
Abstract
The proximity extension assay (PEA) enables large-scale proteomic investigations across numerous proteins and samples. However, discrepancies between measurements, known as batch-effects, potentially skew downstream statistical analyses and increase the risks of false discoveries. While implementing bridging controls (BCs) on each plate has been proposed to mitigate these effects, a clear method for utilizing this strategy remains elusive. Here, we characterized batch effects in PEA proteomics and identified three types: protein-specific, sample-specific, and plate-wide. We developed a robust regression-based method called BAMBOO (Batch Adjustments using Bridging cOntrOls) to correct them. Simulations comparing BAMBOO with established correction techniques (median centering, median of the difference (MOD), and ComBat) revealed that median centering and ComBat were significantly impacted by outliers within the BCs, whereas BAMBOO and MOD were more robust when no plate-wide effects were introduced. Optimal batch correction was achieved with 10–12 BCs. We validated the simulation results using experimental data and found that BAMBOO and MOD had a reduced incidence of false discoveries compared to alternative methods. Our findings emphasize the prevalence of batch effects in PEA proteomic studies and advocate for BAMBOO as a robust and effective tool to enhance the reliability of large-scale analyses in the proteomic field.
Similar content being viewed by others


Introduction
Identifying a phenotype from a set of biomarkers can greatly improve our understanding of biological processes in health and disease. The identification and validation of proteomic biomarkers have become an essential area of research in the field of personalized medicine, as they hold great potential for improving disease detection, monitoring, and therapeutic decision-making1. The challenge is to identify the specific protein(s) or protein pattern(s) associated with a specific phase of a disease.
Proximity extension assays (PEA), like Olink’s (Uppsala, Sweden) target panel, are proteomics measurement techniques that address this challenge. These techniques allow a large number of proteins to be measured in many samples simultaneously. In brief, this technique uses pairs of oligonucleotide-conjugated antibodies. Upon binding with the protein of interest, the matching oligonucleotides on the antibody pairs form an amplicon which can be subsequently amplified and measured using qPCR. It enables accurate and consistent measurements of proteins without cross-reactivity at a relatively low cost in volumes as low as 1 µl of various matrices like serum, plasma, synovial fluid and dried blood spots2,3. The standardization and scalability of PEA techniques are key features, making them a compelling technology for (large) proteomic studies. However, comparing or pooling data from different centers, or data derived from measurements over prolonged periods of time, remains a challenge, due to technical variations and the introduction of inter-plate variability. These so-called batch effects increase the risk of false discoveries in downstream statistical analyses4.
To mitigate batch effects in multicenter studies or repeated measurements of a longer period of time, it has been suggested to include at least 8 so-called “bridging controls” (BCs) in every measurement, referring to the practice of including the same samples (with identical freeze–thaw cycle) on every plate5. The analyses of differences between these technical replicates, allow correction of batch effects across different plates and time points. Various methods have been developed to address batch effects in transcriptomic data and mass spectrometry data, including RUV6, ComBat7,8, median centering method9, and Median of the difference (MOD)10. Although some of these methods have been used to correct for batch effects in PEA studies9,11,12, little is known regarding the nature of these batch effects or the number of bridging controls required for optimal correction. To our knowledge no comprehensive study has been published comparing the accuracy of these existing methods using bridging controls for analyses of PEA data.
In this manuscript, we aimed to characterize batch effects in a proteomic study applying the Olink Target panel. We found 3 distinct batch effects and developed a new correction method called BAMBOO for Batch Adjustment using Bridging cOntrOls. In a simulation study, we compared BAMBOO with 3 existing correction methods and showed that overall BAMBOO is the current most robust method. We also observed that BAMBOO can effectively reduce false discovery rates using experimental data in comparison to other methods.
Results
Identification of 3 types of batch effects
We measured a set of 24 samples on two different plates to identify potential batch effects in PEA studies. To visualize potential batch effects, we plotted both plate measurements against each other. If no batch effect was present, one would expect a perfect agreement between the two measurements of each sample and protein (i.e. all the data on the first diagonal x = y). Based on the differences between the measurements, we were able to identify three distinct types of batch effects.
Firstly, we color-coded the 92 proteins and we observed that protein measurements were grouped together (Fig. 1A). For certain proteins, specifically those noted P1, P2, P3 and P4 in Fig. 1A, there is a general deviation from the first diagonal. This indicates that after measuring these proteins on the second plate the NPX values for the 24 samples were higher or lower compared to the first time. We called this batch effect a "protein specific batch effect".

NPX values of 24 samples measured on two different plates. (A) The protein specific batch effects shown in four proteins (p1, p2, p3, p4). Colors highlight the different proteins, values below LOD in one of the two plates are indicated with an open symbol. (B) Example of the sample-specific batch effect for two samples (blue and orange). (C) Visualization of plate-wide effect as shown by a robust linear regression line (blue) fitted to the data.
Secondly, when we color-coded the 24 samples instead of the proteins (Fig. 1B), a distinct deviation from the first diagonal was observed, most noticeable for the purple and red sample. This disparity strongly suggests that all values for a specific sample can be offset with a certain amount between measurements. We called this effect a "sample specific batch effect".
Lastly, we looked at the measurements of the entire plate (Fig. 1C). A notable deviation from the first diagonal can be observed for lower NPX values. To confirm this, a regression model was fitted to the data and investigated if this regression line was significantly different from the first diagonal. To make sure that the above-mentioned batch effects (protein- and sample-specific) did not influence the regression, we used a robust linear regression l (intercept = -0.5; SE = 0.0178; slope = 1.04; SE = 0.0024). We found that the intercept was significantly different from 0 (p < 0.01) and the slope was significantly different from 1 (p < 0.01). This implies that besides the first two described batch effects, there is an overall deviation from the first diagonal influencing all proteins of all samples on the plate equally. We called this a “plate-wide” batch effect.
BAMBOO: a new batch effect correction method for PEA study
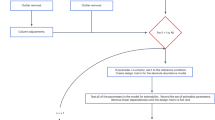
Based on the identified batch effects, we developed a new correction method called BAMBOO for Batch AdjustMents using Bridging cOntrOls. This approach uses bridging controls to adjust measurements from one plate to a reference plate in 4 steps.
The first step is quality filtering, in which the amount of batch effect is determined for each BC \(j\) using the following formula: \({BE}_{j}={\sum }_{i=1}^{{N}_{BC}}{NPX}_{i,1}^{j}-{NPX}_{i,2}^{j}\). Using the Interquartile Range (\(\left[{Q}_{1};{Q}_{3}\right]\)) on the \({BE}_{j}s\), all BCs with a \({BE}_{j}\) lower than \({Q}_{1}-1.5({Q}_{3}-{Q}_{1})\) or higher than \({Q}_{3}+1.5({Q}_{3}-{Q}_{1})\) can be considered as outliers and are removed. In addition, values below the limit of detection (LOD) are removed as they have a higher chance of being on the non-linear phase of the S-curve13. However, if this results in less than 6 BCs measurements for a protein, values below LOD are kept but the protein is flagged to indicate that any statistical result(s) coming from this protein should be interpreted with caution.
In the second step, we estimate the plate-wide batch effects using a robust linear regression model on the bridging control data: \({NPX}_{i ,1 }^{j} = {b}_{0 }+{b}_{1}{NPX}_{i , 2 }^{j}\) , where \({b}_{0}\) and \({b}_{1}\) are used as adjustment factors for plate-wide batch effects.
In the third step, we estimate the adjustment factor for protein specific batch effects (\({AF}_{i})\) as follows: \({AF}_{i} =median({NPXj}_{i, 1 }^{j}- ({b}_{0 }+{b}_{1}{NPX}_{i , 2 }^{j}))\). Lastly, using all the adjustment factors, we adjust the non-bridging control samples to the reference plate: \(adj.NP{X}_{i, 2}^{j} = ({b}_{0 }+{b}_{1}{NPX}_{i, 2 }^{j}) + {AF}_{i}\).
Comparing BAMBOO to other methods: a simulation study
To evaluate our new approach in comparison to existing ones, we performed a simulation study tuning the strength of the different batch effects described above, the number of BCs, the number of outliers within the BCs, plate wide batch effect and other variables (Table 1). We compared BAMBOO with 3 other existing approaches (ComBat, median centering and MOD, see description in methods) and a scenario without any batch effect correction using the following qualitative measures: Accuracy, true positive rate (TPR) and true negative rate (TNR). The values chosen to simulate the different batch effect parameters were in line with what we observed in Fig. 1 with the exception of the plate-wide effect for which we considered the extreme value of 0.1.
First, we compared the accuracy of the 4 batch effect correction methods and the scenario without batch effect correction, without the introducing of a plate-wide effect or outliers in the bridging controls (Fig. 2A). In the scenario without batch effect correction mean accuracy was 84% and was not influenced by the number of bridging controls (not shown). In contrast, all 4 methods show high accuracy (> 95%) however the median centering method resulted in lower accuracy regardless of the number of BCs (96.8% to 97.2%). BAMBOO and MOD showed similar accuracies while ComBat reached slightly higher values. Using more than 10 BCs did not increase the accuracy for BAMBOO, MOD and ComBat.

Accuracies of the four batch correction methods. (A) The accuracies of the four methods without plate-wide batch effects or outliers. (B) Accuracies of the four methods with a plate-wide effect of 0.0025, 0.05 and 0.1. (C) Accuracies of the four methods with 1, 2 or 3 outliers. Lines represent mean of the accuracy over all the simulations.
Since BAMBOO was designed to also correct for plate-wide batch effects, we investigated accuracy when plate-wide effects were introduced. We considered 3 different scenarios: a small, moderate, and large plate-wide effect (Fig. 2B, Supplementary figure 1B). In the scenario with no batch effect correction the mean accuracy dropped to 74%, 58% and 35% respectively, and was not influenced by the number of bridging controls (not shown). In the scenarios with batch effect correction, the median centering method achieved the lowest accuracies (although still acceptable values > 90%) regardless of the scenario and number of BCs used. BAMBOO and ComBat produced similar accuracies when low plate-wide effects were included, while MOD showed lower accuracies overall. When the plate-wide effect was moderate or large, a clear superiority of BAMBOO over ComBat and MOD methods was observed (Fig. 2B, Supplementary figure 1B).
Next, we introduced outliers among the BCs and investigated the accuracy when no plate-wide effect was present. As expected, the introduction of outliers did not influence the accuracy when no batch effect correction was performed. However, when 1, 2 or 3 outliers were included the median centering method and ComBat performed poorly with accuracies as low as 60–80% in cases with less than 10 BCs. In contrast, both BAMBOO and MOD showed high accuracies (> 90%) in all cases (Fig. 2C). Similar results were found when introducing plate-wide effects (small, moderate, and large). Notably, BAMBOO outperformed MOD when large plate-wide effects were present (Supplemental Figure 2).
Since BAMBOO and MOD performed the best based on accuracy to correct batch effects in the presence and absence of outliers, we investigated the TPR and TNR. As we observed that accuracy did not increase with more than 10 BCs, and to limit the number of simulations, we now only considered two scenarios. One with 10 BCs and one with 5 BCs to investigate a more cost-effective study setup (i.e. using less BCs to have more “real” samples measured on each plate).
When using 10 BCs and no plate-wide effect and outliers were simulated, we observed similar TPR and TNR for BAMBOO and MOD (TPR: 99% and TNR: 97%; Figs. 3 and 4). Similarly, when we simulated outliers, both methods performed equally well (TPR > 98% and TNR > 96%). However, when we simulated plate-wide effects with and without outliers, MOD had lower TPR and TNR compared to cases without plate-wide effects while BAMBOO kept similar rates (Figs. 3 and 4). In addition, we observed that in scenarios with plate-wide effects MOD identified false positives that have a larger mean difference compared to the true data and false negatives that have a small mean difference compared to the true data. We observed similar results when we used only 5 BCs (Supplemental Figure 3 and Supplemental Figure 4). Surprisingly, we did not observe differences in TPR and TNR when using 5 BCs or 10 BCs when no outliers and no plate-wide effects were present for both methods. However, MOD had lower TPR and TNR compared to cases with 10 BCs when outliers and/or plate-wide were simulated.

True negative rate for BAMBOO and MOD using 10 bridging controls. The effect size between the two groups after batch correction are on the x-axis and the effect size in the “true” data (i.e. before batch introduction on the simulated data) are on the y-axis. Columns show the number of outliers in the simulations and the rows the strength of the plate-wide effect. False positives (proteins that were not significantly different in the true data and found statistically significant after batch effects correction) are in red and true negatives (proteins that were not significantly different in the true data and still not statistically significant after batch effects correction) are in grey.

True positive rate for BAMBOO and MOD using 10 bridging controls. Left figure (BAMBOO), right figure (MOD). The effect size between the two groups after batch correction are on the x-axis and the effect size in the “true” data (i.e. before batch introduction on the simulated data) are on the y-axis. Columns show the number of outliers in the simulations and the rows the strength of the plate-wide effect. False negatives (proteins that were significantly different in the true data and found not statistically significant after batch effects correction) are in orange and true positives (proteins that were significantly different in the true data and still statistically significant after batch effects correction) are in grey.
In conclusion, both BAMBOO and MOD perform well in removing batch effects when no outlier within the BCs and/or no (or small) plate-wide effect are present. But BAMBOO outperforms MOD when plate-wide effect and/or outliers are introduced and even more when a small number of BCs were measured.
Validation to experimental data: Healthy controls vs viral infected individuals
To validate our method on real sample data, we compared 31 healthy controls (HC, measured on plate A) and 14 viral infected individuals (measured on plate B). These data were obtained from different studies and were measured on separate plates months apart. To correct for the batch effects, a set of 10 BCs were included on both plates. We measured the 92 proteins using the Olink T96 Immuno-Oncology panel.
To visually assess the presence of batch effects, we first plotted the 10 BCs of each plate against each other (Supplemental Figure 5). We observed protein specific batch effects as well as a plate-wide specific batch effect. The presence of these batch effects was confirmed using a hierarchical cluster analysis where we observed a clear separation of both plates (Fig. 5). We corrected the data using 4 different approaches: BAMBOO, MOD, the median centering and ComBat.

Heatmap of the NPX values of HC and viral infection samples measured on plate A and plate B. NPX values were transformed as Z-scores. Each row represents a protein, and each column a sample. Clustering was performed using Wards-d2 clustering algorithm with the Euclidean distance.
v
The batch adjusted data was used to identify differentially expressed proteins (using the Wilcoxon rank sum test with an FDR cut-off at 0.05). When no batch effect correction was applied, 84 of the 92 proteins were called significant. However when batch effect correction was applied all 4 methods found the same 60 proteins to be significantly different between the two groups of individuals (Supplementary Figure 6). 9 proteins were found significantly different after batch effects correction only by one of the 4 methods: 5 after using ComBat, 3 after using median centering method, 1 after using BAMBOO and none after using MOD. The protein found significant after using BAMBOO had more than 6 measured values below LOD and was therefore flagged by BAMBOO to indicate that results should be interpreted with caution (see methods).
For these 9 proteins, we investigated if their discovery could be due to improper or incomplete removal of batch effects. This was done by looking into the paired bridging control measurements after batch effect correction (Fig. 6). For the 5 proteins called significant by only ComBat, 3 proteins showed a clear deviation from the first diagonal indicating improper batch effect correction. Additionally, we observed that 4 of these 5 proteins had one measurement that could be classified as an outlier. For the 3 proteins found significant only by median centering method, we also observed a deviation from the first diagonal for 2 of them. Additionally, we observed that the 3 proteins present a bimodal distribution with a median value that could be defined as an outlier.

NPX values of the BCs for the proteins found significant by only one of the four methods after batch correction. (A) NPX values of the bridging controls for the protein found significant only after batch correction with BAMBOO. (B) NPX values of the bridging controls for the 3 proteins found significant only after batch correction with median centering normalization. (C) NPX values of the bridging controls for the 5 proteins found significant only after batch correction with ComBat.
Based on the analysis of experimental data, we can conclude that ComBat and median centering greatly suffer from the presence of outliers within the BCs and that BAMBOO is able to flag potential false discoveries due to low measured values.
Discussion
Here, we investigated batch effects occurring in proteomic studies using Olink PEA technologies. We developed a new method, called BAMBOO, which can correct for the identified batch effects using a minimal number of bridging controls. We compared this new method to existing alternatives using a simulation study and an experimental dataset. In both cases, BAMBOO corrected well for the batch effects and had potentially less false discoveries.
With the emerging technologies and increasing prevalence of large-scale proteomic studies, efforts to characterize batch effects are crucial. However, in many cases, the methodology for their assessment remains unclear or even unattainable when no bridging controls are included. A recent comprehensive study by Eldjarn et al., investigated the reproducibility of Olink and SomaScan, using the ratio of the coefficient of variation (CV) of repeated measurements to the CV of the assay14. Their findings revealed that the Olink Discovery assays exhibit greater precision than SomaScan. Interestingly, imperfect CV ratios suggested the potential presence of batch effects, in contrast to previous studies with smaller sample sizes15,16,17. In another study, Haslam et al., evaluated Olink’s reproducibility using a triplicate of plasma samples among other analyses18. They found that approximately half of the proteins measured demonstrated excellent stability (Spearman r > 0.75) while about a third exhibited good stability (Spearman 0.40 < r < 0.75). These findings align with our results, indicating that most proteins measured in technical replicates display a good correlation between plates. However, it is noteworthy that some proteins show protein-specific batch effects.
A recent paper by Dammer et al., presents a comprehensive overview of current available methods to correct for batch effects that might be due to variations in sample preparation, batching, platform settings, personnel, and other experimental procedures19. They also proposed a new version of the median polish approach initially described by John Tukey in 197720. This new method is called TAMPOR and it can be used with or without bridging controls. While this method appears efficient to compare data from different platforms, data are transformed by an abundance normalization and therefore lose their original log2 scale (in case of Olink), complicating data interpretation. It seems that methods such as BAMBOO and MOD are preferable for large scale studies as data will keep their original scale and TAMPOR might be preferred when comparing or combining data from different platforms.
We performed a simulation study to compare our new approach, BAMBOO, with existing approaches. The most basic batch correction method, median centering, did not perform well even in scenarios without plate-wide effect and outliers. Subsequently, ComBat, originally designed for processing transcriptomics data performed equally well as BAMBOO in the absence of plate-wide effects and outliers. However, its performance suffered when these factors were present. Although originally developed for microarray data correction, ComBat is widely used for analyses in other fields of omics data, such as Olink studies12,21,22. Lastly, MOD, a simple method in which the median of the paired-differences between bridging controls is used as a correction factor, showed comparable performance to BAMBOO in scenarios with outliers. However, this method was not robust against plate-wide batch effects. We showed that in scenarios with plate-wide effects, MOD identified more false positives with relatively large effect sizes and more false negatives with small effect sizes. It is possible that MOD over-corrects for batch effects and hence leads to more false positives and negatives in statistical analysis.
In our simulation study and the validation using experimental data, we saw that both ComBat and median centering are impacted by outliers. Proteins called significantly different after correction with one of these methods showed a clear deviation from the first diagonal. Even though the experimental data did not contain a complete sample as outliers (all proteins of a sample), some individual proteins could be identified as such (outside the expected range). Interestingly, most studies made on microarray data show that ComBat can deal well with outliers7,8,23. When looking at other types of data, such as imaging, Han et al., showed that using ComBat without identifying outliers could lead to false discoveries24. This suggests that the type of data used for ComBat can also influence its performance.
It is advised to take along at least 8 bridging controls per plate for correcting batch effects5. Logically, the more bridging controls are used, the better batch effects are corrected. However, there is a balance between the number of experimental samples that can be measured and how precisely batch effects need to be corrected, also in terms of the available budget. Our simulation study showed that 10 BCs are sufficient to accurately correct for batch effects even when there is a strong plate-wide effect, when using BAMBOO. However, even in economical scenarios with 5 BCs, we showed that BAMBOO still adjusts well for batch effects, even with strong plate-wide effects (Accuracy > 96%). However, its performances dropped with the addition of one outlier. Hence, we advise to take along 10–12 BCs to account for the removal of potential outliers when using BAMBOO. Additionally, we recommend using a biologically heterogeneous group of samples (i.e. healthy and diseased) to increase the ranges of measurements and making sure that the majority will be above the LOD.
One of the novelties of our approach is to flag proteins for which BCs are below the limit of detection. In this situation, it is difficult to compute adjustment factors as the difference between two plates for those BCs will be null. Hence, those proteins might still show a batch effect in the downstream analyses. Another novelty of our approach is the ability of BAMBOO to detect outliers within the BCs and to exclude them from correction.
In conclusion, we have identified the different batch effects that can be observed in proteomic studies and developed a new method to correct for them and compared it with 3 commonly used methods. We showed that 10–12 bridging controls is the optimal number of BCs to take along to accurately correct for batch effects. ComBat and median centering cannot properly correct for them, and we therefore advise to not use them for PEA studies. One method (MOD) was influenced by plate-wide batch effects and is therefore not recommended to use when such batch effects are present in the data. We therefore advise to use BAMBOO in all studies, which is available on GitHub (https://github.com/CIC-UMCutrecht/BAMBOO/).
Methods
PEA measurements using Olink technology
Relative protein concentrations were measured using PEA technology based Proseek Multiplex panels (Olink Proteomics), performed by the Olink service provider, Arcadia, in the UMC Utrecht, the Netherlands. In short, upon binding of antibody pairs to their respective targets, DNA reporter molecules conjugated to the antibodies give rise to new antigen specific DNA amplicons. Subsequently, amplicons are quantified using real-time PCR. The raw quantification cycle values are normalized and converted into normalized protein expression (NPX) units. The NPX values are expressed on a log2 scale in which one unit increase in NPX values represents a doubling of the protein concentration. Different quality controls were measured on every sample and plate using Olink’s standard quality control protocol25.
Bridging controls and experimental data
To characterize batch effects, we analyzed a selection of 8 healthy controls (HC) samples and 16 samples from patients with autoimmune disease to maximize the ranges of values. All samples were measured twice on separate plates. We obtained informed consent for all HC and patients. The institutional ethics committee of UMC Utrecht (the Netherlands) approved blood draws for all studies (07/125 for HC, NL61114.041.17. for IBD patients and NL47875.041.14 for JDM patients). To evaluate batch correction methods on actual experimental data, we measured serum samples from 14 participants that experienced a virus infection included within the RESCEU project (ref 17/069 and NL60910.041.17), along with 31 serum samples from healthy controls, using Olink’s Target 96 Immuno-oncology panel. For these data all methods were performed in accordance with the relevant guidelines and regulations.
After blood draw, serum samples were allowed to stand for at least 30 min and maximum of 4 h before centrifugation at 3000 RPM for 10 min and stored at -80°C. Sodium heparin plasma samples were obtained by spinning at 1000 g for 10 min. Healthy control serum samples were directly aliquoted into micronic tubes at a volume of 50 µl each and stored at -80°C prior to measurement. Patient samples, used as bridging controls, were initially stored at -80°C, thawed, aliquoted in 20 µl amounts, and refrozen at -80°C before measurement.
Simulated data
To compare our new approach to existing methods, we performed a simulation study. Each simulation involved two plates, each containing 88 samples for measurement of 92 proteins. Each protein \(i (i=1,\dots ,92)\) was assumed to follow a normal distribution \(N({\mu }_{i},{\sigma }_{i})\), where \({\mu }_{i}\sim U(\text{0,15})\) and \({\sigma }_{i}\sim U(\text{0.1,2})\). To introduce biological variability (for instance healthy controls vs. diseased individuals), we assumed that a certain number of proteins had different means (\({\mu }_{i}^{BG}\), where \(BG\) denotes the different biological groups). The number of proteins for which we assumed biological variability, and the differences in mean were tunable parameters (\({N}_{BV}\) and \({\Delta }_{BV}\)) in our simulations. Each sample was defined by randomly drawing values from these 92 normal distributions. To simulate the bringing controls, a number of samples (\({N}_{BC}\)) were identical on both plates.
Subsequently, batch effects were added to the simulated plates. Random noise was added to each protein following a normal distribution \(N(0, {\sigma }_{i}^{noise})\). For a number of proteins (\({N}_{BE}\)), additional noise was added by changing the mean of the distribution from which the random noise was drawn to \(N({\mu }_{i}^{BE},{\sigma }_{i}^{noise}\)). In addition, a selected number of samples (\({N}_{OS}\)) were introduced as potential outliers (\(OS\)) on one of the two simulated plates. Those samples were created by randomly adding or subtracting to the NPX of all protein one value from the following list: \(-3, -2.5, -1.5, 1.5, 2.5, 3\). Finaly, we introduced noise to all values on one plate by using a linear function (intercept β0 and slope β1) as follows: \({NPX}_{D}={\beta }_{0}+{\beta }_{1}NPX\). Table 1 shows the parameter values used for the simulations. Each possible parameter combination was simulated 50 times.
Comparison of the methods
The simulated and experimental plates were corrected for batch effects using 4 different methods: our new method called BAMBOO (Batch Adjustment using Bridging cOntrOls), median centering (also called intensity normalization), the MOD method, and SVA’s ComBat. Median centering calculates the adjustment factor for each protein based on the difference between the medians of the two plates9,25. In the MOD method, which is the method advised by Olink, the adjustment factor is derived from the pair-wise difference between the bridging controls on both plates25. Lastly, in ComBat, a linear model is used to estimate batch-specific means and variances for each protein, this method is based on an Empirical Bayes framework7,8. For this method, one of the covariates was set to the sample identification variable to incorporate the bridging controls.
The quality of batch effect correction in the simulated data was determined by computing the number of True Positives (TP), True Negatives (TN), False Positives (FP) and False Negatives (FN). TP were proteins significant in the dataset without batch effects that remained significant after correction of the batch effect introduced data, while TN were non-significant proteins that remained non-significant after correction. FP were proteins non-significant in the dataset without batch effects but became significant after correction, and FN were significant proteins that lost significance after correction. Based on these values the following qualitative measures were calculated: accuracy (\(acc= \frac{TP + TN}{TP + TN + FP + FN}*100\)), true positive rate (\(TPR= \frac{TP}{TP+ FN}*100\)), and true negative rate (\(TNR= \frac{TN}{TN + FP}*100\)).
Differential proteins were identified using t-tests between the two different biological groups using a statistically significant threshold after FDR correction of 0.05.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Califf, R. M. Biomarker definitions and their applications. Exp. Biol. Med. 243, 213–221 (2018).
Akesson, J. et al. Proteomics reveal biomarkers for diagnosis, disease activity and long-term disability outcomes in multiple sclerosis. Nat. Commun. 14, 6903 (2023).
Deng, J. et al. Multi-omics approach identifies PI3 as a biomarker for disease severity and hyper-keratinization in psoriasis. J. Dermatol. Sci. 111, 101 (2023).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
Olink Proteomics. Strategies for design of protein biomarker studies. White paper (2018). [Online] Available at: https://www.olink.com/content/uploads/2021/09/olink-strategies-for-design-of-protein-biomarker-studies-1098-v2.0.pdf
Risso, D. et al. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Zhang, Y., Parmigiani, G. & Johnson, W. E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genomics Bioinform. 2, Iqaa078 (2020).
Alhamdow, A. et al. Cancer-related proteins in serum are altered in workers occupationally exposed to polycyclic aromatic hydrocarbons: a cross-sectional study. Carcinogenesis 40, 771–781 (2019).
Dubois, E. et al. Assessing normalization methods in mass spectrometry-based proteome profiling of clinical samples. BioSystems 215–216, 104661 (2022).
Shah, R. V. et al. Proteins altered by surgical weight loss highlight biomarkers of insulin resistance in the community. Arterioscler Thromb. Vasc. Biol. 39, 107–115 (2019).
Stanne, T. M. et al. longitudinal study reveals long-term proinflammatory proteomic signature after ischemic stroke across subtypes. Stroke 53, 2847–2858 (2022).
Olink Proteomics. How is the limit of detection (LOD) estimated and handled? [Online] Available at: https://olink.com/faq/how-is-the-limit-of-detection-lod-estimated-and-handled/
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Katz, D. H. et al. Proteomic profiling platforms head to head: Leveraging genetics and clinical traits to compare aptamer-and antibody-based methods. Sci. Adv. 8, 5164 (2022).
Candia, J. et al. Assessment of variability in the SOMAscan assay. Sci. Rep. 7, 14248 (2017).
Raffield, L. M. et al. Comparison of proteomic assessment methods in multiple cohort studies. Proteomics 20, e1900278 (2020).
Haslam, D. E. et al. Stability and reproducibility of proteomic profiles in epidemiological studies: Comparing the Olink and SOMAscan platforms. Proteomics 22, e2100170 (2022).
Dammer, E.B., Seyfried, N.T. & Johnson, E.C.B. Batch correction and harmonization of –Omics datasets with a tunable median polish of ratio. Front. Syst. Biol. 3, (2023).
Tukey, J. W. Exploratory Data Analysis (Addison-Wesley, 1977).
Åkesson, J. et al. Proteomics reveal biomarkers for diagnosis, disease activity and long-term disability outcomes in multiple sclerosis. Nat. Commun. 14, 6903 (2023).
Angerfors, A. et al. Proteomic profiling identifies novel inflammation-related plasma proteins associated with ischemic stroke outcome. J. Neuroinflamm. 20, (2023).
Chen, C. et al. Removing batch effects in analysis of expression microarray data: An evaluation of six batch adjustment methods. PLoS ONE 6, e17238 (2011).
Han, Q. et al. Characterization of the effects of outliers on ComBat harmonization for removing inter-site data heterogeneity in multisite neuroimaging studies. Front. Neurosci. 17, 1146175 (2023).
Olink Proteomics. Data normalization and standardization. White paper (2021). [Online] Available at: https://www.olink.com/content/uploads/2021/09/olink-data-normalization-white-paper-v2.0.pdf
Acknowledgements
The authors would like to acknowledge Louis Bont for providing the viral infected samples used as experimental data.
Author information
Authors and Affiliations
Contributions
HMS contributed to the analysis, interpretation of the data and the development of the software under the mentorship of JD. EMD and SN contributed to the design of the study and acquisition of the data. AS and BO contributed to the acquisition of the data. AP and FvW contributed to the interpretation of the data. All authors contributed to the revision of the manuscript drafted by HMS and JD.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Informed consent was obtained from all individuals included. The different studies were approved by the ethics committee of the UMC Utrecht (the Netherlands): 07/125 for HC, NL61114.041.17. for IBD patients,NL47875.041.14 for JDM patients and NL60910.041.17 for virus infection patients and all methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Smits, H.M., Delemarre, E.M., Pandit, A. et al. The BAMBOO method for correcting batch effects in high throughput proximity extension assays for proteomic studies. Sci Rep 15, 1498 (2025). https://doi.org/10.1038/s41598-024-84320-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-84320-4
