
Abstract
The solubility of commonly used anti-inflammatory drugs has become a significant concern in contemporary medicine. Furthermore, inflammatory arthritis stands out as the most prevalent chronic inflammatory disease globally. The disease’s pathology is characterized by heightened inflammation and oxidative stress, culminating in chronic pain and the loss of joint functionality. Fenofibric acid (FFA) exhibits notable anti-inflammatory potential. This research assesses the anti-inflammatory effects of FFA, both in its standalone form and as inclusion complexes (ICs) with β-cyclodextrin and its methyl derivatives. FFA is encapsulated within the cavities of cyclodextrins (CDs), a fact confirmed by spectral changes observed in FFA. Distinct rock and seed-like morphologies are apparent for FFA with CDs, indicating that the CDs have influenced the surface of the guest. The introduction of CDs significantly enhances the thermal stability of FFA. ICs demonstrate superior results in inflammation activity compared to FFA alone. The efficacy of FFA complexed with CDs in mitigating inflammation positions it as a promising new drug. Additionally, our findings reveal that incorporating FFA into the CD cavity as a drug release system enhances the pharmacological profile of this substance, FFA.
Similar content being viewed by others
Introduction
The body initiates inflammation in response to tissue damage, to restore the integrity of the injured tissue through various mechanisms of induction, regulation, and resolution. Regardless of the triggering stimulus, this response is crucial for reinstating homeostasis and serves a significant physiological function1,2. Non-steroidal anti-inflammatory drugs (NSAIDs) rank among the most widely used medications globally. Primarily employed to alleviate both short- and long-term pain, mitigate inflammation, and lower fever, they achieve these effects by inhibiting the cyclooxygenase enzyme3. However, despite their efficacy, NSAIDs are linked to a diverse array of well-established side effects, largely attributable to their limited solubility and, consequently, exhibit dose-dependent adverse reactions4.
For a drug to be absorbed by the body, it must be in solution. However, the limited solubility of compounds with poor water solubility can lead to reduced drug absorption. This, in turn, necessitates higher dosages and may contribute to the emergence of side effects due to variations in individual absorption. Consequently, addressing the solubility challenges of poorly soluble drugs stands as a critical concern in drug formulation5. Nevertheless, the limited solubility of NSAIDs, often associated with low bioavailability, imposes constraints on their application in topical and parenteral contexts. Strategies to enhance the water solubility of NSAIDs include incorporating surface-active agents and forming water-soluble salts. To augment dissolution and absorption rates, techniques such as improving wettability and micronizing drug particles are frequently employed to boost the bioavailability of poorly water-soluble NSAIDs6,7. A novel approach involves using deep eutectic solvents (DESs) as environmentally friendly solvents for dissolving NSAIDs8,9. While salt formation remains a common and effective method to improve the solubility and dissolution rate of acidic and basic drugs3,10,11, not all active pharmaceutical ingredients (APIs) can yield multidrug salts. Despite these efforts, achieving the desired outcomes has not always been successful. In recent years, there has been a focus on utilizing and developing various macromolecules to enhance the solubility of NSAIDs and mitigate their side effects12,13,14. Nonetheless, a growing body of evidence has indicated that these medications can lead to substantial toxicity15. Consequently, the task of developing novel, safe, and productive anti-inflammatory drugs continues to pose a challenge16.
To address the challenges associated with poor solubility or bioavailability of certain drug molecules, the focus often shifts toward strategies aimed at enhancing their stability and effective delivery. One such approach involves the use of cyclodextrins (CDs), which have gained significant attention for their ability to improve the solubility of poorly soluble drugs17,18,19. CDs are cyclic oligosaccharides that are well-regarded for their capacity to encapsulate hydrophobic drug molecules within their central cavity, thereby enhancing their aqueous solubility and bioavailability. Chemically, CDs are composed of glucose units linked by α-(1,4)-glycosidic bonds, forming a truncated cone-shaped structure. They are derived from the enzymatic degradation of starch by CD glucosyltransferase. The most commonly used types of CDs are α-, β-, and γ-CDs, which consist of six, seven, and eight glucose units, respectively. The size of the CD cavity increases with the number of glucose units, with β-CD being the most widely studied and used due to its optimal cavity size for accommodating a variety of drug molecules. The CD ring exhibits an amphiphilic nature, comprising a hydrophilic outer layer (formed by hydroxyl groups) and a lipophilic cavity20,21 and complexing poorly water-soluble drugs with CDs results in the formation of an “inclusion complex”22,23,24. To enhance CD monomer properties, chemically derived derivatives (e.g., Hydroxypropyl-β-CD (HCD) or Methyl-β-CD, among others) and polymers have demonstrated superior capabilities, such as increased complexation efficiency or release, compared to natural CDs25,26,27. Methyl-β-CD (MCD) offers distinct advantages over other modified BCDs in several key areas, making it a preferred choice for pharmaceutical and biomedical applications: enhanced solubility, and superior drug delivery capabilities28,29,30,31,32. Recently, administering an anti-inflammatory drug as an inclusion complex (ICs) has shown the potential to enhance efficacy and formulation capabilities, representing an intriguing avenue for improving current treatments33,34,35,36,37,38.
Recognizing the significance of ICs in drug development, this investigation seeks to assess the influence of CD inclusion on the biopharmaceutical and pharmacological attributes of fenofibric acid (FFA), a non-steroidal anti-inflammatory drug. A recent study has highlighted the potential benefits of FFA in combating the pathogenesis of SARS-CoV-2 infection and alleviating the severity of COVID-19 due to its anti-inflammatory activities39. To date, several studies in the literature have focused on the formation of ICs of FFA with both BCD and HCD. These studies demonstrate a notable improvement in the solubility of FFA when complexed with BCD, with a moderate enhancement observed using HCD as well40,41,42,43,44. The research has predominantly investigated individual CD complexes, analyzing solid-state complexes to characterize the interactions between FFA and CDs45,46,47. These analyses have conclusively proven the occurrence of complexation between FFA and the respective CDs. In addition to the initial studies, there has been further exploration involving methyl-substituted BCD, which has provided insights into its interaction with FFA48,49. However, a thorough review of the available literature reveals a significant gap: while numerous studies emphasize the complexation process and its impact on the physicochemical properties of FFA, there is a lack of research exploring the biological applications of these complexes, particularly their potential anti-inflammatory activity. This gap highlights an opportunity for further investigation into the therapeutic potential of FFA-CD complexes in biological systems.
Therefore, we have initiated a research project focusing on FFA to develop ICs with CDs. This includes both natural CD (BCD) and modified CD (MCD), enabling a comprehensive comparative analysis of their properties. A detailed chemical characterization of these ICs is being conducted, with a particular emphasis on using ROESY to elucidate the spatial interactions between FFA and the CD molecules. Additionally, theoretical approaches, such as quantum mechanical calculations, are being employed to support and validate the experimental findings. To ensure the biocompatibility of these ICs, cytotoxicity assays are being performed. This step is critical, as confirming the safety of the complexes is a prerequisite for their potential application in anti-inflammatory therapy. By addressing both the chemical and biological aspects of the ICs, this study aims to lay the groundwork for the effective therapeutic use of FFA-CD complexes in managing inflammatory conditions.
Materials and methods
Materials
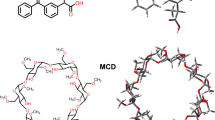
The essential chemicals used in this study include FFA, BCD, and MCD. FFA, with the molecular formula C17H15ClO4, has a molecular weight of 318.75 g/mol and a purity of 99%. BCD, with the molecular formula C42H70O35, has a molecular weight of 1134.98 g/mol and a purity of 98%. MCD, with the molecular formula C54H94O35 and a molecular weight of approximately 1303.31 g/mol, has a purity of 98% and a degree of substitution ranging from 6.0 to 10.0. All chemicals were obtained from Sigma-Aldrich Chemical Co (Seoul, South Korea). Throughout the study, distilled water was used as the solvent. The structural representations of FFA (the guest molecule) and the two CDs (BCD and MCD) are shown in Fig. 1, which illustrates the molecular structures and the key interactions involved in the complexation.

Structures of FFA (A), BCD (B), and MCD (C).
Phase solubility (PS) study
Phase solubility diagrams50 for the ICs were constructed by adding an excess amount of FFA to CD solutions, specifically 16 mg of FFA per mL of solution. The CD concentrations were set at 22 mg of BCD and 23 mg of MCD per mL of solution, with the concentration of CDs progressively increasing during the experiment. To ensure that FFA remained in its unionized form, the pH of the FFA solution was carefully adjusted to 3.0 using an appropriate buffer solution. This pH was kept below the pKa of FFA to maintain its low solubility, as unionized drug molecules generally exhibit poor solubility in water. The prepared solutions were stirred at room temperature for 24 h to allow the system to reach equilibrium. After this period, the solutions were filtered through 0.45 μm filters to remove any undissolved particles. The solubility of FFA was then determined by measuring the absorbance at a wavelength of 295.0 nm, allowing for the quantification of dissolved FFA and the construction of PS diagrams to evaluate the solubility enhancement by the CDs.
Preparation of FFA:CD ICs
The preparation of ICs involving FFA with the hosts, BCD and MCD was carried out by first dissolving FFA at a concentration of 0.47 mM in ethanol. Concurrently, BCD and MCD were each dissolved at a concentration of 0.47 mM in 20 mL of distilled water. The solution of BCD, and MCD were separately subjected to sonication in an ultrasonic bath (Hielscher UP200St, 30 kHz, 100 W, Iran) for 5 min to ensure complete blending of the components. To form the ICs, a solution of FFA was gradually added drop by drop to separate solutions of BCD and MCD, each maintaining an identical concentration, resulting in a 1:1 molar ratio of FFA to the CDs. The resulting mixtures were heated to 60.0 °C while being stirred magnetically to promote complex formation. Upon completion of the mixing process, the resulting ICs exhibited water solubility, evidenced by the clear, homogenous appearance of the solution. To remove the solvent, a water-based removal technique was applied. The solutions were filtered through a 0.45 μm syringe filter to remove any particulate matter. Subsequently, the filtered solutions were frozen at -81.0 °C for 48 h, followed by lyophilization at − 80.5 °C under a pressure of 1.09 Pa for 72 h in a freeze-dryer51,52. This freeze-drying process resulted in the formation of a white powder, representing the ICs. The final products were denoted as IC-1 (FFA:BCD ICs) and IC-2 (FFA:MCD ICs), respectively.
Experimental procedure for anti-inflammatory activity
The synthesized compound and the standard anti-inflammatory drug, diclofenac sodium, were evaluated for their anti-inflammatory activity using the inhibition of albumin denaturation technique, with slight modifications to the established method53. This technique is widely used to assess anti-inflammatory properties based on the ability of test compounds to prevent protein denaturation, a common precursor to inflammatory processes. To prepare the solutions, both the synthesized compound and diclofenac sodium were dissolved in a minimal quantity of dimethylformamide (DMF) to ensure solubility. These stock solutions were then diluted with phosphate buffer (0.2 M, pH 7.4) to achieve the desired concentrations. The final concentration of DMF in all test solutions was maintained below 2.5%, minimizing any potential solvent interference in the assay. For the assay, a 2.5 mL aliquot of the test solution, containing various concentrations of the drug, was mixed with 1 mL of a 1 mM bovine serum albumin (BSA) solution prepared in phosphate buffer. The mixture was incubated at 37 °C for 10 min to allow the drug to interact with the BSA. Denaturation of the protein was induced by exposing the reaction mixture to 70 °C for 10 min in a water bath. Following this heat-induced denaturation, the samples were cooled to room temperature, and the turbidity of the resulting solutions was measured spectrophotometrically at 660 nm. The extent of inhibition of albumin denaturation was determined by comparing the turbidity of the test samples with a reference sample where no drug was added. The percentage inhibition of denaturation was calculated using the following formula:
At Absorbance of test, Ac Absorbance of control.
Results and discussion
Effect of FFA on the addition of BCD, and MCD
The absorption spectra of FFA in aqueous solution (pH ~ 7) are recorded at various concentrations of BCD and MCD, with the results presented in Fig. S1. As shown in Fig. S1A and B, the absorbance of FFA increased with rising concentrations of both BCD and MCD. Importantly, no shifts were observed in the absorption maximum of FFA upon the addition of either CD, suggesting that the structure of the guest molecule remained unchanged during complexation. This indicates that FFA was likely encapsulated within the cavities of the CDs, rather than undergoing a chemical transformation. The observed increase in absorbance can be attributed to the encapsulation of FFA within the hydrophobic cavities of the CDs, which alters the environment of the drug from the more protic aqueous phase to the less protic microenvironment inside the CD cavity. Additionally, some of the observed absorbance increases may also result from FFA adsorption onto the walls of the container. To ensure the stability of the complex, absorbance measurements were repeated at approximately 12 and 48 h after the solution preparation. No significant change in absorbance was observed, indicating that the encapsulation of FFA remained stable over time, and the IC did not decompose, even after prolonged storage. Furthermore, Fig. S1C and D illustrate the effects of adding BCD and MCD on the fluorescence spectra of an aqueous solution of FFA at a constant concentration. As shown, increasing the concentration of CD resulted in a noticeable enhancement of the fluorescence intensity, reaching up to 12 × 10–3 M. This enhancement is likely a result of the complexation between FFA and the CDs. At shorter intermolecular distances, radiation-less energy transfer from the excited state of FFA to the ground state of the CDs occurs, which amplifies the fluorescence intensity54,55,56. This further supports the notion that the complexation of FFA with the CDs not only affects its absorption properties but also enhances its fluorescence behavior.
The binding constant (K) and stoichiometry of the ICs were determined using the Benesi–Hildebrand (BH) equation57,58,59. This method is applied to analyze the variations in the absorbance as well as fluorescence intensities of FFA as the concentrations of CDs are varied. The observed data confirmed the formation of a 1:1 complex in both systems, as illustrated in Fig. 2. To calculate the K for the 1:1 complex, the BH equation was utilized, which allows for the determination of the binding affinity. The binding constant (K) was then determined from the following Eqs. (1), and (2), based on the measured absorbance and fluorescence data. This analysis not only validates the formation of 1:1 ICs but also provides precise values for the binding affinity, further supporting the stability and strength of the interaction between FFA and the CDs.

BH absorbance plots of FFA with BCD (A), MCD (B), BH fluorescence plots of FFA with BCD (C), and MCD (D).
In order to further evaluate the spontaneity of ICs, thermodynamic parameters can be utilized to provide a deeper understanding of the displacement of FFA within the CD cavities and the exchange of water molecules that occurs during complexation. Specifically, the Gibbs free energy change (ΔG) is an essential metric used to assess the spontaneity of the complexation process and to explore the influence of temperature on the stability of the ICs60,61. In this study, under aqueous conditions at neutral pH and room temperature, negative ΔG values were observed for both FFA:BCD and FFA:MCD ICs (refer to Table S1). A negative ΔG indicates that the complexation process is thermodynamically favorable and occurs spontaneously, providing valuable insight into the binding strength between the CD and FFA. Moreover, the negative ΔG values suggest that the complexation is exergonic, meaning that the reaction releases energy and proceeds spontaneously at room temperature. These findings not only support the formation of stable ICs but also highlight the driving forces behind the interaction between FFA and the CDs, shedding light on the nature of the binding process.
Phase solubility (PS) diagrams
PS diagrams are constructed for both ICs to evaluate the solubility behavior of the free FFA drug about varying concentrations of CDs62,63. These diagrams serve as a critical tool in understanding the solubilization potential and binding characteristics of the drug-CD system64. The PS analysis revealed a direct and linear increase in FFA concentration with an increasing concentration of CDs, consistent with an AL-type solubility profile (Fig. 3). This type of linearity is indicative of the formation of a 1:1 stoichiometric complex between FFA and CDs, a characteristic commonly observed in simple host–guest inclusion systems65,66,67. In the absence of CDs, the water solubility of FFA was determined to be approximately 1.5 mM, reflecting its intrinsic solubility in an aqueous medium. However, the addition of CDs significantly enhanced the solubility of FFA. At the highest CD concentrations, the water solubility of FFA was observed to increase approximately 1.55-fold and 1.68-fold in the presence of BCD and MCD, respectively. This enhancement can be attributed to the encapsulation of the FFA molecule within the hydrophobic cavity of the CDs, which mitigates the hydrophobic interactions that limit its solubility in water. Interestingly, the saturation concentration of FFA displayed minimal variation, even at high CD concentrations. This suggests that the dissolution behavior of the FFA:BCD and FFA:MCD systems is governed predominantly by weak intermolecular interactions between FFA and CDs in the buffer solution. The limited strength of these interactions implies that the inclusion process does not significantly alter the physicochemical environment beyond facilitating improved solubility. This observation aligns with prior studies emphasizing the role of weak host–guest interactions in modulating drug solubility in CD systems67. The LOD, and LOQ values are found to be 0.021, and 0.046 mg/mL, respectively.

PS diagram of FFA in the presence of BCD (IC-1), and MCD (IC-2).
Analysis of proton NMR spectrum of IC-1
The 1H NMR spectrum of the IC-1 is shown in Fig. S4. In this spectrum, the aromatic protons from both FFA and BCD are clearly observed. Notably, the protons on the phenyl ring of FFA, which bears a chlorine atom, experience a deshielding effect, with a chemical shift change of 0.002 ppm. Additionally, the methyl proton of FFA undergoes a slight upfield shift of 0.005 ppm. Other protons in the FFA molecule remain unaffected by the complexation. For BCD, the equatorial methine proton at C-1 exhibits a shielding effect, with a chemical shift change of 0.03 ppm. These changes in proton chemical shifts are indicative of the formation of the IC between FFA and BCD. Further confirmation of this complexation is provided by the ROESY analysis, which also supports the interaction between FFA and BCD in IC-1. A detailed list of the proton chemical shifts for FFA, BCD, and IC-1 can be found in Table S2.
Analysis of proton NMR spectrum of IC-2
The 1H NMR spectrum of the IC-2 is shown in Fig. S4. In this spectrum, the aromatic protons of both FFA and MCD are clearly detected. The protons on the phenyl ring of FFA, which contains a chlorine atom, exhibit a deshielding effect, resulting in a chemical shift change of 0.01 ppm. Additionally, the methyl proton of FFA experiences a slight upfield shift of 0.001 ppm, while the other protons in FFA remain unaffected by the complexation. For MCD, all protons, except for the H2 proton, experience shielding effects, with chemical shifts changing between 0.12 and 0.075 ppm. The H2 proton of MCD, on the other hand, undergoes a deshielding shift of 0.2 ppm. Furthermore, the methyl protons on the methoxy groups of MCD are shielded by 0.4 ppm. These changes in proton chemical shifts provide strong evidence for the formation of IC-2 between FFA and MCD. The formation of the ICs is further confirmed by the ROESY analysis, which supports the interaction between FFA and MCD in IC-2. A detailed list of the proton chemical shifts for FFA, MCD, and IC-2 is presented in Table S3.
ROESY analysis of IC-1
The ROESY of IC-1 is shown in Fig. 4A, and the cross peaks are indicated as A, B, C, D, and E (Fig. S5). In the ROESY, we observed the cross-peaks A (6.92/1.59), B (6.92/3.29), and C (6.92/7.72). The above cross peaks reveal correlations between the phenyl protons attached to oxygen and methyl protons connected with the phenyl ring with ether linkage. The proton chemical shifts at 6.92, 7.72, and 1.59 are already assigned to H3’'/H5’', H2’'/H6’' and methyl proton of the acid part attached. Cross peak B suggests the correlation between protons of phenyl ring attached to oxygen and H4 of β-CD. This confirms the formation of the inclusion complex. Additionally, a few more cross-peaks D, E, F, and G at 5.73/6.92, 5.73/4.82, 5.73/3.59 and 5.73/3.30 are obtained. These correlations are due to the H1 of β-CD with phenyl protons, equatorial hydroxyl proton at C-3, methylene protons attached with C-5 and H4 (axial protons) of the B-CD ring. Hence, the chemical shifts at 3.59 ppm correspond to the methylene protons, while 4.82 ppm corresponds to the axial methine proton H4. Hydroxy proton attached with methylene group at C-5 is observed as a doublet with a small coupling constant at 5.68 ppm. This hydroxy proton at 5.68 ppm exhibits cross-peak H at 5.68/7.62. This cross-peak reveals correlations between the hydroxy proton and H3’/ H5’ (the phenyl ring attached with chlorine). Based on the above correlation and shift in the proton chemical shifts structure of the inclusion complex is proposed. The Methyl proton is shielded by 0.005 ppm and the aromatic proton with chlorine moiety is deshielded by 0.02 ppm all other proton chemical shifts are unaffected. This suggests that the phenyl ring bearing chlorine as a substituent is outside of the bucket, and the acid group with methyl group is inside the bucket. Finally, the proposed structure of IC-1 is given in Fig. 4C.

ROESY of IC-1 (A), IC-2 (B), proposed structure for IC-1 (C), and proposed structure for IC-1 (D).
ROESY analysis of IC-2
The ROESY of the IC-2 is shown in Fig. 4B, and the cross peaks are indicated as A, B, C, D, and E (Fig. S6). ROESY shows cross-peaks A, B, and C at 6.92/1.59, 6.92/3.53, and 6.92/7.71 ppm. These cross peaks reveal correlations between the phenyl protons connected by ether linkage and methyl proton of the ether linkage and methylene protons of the MCD moiety. A multiplet centered at 4.82 ppm has already been assigned to the H3 proton of the MCD ring. It also shows a correlation with phenyl proton connected with ether linkage. Additionally, two other cross-peaks D and E at 4.53/4.82 and 4.53/3.49 ppm, show correlations between the equatorial hydroxyl proton at C-3 with protons at H3 (axial protons) and a methoxy methyl group at C-2. Methine proton at C-5 (H5) shows a correlation with methylene (attached to the same carbon within the MCD ring) and methyl of the methoxy group attached to methylene at C-5. In addition to the above correlation two more important cross peaks F and G at 7.71/3.32 and 7/71/ 3.59 ppm reveal the correlation between the protons of the phenyl ring bearing chlorine with methoxy methyl and methylene protons attached with C-5. It confirms the phenyl ring with chlorine is out of the cavity. Based on the NMR results obtained and the above correlations from ROESY strongly suggest that the structure of IC-2 may be with the acid part of the guest inside the cavity and the counterpart is in the out of the bucket as shown in Fig. 4D.
FT-IR spectral analysis
The FT-IR spectrum of FFA exhibits several distinctive features. Notably, broad peaks at 2984 cm-1 are indicative of O–H stretching, pointing to the existence of carboxylic acid groups in the molecular structure. Additionally, peaks at 1700 cm-1 and 1646 cm-1 correspond to C=O stretching (ketone) and the symmetrical stretching of C–C=C (alkenes), respectively. Aromatic rings are confirmed by peaks at 1596 cm-1 and 1500 cm-1, which represent the symmetrical stretching of C–C=C bonds. Vibrations at 759 cm-1 and 674 cm-1 are associated with alkyl halide (C–Cl) groups, providing further evidence of halogenated aromatic structures. Minor peaks around 1649 cm-1 correspond to C=C aromatic stretching, while the C–Cl stretches are observed around 678 cm-1 in the FFA spectrum. Moreover, additional peaks at 2875 cm-1 and 2920 cm-1 are attributed to –CH stretching, suggesting the presence of a surfactant component in FFA68,69,70,71. Figure 5A presents the FT-IR spectra of both FFA and its ICs. The wavenumbers between 950 cm-1 and 1200 cm-1 are typically characteristic of carbohydrates, specifically the functional groups within the carbohydrate structures72. In the fingerprint region of the spectrum, significant absorption peaks at 1157 cm-1, 1085 cm-1, and 1035 cm-1 correspond to the anhydroglucose ring’s C–O stretching vibrations. The peak at 941 cm-1 (Fig. S7) represents the skeletal vibration mode of the α-(1 → 4) glycosidic linkage, a defining feature of cyclodextrins73. A broad band at 3421 cm-1 is observed, signifying the O–H stretching vibration, while the band at 2922 cm-1 is due to C-H stretching vibrations74. The peak at 1635 cm-1 is indicative of the bending vibration of water molecules within the β-CD cavity75. When compared with the spectra of CDs, the FT-IR analysis of the ICs shows no new bond formation and no significant shift in the spectral peaks, indicating that the fundamental chemical structure of FFA remains largely unchanged. The stretching frequencies of the peaks are largely concealed by the CDs’ spectral features, underscoring the CDs’ significant contribution to the inclusion process and their dominant role in forming the inclusion complex with FFA76,77.

FT-IR spectra of FFA, and IC-1, and IC-2 (A), Raman spectra of FFA, and IC-1, and IC-2 (B).
Raman spectral analysis
Figure 5B presents the Raman spectra of FFA and its ICs, highlighting the interactions between FFA and the CD cavities. In the Raman spectrum of pure FFA, four prominent bands are observed, each corresponding to specific molecular vibrations: γ (C = O) at 1620 cm-1 (moderate intensity), γ (acid) at 1635 cm-1, γ (C–O–C) at 805 cm-1, and γ (C–Cl) at 625 cm-1 (broad). Additionally, a broad peak at 3360 cm-1 is observed, which is characteristic of the –OH group in FFA. For both BCD and MCD, a broad peak at 3400 cm-1 is also observed, attributed to the –OH groups in the cyclodextrins (Fig. S8). When the ICs are formed, this broad peak at 3400 cm-1 becomes significantly reduced, indicating that the –OH groups in the CDs are involved in interactions with FFA, likely through hydrogen bonding. Interestingly, while the C=O stretching band is absent in the spectra of both CDs alone, it reappears in the spectra of the ICs. This suggests that the carbonyl group of FFA is now interacting with the CDs, confirming the presence of both FFA and CD components in the ICs. The formation of the ICs with BCD and MCD results in notable shifts in the positions and intensities of the key Raman peaks of FFA, further indicating that the CDs influence the molecular structure and behavior of FFA, likely through encapsulation and complexation as well.
FE-SEM analysis
The surface morphology of the ICs is visualized through FE-SEM analysis. As depicted in Fig. 6, FFA is present in the form of small stone-like crystals with a non-uniform shape, characterized by distinct edges and corners. In contrast, IC-1 exhibits rock-like structures at all magnifications, while IC-2 displays seed-like structures. A notable transformation in surface morphology is observed when comparing FFA with its respective ICs. The original structure of FFA has entirely disappeared, giving rise to a new structural morphology that does not resemble bare CDs78,79. Consequently, the discernible differences in the surface morphology of ICs provide compelling evidence for the complexation of FFA with BCD and MCD80,81.

FE-SEM images of FFA (A), IC-1 (B), and IC-2 (C).
Thermal stability analysis
Thermogravimetry (TG), particularly with continuous temperature rise, is a widely used and effective technique for evaluating the thermal behavior of substances, including pharmaceuticals and their formulations. In this study, TGA was employed to investigate the thermal properties of the ICs. The TG curves for FFA, IC-1 (FFA:BCD), and IC-2 (FFA:MCD) are shown in Fig. 7, providing insight into their thermal stability. For pure FFA, which is known to be a stable organic drug, the TG curve reveals minimal mass loss as the temperature gradually increases up to approximately 220 °C. This indicates that FFA remains thermally stable in this temperature range. However, beyond 220 °C, a gradual weight loss is observed, which signifies the onset of thermal decomposition. The decomposition occurs within the temperature range of 220–300 °C, with more than 90% of the mass lost during this interval, suggesting significant degradation of the drug at higher temperatures. The DTA curves for FFA further support this observation, showing two distinct peaks at 240 °C and 280 °C. These peaks correspond to the endothermic processes associated with the weight loss and decomposition of FFA. The presence of these peaks indicates that FFA remains thermally stable up to around 240 °C, after which it begins to degrade. This analysis highlights the thermal behavior of FFA and provides a baseline for understanding the stability of its inclusion complexes with BCD and MCD.

TGA of FFA (A), IC-1 (B), and IC-2 (C).
For IC-1, the TGA curve remains stable up to 220 °C. A small and negligible reduction in mass occurs up to 100 °C, attributed to the release of moisture content from the ICs. Post 220 °C, there is a sudden weight decrease (around 20%) up to 240 °C, followed by a gradual decrease starting at 275 °C. The temperature range between 280–340 °C gets nearly 50% weight loss due to major FFA decomposition in IC-1. The DTA peaks at 239 °C and 320 °C support the observed weight losses. In the case of IC-2, a stable curve is observed up to 245 °C, with negligible weight loss (less than 1%) before 100 °C, likely due to moisture content removal. Post 245 °C, there is a gradual weight decrease. The DTA peak appears at 282 °C, supporting the thermal behavior of IC-2. Both ICs demonstrate resistance to temperature increases up to 220 °C and 245 °C for IC-1 and IC-2, respectively. Thermal stability is enhanced for FFA after modification with ICs, with IC-2 exhibiting significantly improved stability compared to IC-182. This suggests a higher binding interaction of FFA with MCD than BCD, reflecting enhanced thermal stability and the ability to withstand temperature variations to a greater extent83,84.
Interaction of FFA with CDs in the virtual state
Utilizing quantum mechanical calculations at the B3LYP/6-31G(D, P) level of theory, implemented within the Gaussian 16 package, allows for an in-depth investigation of the ICs formed between FFA and CDs. This computational approach provides detailed insights into the molecular interactions, enabling a comprehensive understanding of the electronic structures and properties of the ICs. These insights shed light on the intricate behavior and interaction mechanisms exhibited by these molecular systems. The study focuses on analyzing the interaction between FFAs and CDs within ICs, emphasizing the role of hydrogen bonding interactions and energy parameters in determining the stability and orientation of these complexes. For IC-1 (Fig. S9), composed of C1-C4, the orientation of C4 emerges as the favorable mode of inclusion. This preference is attributed to the shorter hydrogen bonds and higher complexation energy, as compared to other orientations (Fig. 8A). Similarly, IC-2 (Fig. S9), formed between C5 and C6, identifies C5 as the favorable mode of inclusion. This conclusion is based on the higher number of hydrogen bonds and the greatest complexation energy relative to the alternative orientation, C6 (Fig. 8B). To substantiate these findings, Table 1 presents a comprehensive summary of the hydrogen bonding interactions and associated energy parameters, providing an extensive dataset for further interpretation and validation. These results highlight the critical role of hydrogen bonding and complexation energy in dictating the structural preferences and stability of the ICs, offering valuable insights into their molecular dynamics and potential applications.

Hydrogen bonding with favorable orientation of IC-1 (A), and IC-2 (B).
Anti-inflammation activity by BSA denaturation assay
The anti-inflammatory activity of the studied ICs was initially assessed in vitro using the albumin denaturation assay. This assay is widely employed for such studies because protein denaturation is a critical pathological process associated with inflammation and conditions such as rheumatoid arthritis85,86,87. Given the well-documented anti-inflammatory properties of free FFA, the assay was utilized to evaluate the activity of FFA in their free and complex forms, specifically in IC-1 and IC-288,89,90. The results, depicted in Fig. 9A, show that all samples (FFA, IC-1, and IC-2) exhibit enhanced anti-inflammatory activity at concentrations up to 400 μg/ml. Interestingly, the release of FFA from the ICs was higher than that from bare FFA, suggesting an improved delivery mechanism facilitated by the ICs. Both IC-1 and IC-2 demonstrated notable protective effects on BSA, preventing its denaturation with inhibition rates exceeding 76%. Among the samples, IC-2 exhibited the highest protective effect, achieving an inhibition rate of 78%, which correlates with its superior FFA release rate (IC-2 > IC-1 > FFA). This enhanced activity can be attributed to the strong inclusion complex formed in IC-2, as evidenced by its high binding constant. The differing complexation abilities and release profiles of ICs can be attributed to the structural characteristics of the CD used. MCD exhibited a higher drug release rate than BCD. This discrepancy arises from the nature of the interaction between FFA and the host molecule. In MCD, FFA primarily interacts with the outer rim, resulting in a less constrained complex that facilitates faster release. In contrast, BCD captures FFA within its interior cavity, forming a more stable and robust complex that restricts rapid release91.

Anti-inflammation by protein denaturation assay (A) and HRBC assay (B) of FFA, IC-1, and IC-2.
Anti-inflammation activity by HRBC assay
To further evaluate the anti-inflammatory properties of the studied ICs, the HRBC membrane stabilization assay was employed. This assay assesses the ability of the samples to protect erythrocyte membranes against hemolysis induced by hypotonic stress or heat, which is a marker of anti-inflammatory activity. Hemolysis, indicated by the lysis of red blood cells and subsequent release of hemoglobin into the medium, was calculated, with 100% lysis set as the control (using distilled water as the hypotonic solution). The percentage inhibition of hemolysis was determined using a standard equation as described in earlier studies92,93. The results, illustrated in Fig. 9B, demonstrate a concentration-dependent increase in activity for all samples, FFA, IC-1, and IC-2 up to a maximum concentration of 500 μg/ml. Notably, the release of FFA from the ICs exceeded that from bare FFA, suggesting that the inclusion complexes enhanced the bioavailability of the FFA. Both IC-1 and IC-2 exhibited significant protection of erythrocyte membranes against heat-induced hemolysis, achieving inhibition rates of over 80%, with each complex reaching 81% at the highest concentration tested. This protective effect aligns with the FFA release rate (IC-2 > IC-1 > FFA), highlighting the role of efficient drug delivery in enhancing biological activity. The anti-inflammatory efficacy of IC-2, as reflected by its inhibition of hemolysis, correlates strongly with its higher complexation constant. This indicates that the stronger and more stable interaction between FFA and the CD in IC-2 plays a critical role in modulating its activity. The difference in complexation ability between MCD and BCD was also evident. MCD facilitated a higher release of FFA compared to BCD, which can be attributed to the interaction mechanism. In MCD, FFA primarily associates with the outer rim of the CD, resulting in a less constrained IC that allows faster release. Conversely, in BCD, FFA is encapsulated within the interior cavity, forming a more stable but slower-releasing complex94.
Molecular docking study of FFA with cyclooxygenase 1, and 2 (1PGG and 4COX)
Given the enhanced in-vitro anti-inflammatory effects observed for FFA following the formation of its ICs, it is crucial to further explore the binding interactions between FFA and key cyclooxygenase enzymes. To achieve this, molecular docking analysis is employed as an effective computational approach. Molecular docking is a widely used tool in drug development, particularly for identifying potential therapeutic targets and predicting atomic-level interactions between chemical ligands and their biological targets. This method enables a detailed understanding of biochemical processes and the molecular behaviors occurring at the binding sites of enzymes or receptors. The molecular docking process involves two primary steps: first, predicting the conformation, location, and orientation of the ligand (in this case, FFA) within the enzyme’s binding site, and second, determining the binding affinity of the interaction. This computational technique has been applied in the present study to investigate how FFA interacts with enzymes involved in inflammation, particularly cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), which are key enzymes in the inflammatory pathway. The interaction of FFA with COX-1 and COX-2 enzymes was analyzed using docking simulations, with the PDB files for COX-1 (1PGG.pdb) and COX-2 (4COX.pdb) serving as the targets. The three-dimensional interactions between FFA and these enzymes are illustrated in Fig. 10, and the corresponding binding interaction parameters are summarized in Table 2. In the docking simulation with COX-1 (1PGG.pdb), FFA exhibited a strong binding affinity, with a calculated binding energy of -9.07 kcal/mol and an inhibition constant (Ki) of 226.56 nM. The drug formed seven hydrogen bonds with various amino acid residues of COX-1. These included six conventional hydrogen bonds with the following residues: THR206, HIS203, TRP387, THR207, THR206 (repeated), and ASN382. The bond distances for these interactions ranged from 2.63 to 3.31 Å. Additionally, FFA interacted with PHE210 through a pi-donor hydrogen bonding interaction, with a distance of 3.93 Å. These interactions highlight the strong affinity of FFA for COX-1 and its potential as an effective inhibitor of this enzyme. When FFA was docked with COX-2 (4COX.pdb), the binding energy and inhibition constant were even more favorable than those observed with COX-1, suggesting that FFA may bind more strongly to COX-2. The binding energy was calculated to be -9.16 kcal/mol, and the inhibition constant was 193.46 nM. In this case, FFA formed two hydrogen bonds with the amino acid residues of COX-2. Specifically, it formed a conventional hydrogen bond with TYR355, with a distance of 2.92 Å, and a carbon-hydrogen bond with SER353, with a distance of 3.42 Å. These interactions suggest that FFA could be a potent inhibitor of COX-2, further supporting its anti-inflammatory potential.

Three-dimensional interactions of the FFA with enzyme 1PGG.pdb (A), and 4COX.pdb (B).
MTT assay
The biocompatibility of FFA and its ICs with BCD and MCD was evaluated by investigating their impact on cell proliferation. The experiments involved treating normal cell lines with varying concentrations of the samples over incubation periods of 6 h and 24 h to observe their effects on cellular health and viability95. The analysis revealed that treatments with FFA, IC-1 (FFA:BCD), and IC-2 (FFA:MCD) had no adverse effect on cell viability in normal cell lines. This observation was consistent across all tested concentrations, as indicated by the data presented in Fig. 11 and Table S5. The frequency of cell death in treated cultures was comparable to that of untreated control cells, suggesting that these formulations do not induce cytotoxicity under normal conditions. While FFA generally maintained cell viability, some signs of cell death were observed at higher concentrations (100 µg/ml and 200 µg/ml) after 6 h and 24 h of treatment. This effect was more pronounced in HaCaT cells, where higher doses of FFA seemed to compromise cellular integrity. Morphological changes, such as elongation, were observed in some cells; however, this had a minimal impact on overall cell function and did not significantly contribute to cell death (Fig. 12). The IC-1 and IC-2 showed enhanced cell viability compared to free FFA, even at higher concentrations. The complexation of FFA with BCD and MCD likely improves its aqueous solubility and reduces direct cellular exposure to the drug, mitigating potential cytotoxic effects. IC-treated cells exhibited a healthier growth profile, confirming that the encapsulation process enhances biocompatibility. ICs lower the direct toxicity of FFA, likely due to controlled release and improved solubility. CD-complexed FFA promotes cell proliferation and minimizes stress on the cells, maintaining a favorable growth environment. The use of BCD and MCD as carriers ensures that FFA can be used at higher doses without compromising cellular integrity. The findings underscore the importance of inclusion complexation in enhancing the biocompatibility of FFA. While free FFA can induce cytotoxicity at elevated concentrations, its encapsulation in BCD and MCD protects cells, resulting in superior safety profiles. MCD has been reported to exhibit some degree of cytotoxicity, leading to cell death in certain assays96. However, in our study, when MCD is utilized as a host molecule to form the IC-2, no evidence of cell death was observed. The results from our assays clearly demonstrated that the IC-2 exhibited no adverse impact on cell viability, suggesting that the encapsulation of the guest molecule within the MCD cavity mitigates its potential cytotoxic effects. These insights are crucial for the development of FFA-based formulations, particularly for therapeutic applications requiring high-dose or long-term treatments. The study establishes that IC formation not only improves biocompatibility but also enhances therapeutic potential by maintaining cellular health.

HaCaT viability by increasing concentrations of FFA, IC-1, and IC-2 at 6 h (A), and 24 h (B).

Cell viability of FFA ((A) at 6 h, (B) at 24 h), IC-1 ((C) at 6 h, (D) at 24 h), and IC-2 ((E) at 6 h, (F) at 24 h).
LDH assay
The LDH release assay is a widely utilized and reliable method to evaluate cellular membrane integrity and cytotoxic effects of various treatments. LDH, an intracellular enzyme, is released into the extracellular environment when the cell membrane is compromised due to stress or damage. In this study, the LDH assay was employed to assess the potential cytotoxic effects of Flufenamic Acid (FFA) and its inclusion complexes, IC-1 (FFA-BCD) and IC-2 (FFA-MCD), on HaCaT cells over incubation periods of 6 h and 24 h. The results demonstrated no significant changes in LDH release levels in HaCaT cells treated with FFA, IC-1, or IC-2 compared to untreated controls. This consistency was observed across both short-term (6-h) and extended (24-h) exposure periods, indicating the absence of membrane damage (Fig. S12 and Table S6). The absence of elevated LDH levels suggests that the tested samples, including free FFA and its ICs, do not compromise the integrity of cellular membranes. This finding aligns with the biocompatibility profiles observed in cell viability studies, further supporting the non-toxic nature of these compounds in normal cell models. The IC-1 and IC-2 may further reduce the direct interaction of FFA with cellular membranes by encapsulating the drug. This protective effect ensures sustained membrane stability, even at potentially higher doses. Although free FFA is effective, it may exhibit direct membrane interactions at higher doses or extended exposure times. However, the encapsulation within BCD and MCD ensures better compatibility by modulating the drug’s release and reducing its direct interaction with cell membranes. The LDH release assay provides compelling evidence that FFA, along with its inclusion complexes IC-1 and IC-2, exhibits negligible cytotoxic effects on HaCaT cells. The lack of significant LDH release underlines the ability of these compounds to maintain membrane integrity, ensuring their safety for potential therapeutic applications. The results reinforce the advantage of CD-based encapsulation in enhancing biocompatibility, making these complexes valuable in developing non-toxic and efficient drug formulations.
Conclusions
In the present work, we have successfully synthesized and characterized the ICs of FFA with BCD, and MCD. The BH plots and phase solubility study showed the FFA formed a 1:1 stoichiometric ratio with CDs. 1H NMR, ROESY, and FT-IR results confirmed the formation of the ICs. The surface morphological differences after the formation of ICs have been taken as the finest evidence. The optimized structural analysis of IC-1 and IC-2 indicates a favorable orientation and appears consistent with experimental analysis. Additionally, we assessed its in-vitro anti-inflammatory activity through BSA denaturation and HRBC assay. Furthermore, our study illustrates that the inclusion of BCD and MCD is an effective strategy for augmenting the anti-inflammatory properties of FFA by shielding against protein denaturation. The enhancement of FFA solubility through the formation of ICs represents a novel and successful approach. This optimized approach to preparing ICs could potentially cover the way for the reevaluation of other poorly water-soluble NSAIDs. Importantly, our samples do not produce any toxicity upto 200 μg/ml. Further investigations have to be carried out to determine whether this approach developed in our study could enhance oral bioavailability and prove beneficial in the management of chronic inflammation.
Data availability
The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Netea, M. G. et al. A guiding map for inflammation. Nat. Immunol. 18, 826–831 (2017).
Francisco Javier, A.-M. et al. Enhanced NSAIDs solubility in drug-drug formulations with ciprofloxacin. Int. J. Mol. Sci. 24, 3305 (2023).
NHS Non-Steroidal Anti-Inflammmatory Drugs. https://www.nhs.uk/conditions/nsaids/ (Accessed 22 December 2022).
Chihiro, T. et al. Effect of cyclodextrin complex formation on solubility changes of each drug due to intermolecular interactions between acidic NSAIDs and basic H2 blockers. Mol. Pharm. 20, 5032–5042 (2023).
Makiko, F., Naohide, H. & Kumi, S. Effect of fatty acid Esters on permeation of ketoprofen through hairless rat skin. Int. J. Pharm. 205, 117–125 (2000).
Vergote, G. J., Vervaet, C. & Driessche, I. V. An oral controlled release matrix pellet formulation containing nanocrystalline ketoprofen. Int. J. Pharm. 219, 81–87 (2001).
Zhang, Q. H., Vigier, K. D. O., Royer, S. & Jérôme, F. Deep eutectic solvents: syntheses, properties and applications. Chem. Soc. Rev. 41, 7108–7146 (2012).
Chao, L., Jun, C., Ning, W. & Erzheng, S. Significantly improving the solubility of nonsteroidal anti-inflammatory drugs in deep eutectic solvents for potential non-aqueous liquid Administration. Med. Chem. Comm. 7, 955–959 (2016).
Bighley, L. D., Berge, S. M. & Monkhouse, D. C. Salt forms of drugs and absorption. Encycl. Pharm. Technol. 13, 453–499 (1996).
Serajuddin, A. T. M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 59, 603–616 (2007).
Perioli, L., Ambrogi, V. & Bernardini, C. Potential prodrugs of non-steroidal anti-inflammatory agents for targeted drug delivery to the CNS. Eur. J. Med. Chem. 39, 715–727 (2004).
Chie, K., Kenji, K. & Kazuo, M. Synthesis of polyamidoamine dendrimers having poly(ethylene glycol) grafts and their ability to encapsulate anticancer drugs. Bioconjug. Chem. 17, 910–917 (2000).
Fatma Ebru, K. & Mehmet, S. Solubility enhancement of non-steroidal anti-inflammatory drugs (NSAIDs) using polypolypropylene oxide core PAMAM dendrimers. Int. J. Pharm. 451, 18–22 (2013).
Arias, L. H. M., González, A. M., Fadrique, R. S. & Vazquez, E. S. Cardiovascular risk of nonsteroidal anti-inflammatory drugs and classical and selective cyclooxygenase-2 inhibitors: a meta-analysis of observational studies. J. Clin. Pharmacol. 59, 55–73 (2019).
Alessandri, A. L. et al. Resolution of inflammation: Mechanisms and opportunity for drug development. Pharmacol. Ther. 139, 189–212 (2013).
Jansook, P., Ogawa, N. & Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 535, 272–284 (2018).
Brewster, M. E. & Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Del. Rev. 59, 645–666 (2007).
Santos, C. I. A. V., Ribeiro, A. C. F. & Esteso, M. A. Drug delivery systems: Study of inclusion complex formation between methylxanthines and cyclodextrins and their thermodynamic and transport properties. Biomolecules 9, 196 (2019).
Kurkov, S. V. & Loftsson, T. Cyclodextrins. Int. J. Pharm. 453, 167–180 (2013).
Matencio, A., Navarro-Orcajada, S., García-Carmona, F. & López-Nicolás, J. M. Applications of cyclodextrins in food science. A review. Trends Food Sci. Technol. 104, 132–143 (2020).
Matencio, A., Bermejo-Gimeno, M. J., García-Carmona, F. & López-Nicolás, J. M. Separating and identifying the four stereoisomers of methyl jasmonate by RP-HPLC and using cyclodextrins in a novel way. Phytochem. Anal. 28, 151–158 (2017).
Matencio, A. et al. Physicochemical, thermodynamical, structural and computational evaluation of kynurenic acid/cyclodextrin complexes. Food Chem. 356, 129639 (2021).
López-Nicolás, J. M. & García-Carmona, F. Effect of hydroxypropyl-β-cyclodextrin on the aggregation of (e)-resveratrol in different protonation states of the guest molecule. Food Chem. 118, 648–655 (2010).
Salazar, S., Guerra, D., Yutronic, N. & Jara, P. Removal of aromatic chlorinated pesticides from aqueous solution using β-cyclodextrin polymers decorated with Fe3O4 nanoparticles. Polymers 10, 1038 (2018).
Krabicová, I. et al. History of cyclodextrin nanosponges. Polymers 12, 1122 (2020).
Matencio, A. et al. Study of oxyresveratrol complexes with insoluble cyclodextrin based nanosponges: developing a novel way to obtain their complexation constants and application in an anticancer study. Carb. Pol. 231, 115763 (2020).
David, M., Frédéric, M., Jean-Marc, D., Dominique, C. & Serge, P. Benefits of methylated cyclodextrins in the development of midazolam pharmaceutical formulations. J. Pharm. Sci. 102, 2102–2111 (2013).
Young-Ae, C. et al. Methyl-beta-cyclodextrin inhibits cell growth and cell cycle arrest via a prostaglandin E(2) independent pathway. Exp. Mol. Med. 36, 78–84 (2004).
Éva, F., Julianna, S., Katalin, C., Milo, M. & Lajos, S. Methyl-beta-cyclodextrins: The role of number and types of substituents in solubilizing power. J. Pharm. Sci. 103, 1443–1452 (2014).
Milena, G. T. et al. Theoretical and experimental study of inclusion complexes formed by isoniazid and modified β-cyclodextrins: 1H NMR structural determination and antibacterial activity evaluation. J. Phys. Chem. B 118, 81–93 (2014).
Pauline, S. S. et al. Methyl-β-cyclodextrin inclusion complex with β-caryophyllene: preparation, characterization, and improvement of pharmacological activities. ACS Omega 2, 9080–9094 (2017).
Rajaram, R. et al. Enhancing ketoprofen’s solubility and anti-inflammatory efficacy with safe methyl-β-cyclodextrin complexation. Sci. Rep. 14, 21516 (2024).
Paola, F. & Iolanda, D. M. Preparation of non-steroidal anti-inflammatory drug/β-cyclodextrin inclusion complexes by supercritical antisolvent process. J. CO2 Util. 44, 101397 (2021).
Luana, H. et al. Pharmacological effects of a complex α-bisabolol/β-cyclodextrin in a mice arthritis model with involvement of IL-1β, IL-6 and MAPK. Biomed. Pharmacother. 151, 113142 (2022).
Seiga, S., Yoshinori, H., Yusei, K. & Noriyoshi, A. Effect of temperature on the structure and drug-release behaviour of inclusion complex of b-cyclodextrin with cyclophosphamide: a molecular dynamics study. Soft Matter. 19, 2902–2907 (2023).
Yi, R. et al. Pterostilbene complexed with cyclodextrin exerts antimicrobial and anti-inflammatory effects. Sci. Rep. 10, 9072 (2020).
Lanlan, L., Bingyu, C., Gang, L., Sheng, C. & Jianxiang, Z. Anti-inflammatory nanotherapies based on bioactive cyclodextrin. Materials 3, 2300106 (2023).
Shadi Salem, A. et al. Fenofibrate for COVID-19 and related complications as an approach to improve treatment outcomes: the missed key for Holy Grail. Inflamm. Res. 71, 1159–1167 (2022).
Ding, X., Zheng, M., Lu, J. & Zhu, X. Preparation and evaluation of binary and ternary inclusion complexes of fenofibrate/hydroxypropyl-β-cyclodextrin. J. Incl. Phenom. Macrocyc. Chem. 91, 17–24 (2018).
Aigner, Z., Bencz, I. & Kata, M. Increasing the solubility characteristics of fenofibrate with cyclodextrin. J. Incl. Phenom. Macrocyc. Chem. 20, 241–252 (1994).
Jagdale, S. K., Dehghan, M. H. & Paul, N. S. Enhancement of dissolution of fenofibrate using complexation with hydroxy propyl β-cyclodextrin. Turk. J. Pharm. Sci. 16, 48–53 (2019).
Mahapatra, A. K., Murthy, P. N., Patra, R. K., Panda, S. & Rautray, R. K. Comparative interaction of β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin with fenofibrate: Phase-solubility behavior and dissolution rates. Lat. Am. J. Pharm. 31, 1302–1309 (2012).
Patel, A. R. & Vavia, P. R. Effect of hydrophilic polymer on solubilization of fenofibrate by cyclodextrin complexation. J. Incl. Phenom. Macrocyc. Chem. 56, 247–251 (2006).
Ye, J.-J., Lu, J.-M., Zheng, M.-K., Ding, X.-Z. & Zhu, X.-Y. The effect of water-soluble excipients on the inclusion complex of fenofibrate and hydroxypropyl-β-cyclodextrin. Chin. Pharm. J. 54, 790–795 (2019).
Palmier, G. F. & Giovannucci, G. Inclusion complexation of fenofibrate with β-cyclodextrin and hydroxypropyl-β-cyclodextrin evaluation of interactions in solution and solid complex characterization. S.T.P. Pharm. Sci. 7, 174–181 (1997).
Ajmera, A. et al. Dissolution rate enhancement of atorvastatin, fenofibrate and ezetimibe by inclusion complex with β-cyclodextrin. Asian J. Pharm. Clin. Res. 5, 73–76 (2012).
Khin, S. Y. et al. Development of fenofibrate/randomly methylated β-cyclodextrin-loaded Eudragit® RL 100 nanoparticles for ocular delivery. Molecules 27, 4755 (2022).
Kondoros, B. A. et al. Cyclodextrin complexation of fenofibrate by co-grinding method and monitoring the process using complementary analytical tools. Pharmaceutics 14, 1329 (2022).
Henriette, S. W., Kim, L. L. & Flemming, M. Cyclodextrin controlled release of poorly water-soluble drugs from hydrogels. Drug Deliv. 15, 69–80 (2008).
Kfoury, M. et al. Solubility, photostability and antifungal activity of phenylpropanoids encapsulated in cyclodextrins. Food Chem. 196, 518–525 (2016).
Seyed Mohammad, T. G. & Seyed Hadi, R. Characterizing the natural canthaxanthin/2-hydroxypropyl-β-cyclodextrin inclusion complex. Carb. Pol. 101, 1147–1153 (2014).
Rajendran, S., Govindharasu, B., Maruthan, K., Jayachitra, A. & Vediappen, P. Multiple biological activities and molecular docking studies of newly synthesized 3-(pyridin-4-yl)-1H-pyrazole-5-carboxamide chalcone hybrids. Bioorg. Med. Chem. Lett. 26, 5624–5630 (2016).
Loukas, Y. L. Multiple complex formation of fluorescent compounds with cyclodextrins: efficient determination and evaluation of the binding constant with improved fluorometric studies. J. Phys. Chem. B 101, 4863–4866 (1997).
Chandrabose, L. et al. Characterization and molecular docking analysis for the supramolecular interaction of lidocaine with β-cyclodextrin. Polycycl. Arom. Comp. 43, 1202–1218 (2023).
Murugan, M. et al. Effect of pH and structural orientation on supramolecular complexation of chloroquine in β-cyclodextrin medium. J. Mol. Liq. 325, 115157 (2021).
Benesi, A. & Hildebrand, J. H. The Benesi-Hildebrand method for determination of Kf for DA association and ε values for DA CT absorption. J. Am. Chem. Soc. 71, 2703–2707 (1949).
Kuntz, I. D. Jr., Gasparro, F. P., Johnston, M. D. Jr. & Taylor, R. P. Molecular interactions and the benesi-hildebrand equation. J. Am. Chem. Soc. 90, 4778–4781 (1968).
Otto, E. Calculating equilibrium constants from spectral data: Reliability of the Benesi-Hildebrand method and its modifications. Chemomet. Intell. Lab. Syst. 39, 85–93 (1997).
Iglesias, E. Inclusion complexation of novocaine by beta-cyclodextrin in aqueous solutions. J. Org. Chem. 71, 4383–4392 (2006).
Murugan, M., Anitha, A., Sivakumar, K. & Rajamohan, R. Effect of pH and structural orientation on supramolecular complexation of chloroquine in β-cyclodextrin medium. J. Mol. Liq. 325, 115157 (2021).
Sarah, J. N., Barbara, R.-S. & Naír, R. Phase solubility diagrams of cocrystals are explained by solubility product and solution complexation. Crystal Growth Des. 6, 592–600 (2006).
Shota, S. et al. Relationship between phase solubility diagrams and crystalline structures during dissolution of cimetidine/cyclodextrin complex crystals. J. Pharm. Sci. 109, 2206–2212 (2020).
Higuchi, T. & Connors, K. A. Phase solubility technique. Adv. Anal. Chem. Inst. 4, 117–212 (1965).
Rajaram, R. et al. Formulation of ensulizole with beta-cyclodextrins for improved sunscreen activity and biocompatibility. J. Pharm. Sci. 113, 1536-1545 (2023).
Rajaram, R. et al. Enhanced photostability and biocompatibility of sunscreen formulation of 2-phenylbenzimidazole-5-sulfonic acid with methyl-beta-cyclodextrin. J. Mol. Liq. 390, 123013 (2023).
Shota, S. et al. Relationship between phase solubility diagrams and crystalline structures during dissolution of cimetidine/cyclodextrin complex crystals. J. Pharm. Sci. 109, 2206–2212 (2020).
Senapati, P. C., Sahoo, S. K. & Sahu, A. N. Mixed surfactant based (SNEDDS) self-nanoemulsifying drug delivery system presenting efavirenz for enhancement of oral bioavailability. Biomed. Pharmacother. 80, 42–51 (2016).
Wira, N. S., Yeyet, C. S., Jessie, S. P. & Diky, M. Development and characterization of self-nanoemulsifying drug delivery system (SNEDDS) formulation for enhancing dissolution of fenofibric acid. J. Res. Pharm. 24, 738–747 (2020).
Sailaja, U., Shahin Thayyil, M., Krishna Kumar, N. S. & Govindaraj, G. Molecular dynamics of amorphous pharmaceutical fenofibrate studied by broadband dielectric spectroscopy. J. Pharm. Anal. 6, 165–170 (2016).
Erizal, Z. et al. Eutectic mixture of fenofibric acid and syringic acid: Improvement of dissolution rate and its antihyper lipidemic activity. ChemistrySelect 8, e202300044 (2023).
Barros, A. S. Use of FT-IR spectroscopy as a tool for the analysis of polysaccharide food additives. Carb. Pol. 51, 383–389 (2003).
Miao, M. et al. Structure and physicochemical properties of octenyl succinic esters of sugary maize soluble starch and waxy maize starch. Food Chem. 151, 154–160 (2014).
Dou, Z., Chen, C. & Fu, X. The effect of ultrasound irradiation on the physicochemical properties and α-glucosidase inhibitory effect of blackberry fruit polysaccharide. Food Hydrocoll. 96, 568–576 (2019).
Song, X., He, G., Ruan, H. & Chen, Q. Preparation and properties of octenyl succinic anhydride modified EarlyIndica rice starch. Starch Stärke 58, 109–117 (2006).
Cheng, J.-H. et al. Preparation and properties of octenyl succinate β-cyclodextrin and its application as an emulsion stabilizer. Food Chem. 218, 116–121 (2017).
Dongmei, K. et al. Preparation and characterization of octenyl succinate β-cyclodextrin and vitamin e inclusion complex and its application in emulsion. Molecules 25, 654 (2020).
Rajaram, R. et al. Water-soluble inclusion complexes for a novel anti-viral agent with low toxicity; Oseltamivir with the β-cyclodextrins. J. Mol. Liq. 366, 120297 (2022).
Rajaram, R. et al. A novel and water-soluble material for coronavirus inactivation from oseltamivir in the cavity of methyl and sulfated-β-cyclodextrins through inclusion complexation. J. Pharm. Biomed. Anal. 221, 115057 (2022).
Bulani, V. D. et al. Inclusion complex of ellagic acid with beta-cyclodextrin: Characterization and in vitro anti-inflammatory evaluation. J. Mol. Struct. 1105, 308–315 (2016).
Zou, A. et al. Folate receptor targeted bufalin/-cyclodextrin supramolecular inclusion complex for enhanced solubility and anti-tumor efficiency of bufalin. Mater. Sci. Eng. C 78, 609–618 (2017).
Shivani, U. et al. Encompassment of benzyl isothiocyanate in cyclodextrin using ultrasonication methodology to enhance its stability for biological applications. Ultrasonics Sonochem. 39, 25–33 (2017).
Li, W. et al. Preparation and characterization of inclusion complex of benzyl isothiocyanate extracted from papaya seed with β-cyclodextrin. Food Chem. 184, 99–104 (2015).
Macedo, O. F. et al. Physicochemical study and characterization of the trimethoprim/2-hydroxypropyl–cyclodextrin inclusion complex. Spectrochim. Acta Part A 86, 101–106 (2012).
Vipin, D. B. et al. Characterisation and anti-inflammatory evaluation of the inclusion complex of ellagic acid with hydroxypropyl-β-cyclodextrin. J. Incl. Phenom. Macrocyc. Chem. 82, 361–372 (2015).
Kumari, K. D. K. P., Weerakoon, T. C. S., Handunnetti, S. M., Samarasinghe, K. & Suresh, T. S. Anti-inflammatory activity of dried flower extracts of Aegle marmelos in Wistar rats. J. Ethnopharmacol. 151, 1202–1208 (2014).
Dounia, S. et al. Solubility enhancement of mefenamic acid by inclusion complex with b-cyclodextrin: in silico modeling, formulation, characterisation, and in vitro studies. J. Enz. Inh. Med. Chem. 36, 605–617 (2021).
Assas, N., Elbahri, Z., Baitiche, M. & Djerboua, F. Effects of some process parameters on the niflumic acid controlled release polymeric microspheres: optimization using designs of experiments. Asia Pac. J. Chem. Eng. 14, e2283 (2019).
Mizushima, Y. & Kobayashi, M. Interaction of anti-inflammatory drugs with serum proteins, especially with some biologically active proteins. J. Pharm. Pharmacol. 20, 169–173 (1968).
Luana, H. et al. Pharmacological effects of a complex α-bisabolol/β-cyclodextrin in a mice arthritis model with involvement of IL-1β, IL-6 and MAPK. Biomed. Pharmacother. 151, 113142 (2022).
Lynne, M. C. & Evelyn, F. An initial investigation into the anti-inflammatory activity and antioxidant capacity of alpha-cyclodextrin-complexed Manuka honey. J. Complement. Integr Med. 9, 25 (2012).
Kundu, M. & Roy, M. N. Subsistence of inclusion complex via assembly of a drug into cyclic oligosaccharide: Its formation, mechanism, behavior and importance. Spectrochim. Acta Part A 183, 332–338 (2017).
Das, K., Datta, B., Rajbanshi, B. & Roy, M. N. Evidences for inclusion and encapsulation of an ionic liquid with β-CD and 18-C-6 in aqueous environments by physicochemical investigation. J. Phys. Chem. B 122, 1679–1694 (2018).
Eloísa, P. B. S. S. S. et al. Nerolidol-beta-cyclodextrin inclusion complex enhances anti-inflammatory activity in arthritis model and improves gastric protection. Life Sci. 265, 118742 (2021).
Khaled, M. E. et al. Dual targeting system by supramolecular complex of folate-conjugated methyl-β-cyclodextrin with adamantane-grafted hyaluronic acid for the treatment of colorectal cancer. Int. J. Biol. Macromol. 113, 386–394 (2018).
Joel, E. U. et al. Characterization of Methyl-β-cyclodextrin toxicity in NGF-differentiated PC12 cell death. Neurotoxicology 28, 613–621 (2007).
Author information
Authors and Affiliations
Contributions
R.R: Conceptualization, methodology, resources, formal analysis, writing—original draft. P.M: Formal analysis. K.M: Formal analysis. M.M: Software, formal analysis. P.D.S: Methodology, and formal analysis. J.H.S: Formal analysis. T.M: Formal Analysis, Writing—review & editing. Y.R.L: Writing—review & editing. Supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rajamohan, R., Muthuraja, P., Murugavel, K. et al. Significantly improving the solubility and anti-inflammatory activity of fenofibric acid with native and methyl-substituted beta-cyclodextrins via complexation. Sci Rep 15, 853 (2025). https://doi.org/10.1038/s41598-024-84745-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-84745-x
Keywords
This article is cited by
-
Hydroxypropyl-β-Cyclodextrin-Enhanced Azelaic Acid Hydrogel for Acne Treatment: Evaluation of Antimicrobial, Anti-inflammatory, and Skin Penetration Properties
Journal of Pharmaceutical Innovation (2025)
