting in the bidentate mode (see supporting information for the IR spectra of ligand and complexes)36,39.
The thermogravimetric analysis (TGA) was used to evaluate the physical behavior of the ligand and complexes, between 10 and 600 °C in N2 atmosphere with a heating rate of 20 °C/min. While the TGA of [Et3NH][PTHIQDTC] shows two weight loss events at 140–100 °C (approx. 50%) and 260–160 °C (approx. 50%) without any residue, the TGA curve for Zn(II) complex shows a single weight loss event (mass loss approx. %78.6) in the temperature range of 400–300 °C, which may be attributed to the loss of the ligand and the remaining residue (approx. 21%) is close to the calculated mass percentage of ZnS2 (20.2%) (Fig. 3). Similar behavior was observed for the Ni(II) complex. The Sn(II) complex starts to lose weight from 300 to 100 °C (approx. 72.8%) and the remaining residue (approx. 27.2%) is completely in agreement with the theoretical percentage of SnS2 (26.7%). TGA curve of the Hg(II) complex shows approximately complete weight loss due to the low sublimation temperature of HgS40. Finally, the TGA curve of the Pb(II) complex shows that the weight loss starts at around 100 °C and the remaining residue (approx. 31.3%) is close to the calculated mass percentage for PbS (30.8%) (See supporting information for the TGA curve of other complexes).

TGA curve for Zn(II) complex (5).
NMR analysis of the ligand and complexes completely confirm their structures. Due to the presence of a chiral center in the ligand, the1H NMR spectrum of the ligand shows four peaks for two methylene groups at 2.69, 3.13, 3.47, and 5.39 ppm (Fig. S1 in supporting information). The peak at 5.39 is shielded to around 4.5 ppm in the complexes and the chemical shifts of the other three peaks mostly remained intact during the complexation. The hydrogen of the chiral center in the ligand appeared as singlet at 7.39 ppm, while this signal was deshielded to 7.56 ppm for the Pb(II) complex and shielded to 6.88 ppm for Ni(II) complex. For other complexes, this proton appeared without significant changes in the chemical shift. The protons of benzene rings were observed around 7.0–7.5 ppm. As a consequence of the planarity of the S2CN group and restricted rotation around the C–N bond, due to partial double bond character, the proton of the chiral center lies close to the S2CN plane; the crystal structure of 6 shows two C–N–C–H torsion angles as − 17.5(11) and − 33.3(9)°. The N13CS2 carbon is the most important13C NMR signal in these complexes, which is observed in the range of 201–213 ppm and is in good agreement with data reported in the literature for the similar compounds36,37,38. The carbon of the dithiocarbamate group in ligand was observed in 210.7 ppm. The methylene carbons in the complexes were observed around 45.0 (CH2CH2N) and 27.0 (CH2CH2N) ppm. The carbon of the chiral center in ligand was assigned at 63.0 ppm in13C NMR analysis. This carbon was shielded to 60.0 ppm (1) and 62.5 ppm (4) and deshielded to 65.0 (5), 66.1 (2) and 66.5 (3) in complexes. The carbons of the aromatic rings in the complexes were appeared at 125.0-140.0 ppm, while for the ligand these carbons were appeared at 125.0–142.0 ppm (See supporting information for NMR spectra).
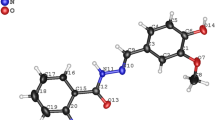
Description of the molecular structure of [Zn(PTHIQDTC)2(DMSO)] (6)
Crystallographic data and data collection parameters for complex 6 are shown in Table 1. Some selected bond lengths and angles of 6 are listed in Table 2. This complex crystallizes in the monoclinic crystal system with P21 space group. The coordination polyhedral and molecular structure of [Zn(PTHIQDTC)2(DMSO)] (6) with the atomic labeling scheme are shown in Figs. 4 and 5, respectively. As shown in these figures, the asymmetric unit of 6 contains one monomeric five–coordinate Zn(II) complex. In this complex, the Zn(II) cation is coordinated by two anionic PTHIQDTC ligands via four S atoms and one dimethyl sulfoxide molecule via O atom. The complex exhibits distorted trigonal-bipyramidal geometry around the Zn(II) cation, with atoms S2 and S3 in axial positions (S2-Zn1-S3 167.28(13)°) and S1, S4 and O1 in equatorial positions (S1-Zn1-S4 135.05(12)°, S1-Zn1-O1 111.6(3)° and S4-Zn1-O1 113.3(3)°). The Zn atom is 0.04 Å out of the equatorial plane. The axial Zn–S bond lengths (Zn1-S2 = 2.634(4)Å and Zn1-S3 = 2.534(4)Å) are slightly longer than related equatorial Zn-S bond lengths (Zn1-S1 = 2.334(3)Å and Zn1-S4 = 2.337(3)Å). The Zn1-O1 bond length is 2.024(9)Å. The Zn–S and Zn–O bond lengths and angles (Table 2) are within normal ranges, as in [Zn{NH(CH2)4O}{S2CN(C2H5)2}2].CCl441, [(Et2NCS2)2Zn(py)]42 and [ZnCl2(bipy)(DMSO)]43 (py is pyridine and bipy is 2,2′-bipyridine). The coordination geometry around each Zn(II) cation is five-coordinated in a distorted trigonal-bipyramidal configuration (τ5 = 0.897; τ5 = (β-α)/60, where β = S2–Zn1–S3 167.28(13)° and α = O1–Zn1–S4 113.3(3)°). The τ parameter being 0 and 1 for perfect square-pyramidal and trigonal-bipyramidal geometries, respectively44,45,46,47,48.
A c-direction side view of the crystal packing of 6 is shown in Fig. 6. As depicted in this figure, the intermolecular C–H…π hydrogen bonds interactions (Table 3) between the aliphatic –CH2– and aromatic –CH groups with phenyl rings are effective factors in the stabilization of the crystal structure and the formation of a 3D supramolecular solid structure.

The coordination polyhedral of [Zn(PTHIQDTC)2(DMSO)] (6). The figure was drawn by Mercury 2.4 program. (https://mercury1.software.informer.com/2.4/)29.

The molecular structure of [Zn(PTHIQDTC)2(DMSO)] (6), with the atom-numbering scheme and 30% probability displacement ellipsoids. The figure was drawn by Mercury 2.4 program. (https://mercury1.software.informer.com/2.4/)29.

Crystal packing diagram for [Zn(PTHIQDTC)2(DMSO)] (6). Intermolecular C–H…π hydrogen bonds are shown as dashed lines. Symmetry codes: (i) − x,1/2 + y,− z and (ii) 1 − x,1/2 + y,1 − z. The figure was drawn by Mercury 2.4 program. (https://mercury1.software.informer.com/2.4/)29.
Conclusion
In conclusion, a chiral ligand of triethylammonium 1-phenyl-1,2,3,4-tetrahydroisoquinoline dithiocarbamate [Et3NH][PTHIQDTC] has been synthesized. Then, five metal dithiocarbamate complexes [M(PTHIQDTC)2] (M = Ni(II) (1), Sn(II) (2), Hg(II) (3), Pb(II) (4) and Zn(II) (5)) with this chiral ligand have been synthesized. All these compounds were fully characterized by IR, 1H and13C NMR, elemental analysis and mass spectrometry. The IR analysis completely confirms the bidentate coordination mode of the ligand to the metals. The thermal stability of title complexes were studied by thermal gravimetric analyses (TGA) and shows higher stability related to the ligand. The chiral center in the ligand was remained intact during ligand synthesis and complexation and no racemization was observed. Complex [Zn(PTHIQDTC)2(DMSO)] (6) was prepared from recrystallization of [Zn(PTHIQDTC)2] (5) in DMSO by diffusion method with methanol. In complex 6, the Zn(II) cation is coordinated by two anionic PTHIQDTC ligands via four S atoms and one dimethyl sulfoxide molecule via O atom. The complex exhibits distorted trigonal-bipyramidal geometry. Also, in this complex, the intermolecular C-H…π hydrogen bonds interactions are effective in the stabilization of the crystal structure and the formation of the 3D supramolecular complex. The prepared complexes can be applied as potential catalysts for asymmetric synthesis. In addition, these complexes are potential candidates for biological studies due to the presence of both dithiocarbamate and (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline (PTHIQ) core in a single structure.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information file].
References
Campanale, C. et al. Properties, methodological approaches and challenges to their control. Toxics. 11, 851–878. https://doi.org/10.3390/toxics11100851 (2023).
Pirozzi, A. V. A., Stellavato, A., La Gatta, A., Lamberti, M. & Schiraldi, C. Mancozeb, a fungicide routinely used in agriculture, worsens nonalcoholic fatty liver disease in the human HepG2 cell model. Toxicol. Lett. 13, 1–4. https://doi.org/10.1016/j.toxlet.2016.03.004 (2016).
Fanjul-Bolado, P., Fogel, R., Limson, J., Purcarea, C. & Vasilescu (eds) (c), A. Advances in the detection of dithiocarbamate fungicides: Opportunities for biosensors. Biosensors 11, 12–37. https://doi.org/10.3390/bios11010012 (2021).
Ronconi, L. et al. Gold(III) dithiocarbamate derivatives for the treatment of cancer: Solution chemistry, DNA binding, and hemolytic properties. J. Med. Chem. 49, 1648–1657. https://doi.org/10.1021/jm0509288 (2006).
Negi, M., Dixit, T. & Venkatesh, V. Ligand dictated photosensitization of Iridium(III) dithiocarbamate complexes for photodynamic therapy. Inorg. Chem. 62, 20080–20095. https://doi.org/10.1021/acs.inorgchem.3c02942 (2023).
Ronconi, L. et al. Gold dithiocarbamate derivatives as potential antineoplastic agents: design, spectroscopic properties, and in vitro antitumor activity. Inorg. Chem. 44, 1867–1881. https://doi.org/10.1021/ic048260v (2005).
Stefan, C. et al. Mechanism of dithiocarbamate inhibition of apoptosis: Thiol oxidation by dithiocarbamate disulfides directly inhibits processing of the caspase-3 proenzyme. Chem. Res. Toxicol. 10, 636–643. https://doi.org/10.1021/tx970006a (1997).
Berry, D. J., de Rosales, R. T. M., Charoenphun, P. & Blower, P. J. Dithiocarbamate complexes as radiopharmaceuticals for medical imaging. Mini Rev. Med. Chem. 12, 1174–1183. https://doi.org/10.2174/138955712802762112 (2012).
Bolzati, C. et al. Biological in Vitro and in vivo studies of a series of New Asymmetrical Cationic [99mTc(N)(DTC-Ln)(PNP)]+ complex (DTC-Ln = alicyclic dithiocarbamate and PNP = diphosphinoamine). Bioconjug. Chem. 21, 928–939. https://doi.org/10.1021/bc900493e (2010).
Jiang, S. et al. Antibacterial activities of Novel Dithiocarbamate-containing 4H-Chromen-4-one derivatives. J. Agric. Food Chem. 68, 5641–5647. https://doi.org/10.1021/acs.jafc.0c01652 (2020).
Sun, J. et al. Fluorescence turn-on detection of gaseous nitric oxide using ferric dithiocarbamate complex functionalized quantum dots. Anal. Chem. 86, 5628–5632. https://doi.org/10.1021/ac501315p (2014).
Sarker, J. C. & Hogarth, G. Dithiocarbamate complexes as single source precursors to nanoscale binary, ternary and quaternary metal sulfides. Chem. Rev. 121, 6057–6123. https://doi.org/10.1021/acs.chemrev.0c01183 (2021).
Ajibade, P. A., Mbuyazi, T. B. & Paca, A. M. Synthesis and crystal structures of bis(diallydithiocarbamato)zinc(II) and silver(I) complexes: precursors for zinc sulfide and silver sulfide nanophotocatalysts. ACS Omega. 8, 24750–24760. https://doi.org/10.1021/acsomega.2c07490 (2023).
Tzeng, B. C. et al. Luminescent agents (luminescent pt(II) complexes containing (1-Aza-15-crown-5)dithiocarbamate and (1-Aza-18-crown-6)dithiocarbamate: Mechanochromic and solvent-induced luminescence. Inorg. Chem. 62, 916–929. https://doi.org/10.1021/acs.inorgchem.2c03726 (2023).
Morina, A., Neville, A., Priest, M. & Green, J. H. ZDDP and MoDTC interactions in boundary lubrication—the effect of temperature and ZDDP/MoDTC ratio. Tribol Int. 39, 1545–1557. https://doi.org/10.1016/j.triboint.2006.03.001 (2006).
Hogarth, G. & Transition Metal, D. 1978–2003. Prog. Inorg. Chem. 53, 71. https://doi.org/10.1002/0471725587.ch2 (2005).
Anamika, Yadav, C. L., Drew, M. G. B., Kumar, K. & Singh, N. Ferrocene-functionalized dithiocarbamate Zinc(II) complexes as efficient bifunctional catalysts for the one-pot synthesis of chromene and imidazopyrimidine derivatives via knoevenagel condensation reaction. Inorg. Chem. 60, 6446–6462. https://doi.org/10.1021/acs.inorgchem.1c00162 (2021).
Vale, J. A., Faustino, W. M., Menezesa, P. H. & de Sá, G. F. Eu(III) dithiocarbamate complex and N-p-tolylsulfonylphenylalanine as a novel chiral catalyst for the asymmetric synthesis of cyanohydrins. Chem. Commun. 3340–3342. https://doi.org/10.1039/B607741M (2006).
Moad, G. A. Critical survey of Dithiocarbamate reversible addition-fragmentation chain transfer (RAFT) agents in radical polymerization. J. Polym. Sci. Part. A Polym. Chem. 57, 216–227. https://doi.org/10.1002/pola.29199 (2019).
Shi, F. et al. Mechanism of the zinc dithiocarbamate-activated rubber vulcanization process: a density functional theory study. ACS Appl. Polym. Mater. 3, 5188–5196. https://doi.org/10.1021/acsapm.1c00902 (2021).
Bond, M. & Wallace, G. G. Determination of copper as a dithiocarbamate complex by reverse-phase liquid chromatography with electrochemical detection. Anal. Chem. 53, 1209–1213. https://doi.org/10.1021/ac00231a018 (1981).
Cheng, R., Ou, S., Xiang, B., Li, Y. & Liao, Q. Dye removal (equilibrium and molecular mechanism of anionic dyes adsorption onto Copper(II) complex of dithiocarbamate-modified starch. Langmuir. 26, 752–758. https://doi.org/10.1021/la9039489 (2010).
Singh, A. et al. Synthesis, crystal structure and screening for anticonvulsant and antianxiety activities of three new Ni(II), Cu(II), and Zn(II) dithiocarbamate complexes. J. Mol. Struct. 1298, 137052. https://doi.org/10.1016/j.molstruc.2023.137052 (2024).
Singh, A., Prasad, L. B., Shiv, K., Kumar, R. & Garai, S. Synthesis, characterization, and in vitro antibacterial and cytotoxic study of Co(II), ni(II), Cu(II), and zn(II) complexes of N-(4-methoxybenzyl) N-(phenylethyl) dithiocarbamate ligand. J. Mol. Struct. 1288, 135835. https://doi.org/10.1016/j.molstruc.2023.135835 (2023).
Hrubaru, M. M. et al. (II), pd(II) and pt(II) complexes of N,N-bis(3,3-dimethyl-allyl)-dithiocarbamate: synthesis, spectroscopic characterization, antimicrobial and molecular docking studies. J. Mol. Struct. 1250, 131649. https://doi.org/10.1016/j.molstruc.2021.131649 (2022).
Naito, R. et al. Synthesis and antimuscarinic properties of quinuclidin-3-yl 1,2,3,4-tetrahydroisoquinoline-2-carboxylate derivatives as novel muscarinic receptor antagonists. J. Med. Chem. 48, 6597–6606. https://doi.org/10.1021/jm050099q (2005).
Stoe, C. X–AREA: Program for the Acquisition and Analysis of Data, Version 1.30 (Stoe Cie GmbH Darmatadt, 2005).
X–RED. Program for Data Reduction and Absorption Correction, Vesion 1.28b (Stoe & Cie GmbH, 2005).
X–SHAPE. Program for crystal optimization for numerical absorption correction, vesion 2.05: Stoe & Cie GmbH: Darmstadt, Germany, 2004. No Title X–SHAPE: Program for Crystal Optimization for Numerical Absorption Correction, Vesion 2.05: Stoe & Cie GmbH: Darmstadt, Germany, (2004).
Sheldrick, G. M. SHELX-97, Program for the Solution and Refinement of Crystal Structures (Univ. Göttingen, 1997).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. Commun. 71, 3–8 (2015).
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341 (2009).
Mercury 141. Copyright Cambridge Crystallographic Data Center. Cambridge, 2001–2005.
Amani, V. Coordinated versus proton transfer gold(III) complexes containingsubstituted-phenanthroline ligands. J. Mol. Struct. 1194, 78–85. https://doi.org/10.1016/j.molstruc.2019.05.089 (2019).
Choopani Jouybari, H., Alizadeh, R., Banisaeed, H. & Amani, V. Synthesis, luminescence studies and structural characterization of new Co(II) and Cu(II) complexes based on phenanthridine as a ligand. Inorg. Chim. Acta. 506, 119553. https://doi.org/10.1016/j.ica.2020.119553 (2020).
Ziyaei Halimehjani, A., Torabi, S., Amani, V., Notash, B. & Saidi, M. R. Synthesis and characterization of metal dithiocarbamate derivatives of 3-((pyridin-2-yl)methylamino) propanenitrile: Crystal structure of [3-((pyridin-2-yl)methylamino)propanenitrile dithiocarbamato] nickel(II). Polyhedron 102, 643–648. https://doi.org/10.1016/j.poly.2015.10.058 (2015).
Ziyaei Halimehjani, A., Marjani, K., Ashouri, A. & Amani, V. Synthesis and characterization of transition metal dithiocarbamate derivatives of 1-aminoadamantane: Crystal structure of (N-adamantyldithiocarbamato) nickel (II). Inorg. Chem. Acta. 373, 282–285. https://doi.org/10.1016/j.ica.2011.02.089 (2011).
Yin, H. D., Zhai, J., Sun, Y. Y. & Wang, D. Q. Polyhedron 27, 663–670. https://doi.org/10.1016/j.poly.2007.10.019 (2008).
Bonati, F. & Ugo, R. Organotin(IV) N,N-disubstituted dithiocarbamates. J. Organomet. Chem. 10, 257–268. https://doi.org/10.1016/S0022-328X(00)93085-7 (1967).
Rinse, J. The vapour pressure, dissociation, and transition point of mercury sulphide. Recl. Trav Chim. Pays-Bas. 47, 28–32. https://doi.org/10.1002/recl.19280470105 (1928).
Azad Malik, M., Motevalli, M. & O’Brien, P. Chalcogenolato-di-thiocarbamato-complexes of zinc: the X-ray single crystal structures of pyridine adducts. Polyhedron 18, 1259–1264. https://doi.org/10.1016/S0277-5387(98)00427-6 (1999).
Lutsenko, I. A., Korneeva, E. V. & Ivanov, A. V. Supramolecular complexes M{NH(CH2)4O}{S2CN(C2H5)2}2].CCl4 (M = Zn or 63Cu(II)): synthesis, structures, spectral and thermal properties. Russ. J. Coord. Chem. 42, 494–501. https://doi.org/10.1134/S1070328416080042 (2016).
Marjani, K., Mousavi, M., Khavasi, H. R., Ansari, M. & Qumi, H. R. (2,2’-Bipyridine-k2N,N’)dichlorido(dimethyl sulfoxide-kO)zinc(II), Acta Crystalloger. E63, m2645. https://doi.org/10.1107/S1600536807047320 (2007).
Addison, A. W., Rao, T. N., Reedijk, J., van Rijn, J. & Verschoor, G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. Dalton Trans. 1349–1356. https://doi.org/10.1039/DT9840001349 (1984).
Klein, A. et al. Five coordinate platinum(II) in [Pt(bpy)(cod)(me)][SbF6]: a structural and spectroscopic study. Inorganics 3, 118–138. https://doi.org/10.3390/inorganics3020118 (2015).
Abedi, A., MehriLighvan, Z., Yasan, M. & Amani, V. Novel cu(II) complexes of bithiazole: structure and biological study. J. Iran. Chem. Soc. 14, 491–502. https://doi.org/10.1007/s13738-016-0997-5 (2017).
Ostad, S. N., Abedi, A., Amani, V., Karimi, P. & Heydarnezhad, S. Influence of methyl group position in bipyridine ligand on structure and luminescence of related zinc(II) nitrate complexes. J. Iran. Chem. Soc. 13, 1417–1427. https://doi.org/10.1007/s13738-016-0857-3 (2016).
Amani, V. A. New imide-pyrazine Zinc(II) binuclear complex; in situ synthesis and crystal structure determination. J. Inorg. Chem. Res. 6, 155–160. https://doi.org/10.22036/J10.22036.2023.398659.1143 (2022).
Acknowledgements
We would like to thank the research council of the Sharif University of Technology for financial support of this work. In addition, we thank Farhangian University and Kharazmi University for support.
Author information
Authors and Affiliations
Contributions
A.Z.H. supervision, conceptualization, writing-review & editing; V.A. supervision, writing, editing, taking single crystal, F.N. running reactions, B.N. collecting X-ray data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Halimehjani, A.Z., Amani, V., Naseri, F. et al. Synthesis and characterization of metal dithiocarbamate derivatives of (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline. Sci Rep 14, 29718 (2024). https://doi.org/10.1038/s41598-024-81589-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-81589-3
