
Abstract
The mechanistic role of gut microbiota in metabolic dysfunction-associated steatotic liver disease (MASLD) and chronic kidney disease (CKD) is increasingly recognized. Despite their close association, comparative data regarding gut dysbiosis in these disorders are limited. This study included 22 healthy controls and 180 patients (90 MASLD, 60 CKD, and 30 both diseases with sex- and age-matched). Fecal bacterial 16 S ribosomal RNA sequencing and butyryl-CoA: acetate CoA transferase (BCoAT) gene expression were analyzed. Plasma intestinal fatty acid binding protein (I-FABP), representing intestinal barrier dysfunction, was assessed using the ELISA method. Our data showed that alpha and beta diversities of gut microbiota differed between MASLD and healthy controls. However, only beta diversities were different between CKD and healthy individuals. The MASLD and CKD groups displayed fewer SCFA-producing genera, particularly Bifidobacterium, than healthy controls. Fecal BCoAT levels were inversely correlated with eGFR and I-FABP levels. Patients with CKD had significantly enriched pathogenic bacteria, reduced BCoAT, and increased I-FABP levels versus MASLD. Combining significant bacterial genera discriminated MASLD from CKD with high diagnostic accuracy (AUC of 0.90). Among patients with both diseases, gut microbial alterations showed mixed characteristics of MASLD and CKD. These data highlighted the shared and distinct gut dysbiosis and related biomarkers, which could provide a better understanding of MASLD and CKD pathogenesis.
Similar content being viewed by others
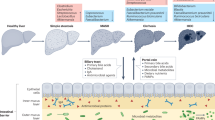


Introduction
Non-communicable diseases (NCDs), such as metabolic dysfunction-associated steatotic liver disease (MASLD) and chronic kidney disease (CKD), have become an epidemic worldwide. MASLD, previously known as non-alcoholic fatty liver disease (NAFLD), has emerged as one of the most common chronic liver diseases, affecting up to 30% of the global general population1. The hepatic features of MASLD range from simple steatosis, specified as excessive fat accumulation in the liver, to steatohepatitis, cirrhosis, and hepatocellular carcinoma (HCC)2. Currently, MASLD is also considered to be a multisystem disease that significantly impacts several extrahepatic manifestations, such as malignancies, cardiovascular disease (CVD), and CKD3. CKD, defined as the sustained reduction in estimated glomerular filtration rate (eGFR), is a leading health concern with an estimated > 10% of the worldwide population4. CKD is an irreversible and progressive disorder that leads to end-stage renal disease (ESRD) and, importantly, is also considered to be a risk factor for CVD.
Previous evidence indicates a close relationship between MASLD and CKD, as the occurrence of CKD is estimated at 20–55% and 5–30% in patients with or without MASLD, respectively5. An updated meta-analysis from observational longitudinal studies has also indicated that MASLD is significantly related to long-term risk of incident CKD stage ≥ 3, with approximately 1.5-fold6. Additionally, a recent large, real-world cohort confirmed a 1.80-fold increased risk of developing long-term CKD in patients with MASLD7. These data emphasize that MASLD is tightly associated with CKD risk and should require early detection to minimize long-term complications related to both diseases. Although the pathophysiological mechanisms that link MAFLD and CKD are not entirely known, it has been well-recognized that these two disorders share many cardiometabolic risk factors, including insulin resistance, type 2 diabetes (T2DM), dyslipidemia, and obesity. Moreover, the close association between MASLD and CKD could be multifactorial beyond conventional factors and thus needs further investigation.
Accumulating data have indicated that gut microbiota and its metabolites, particularly short-chain fatty acids (SCFAs), play an essential role in modulating the development and progression of MAFLD and CKD via the cross-talk of the gut-liver axis and the gut-kidney axis, respectively8,9,10,11. In this context, alterations in gut microbial diversity and composition (gut dysbiosis) and depleted SCFA production associated with increased intestinal permeability (leaky gut) and systemic inflammation could influence the pathogenesis of MAFLD and CKD8,9,10,11. Compared to healthy subjects, an increased abundance of potentially harmful bacteria and decreasing beneficial microbes is consistently observed among patients with MASLD at phylum, family, and genus12. Likewise, significant gut dysbiosis is detected in patients with CKD, leading to metabolic dysregulation and adverse consequences, particularly in ESRD13. However, current data regarding the direct comparison of gut microbiota alteration between patients with MAFLD and CKD are limited.
In this study, we aimed to characterize the diversities and composition of gut microbiota in patients with MASLD compared to CKD and healthy subjects. Additionally, we analyzed fecal gene expression levels of butyryl-CoA: acetate CoA transferase (BCoAT), representing SCFAs, and plasma intestinal fatty acid binding protein (I-FABP), a surrogate marker of intestinal damage, in parallel. We also identified gut dysbiosis and the related biomarkers in patients who had both MASLD and CKD (both diseases), which has never been explored. These results could provide valuable information for a better understanding of the mechanisms by which gut dysbiosis affects the pathogenesis of MASLD and CKD.
Results
Baseline characteristics of the patients
Twenty-two healthy controls and 180 patients were enrolled, including 90 patients with MASLD, 60 with CKD, and 30 with both diseases. Table 1 shows the baseline clinical characteristics of the patients in this study. Among the patient groups, there was no difference in gender, age, presence of T2DM, hypertension (HT), dyslipidemia, serum albumin, platelet count, and urine albumin creatinine ratio (UACR). In contrast, other parameters, including body mass index (BMI), serum creatinine, eGFR, uric acid, hematocrit, aspartate aminotransferase (AST), alanine aminotransferase (ALT), liver stiffness measurement (LSM) and controlled attenuation parameter (CAP) score were significantly different.
The alpha diversities of gut microbiota
Figure 1 demonstrates the richness and evenness of taxa in the patient groups using the Chao1, Shannon, and Simpson indices compared to healthy controls. Based on the Kruskal–Wallis multiple group statistical test, there were significant differences in these indexes between studied groups (Chao1, p = 0.0059; Shannon, p = 0.00057 and Simpson, p = 0.0213), as the levels of alpha diversities were highest in healthy controls.

Alpha-diversity differences among groups (a) Chao1, (b) Shannon, (c) Simpson. Statistical measures were median, interquartile range, and outliers. The statistical analysis was performed using the Kruskal–Wallis test.
For the post hoc pairwise comparison, significant differences in these indexes were constantly found between healthy controls vs. MASLD, healthy controls vs. both diseases and MASLD vs. CKD. Additionally, there was no significant difference in all indexes between healthy controls vs. CKD and MASLD vs. both diseases. When comparing both diseases and CKD, there was a significant difference between Shannon and Simpson but not in the Chao1 index (Supplementary File 1).
The beta diversities of gut microbiota
Beta diversity profiling used Bray-Curtis, Jaccard, unweighted, and weighted UniFrac distance metrics to determine the similarities and differences in microbial composition between groups. Overall, all indices were significantly clustered and differed among the patient groups and healthy controls (PERMANOVA, p = 0.001, Fig. 2). The pairwise analysis further demonstrated significant differences in bacterial communities between healthy controls vs. MASLD, healthy controls vs. CKD, healthy controls vs. both diseases and MASLD vs. CKD. In contrast, there was no significant difference in beta diversities between MASLD vs. both diseases (Bray-Curtis, Jaccard, unweighted UniFrac indices), both diseases vs. CKD (all indices), and both diseases vs. healthy controls (weighted UniFrac index), as shown in Supplementary Tables 1 and 2.

The principal coordinate analysis (PCoA) of beta-diversity profiling among groups (a) Bray-Curtis index, (b) Jaccard index, (c) Unweighted Unifrac index, (d) Weighted Unifrac index. The differences in beta diversity were tested by Permutational Multivariate Analysis of Variance (PERMANOVA).
(a).
Gut bacterial compositional analysis
We further examined the relative abundance at the phylum level of the patient groups compared with healthy controls (Fig. 3a). In healthy subjects, the taxa with high abundance included Firmicutes, Bacteroidota, Actinomycetota, Proteobacteriota, Fusobacteria, and Verrucomicrobiota. We calculated the Firmicutes/Bacteroidota (F/B ratio) in patients with MASLD, CKD, and both diseases (Fig. 3b). The F/B ratio was found to be 2.71 ± 2.24 for MASLD, 5.54 ± 15.57 for CKD, and 3.82 ± 6.36 for both diseases. The results showed that there was no statistically significant difference (p > 0.05) in the F/B ratio between the CKD and MASLD groups compared to healthy controls (2.29 ± 1.66). However, the ratio tended to be higher in the CKD and both disease groups. These findings suggest no significant elevation in the F/B ratio in MASLD and CKD patients. In addition, among the CKD patient populations, Actinomycetota decreased while Proteobacteriota were more enriched than healthy controls (Fig. 3c).

Gut microbial composition (a) at the phylum level, (b) F/B ratio and (c) the linear discriminant analysis effect size (LEfSe) in comparison of bacterial microbiota between patient groups and healthy control at the phyla level.
Figure 4 demonstrates the relative abundance at the genus level, and Fig. 5 demonstrates the linear discriminant analysis effect size (LEfSe) in comparison of bacterial microbiota between patient groups and healthy controls at the genus level. Healthy individuals had higher levels of SCFA-producing genera such as Bifidobacterium, Ruminococcus, Eubacterium, Coprococcus, Erysipelotrichaceae, and Christensenellaceae R7 group compared to patient groups. On the other hand, patients with MASLD, CKD, and both diseases had dominant levels of the genera Ruminococcus gnavus, Lachnoclostridium, and Escherichia Shigella compared to healthy subjects. To identify significant differences in gut microbial genera among groups, we performed a differential abundance analysis using LEfSe. Notably, out of the top 50 genus levels, 22 genera showed the highest relative abundance and significant differences between groups (Table 2). A Venn diagram highlighting converging and diverging gut microbial signatures in patients with MASLD, CKD, and both diseases is illustrated in Supplementary Fig. 1 and Supplementary File 2.

Gut microbial composition at the genus level.

The linear discriminant analysis effect size (LEfSe) in comparison of bacterial microbiota between patient groups and healthy control at the genus level (a) Healthy controls and MASLD, (b) Healthy controls and CKD, (c) Healthy controls and both diseases, (d) MASLD and CKD, (e) MASLD and both diseases, (f) CKD and both diseases.
Fecal BCoAT expression in the studied groups
To investigate butyrate production by gut microbiota, we further measure gene expression levels of BCoAT using real-time PCR in fecal samples. Overall, our result demonstrated a statistical significance in BCoAT expression among the studied groups (one-way ANOVA, p = 0.007). Comparing between groups, healthy controls and patients with MASLD exhibited comparable fecal BCoAT expression (0.033 ± 0.020 vs. 0.029 ± 0.055, p = 0.797), but significantly higher those in patients with CKD (0.009 ± 0.006, p < 0.001) and both diseases (0.010 ± 0.006, p = 0.001). In addition, its expression level was similar between patients with CKD and both diseases (p = 0.671) (Fig. 6a).

Comparing biomarkers between studied groups (a) fecal BCoAT expression, (b) plasma I-FABP levels.
Plasma I-FABP levels in the studied groups
Plasma I-FABP levels were examined using the ELISA method to investigate the biomarkers for gut epithelial permeability. Overall, our result demonstrated a statistical significance in plasma I-FABP levels among the studied groups (one-way ANOVA, p < 0.001) (Fig. 6b). Comparing between groups, patients with CKD had significantly higher I-FABP levels than healthy subjects (2,596.5 ± 1,879.2 vs. 432.3 ± 263.5 ng/ml, respectively, p < 0.001) and patients with MASLD (977.3 ± 992.8, p < 0.001), but comparable with patients with both diseases (1850.8 ± 1241.6, p = 0.06). The level in patients with both diseases was also higher than in patients with MASLD (p = 0.002) and healthy controls (p < 0.001). Patients with MASLD also had higher I-FABP concentrations than healthy controls (p = 0.045).
Correlations between clinical parameters and microbial genera
Figure 7 demonstrates a correlation between microbial genera and various clinical parameters in all the study participants. Overall, the higher relative abundance of SCFA-producing bacteria, including Bifidobacterium, Fusicatenibacter, and Dorea, were positively correlated with higher eGFR. In contrast, higher abundances of Collinsella, Alistipes, and Escherichia_Shigella were correlated with decreased eGFR values. Additionally, Ruminococcus gnavus and Bacteroides were correlated with elevated ALT levels, while Collinsella was positively correlated with the severity of liver steatosis, as measured by CAP.

Correlation between gut genera and clinical parameters in all the study participants.
In addition, fecal BCoAT level exhibited a positive correlation with eGFR value (r = 0.304, p < 0.001) but had a negative correlation with serum creatinine (r = − 0.229, p = 0.002) and plasma I-FABP (r = − 0.163, p = 0.038). Moreover, I-FABP showed a positive correlation with serum creatinine (r = 0.634, p < 0.001) and serum uric acid (r = 0.295, p < 0.001), but had a negative correlation with eGFR value (r = − 0.601, p < 0.001).
Performance of discriminating MASLD from CKD using significant bacteria
The ROC analysis using the Random Forest classifier based on the top 10 genera was performed to evaluate its diagnostic performance in differentiating the MASLD and CKD groups. This method has been indicated as an accurate ‘classification’ tool for biomedical research14. Our data demonstrated that the diagnostic value obtained from the selected genera exhibited an area under the ROC curve (AUROC) of 0.90 (Fig. 8), suggesting that gut microbial composition at the genus level could effectively discriminate MASLD from CKD. Moreover, we carried out pairwise tests to compare important microbial features at the genus level between the other paired groups studied, as shown in Fig. 5.

The ROC curve analysis based on significant bacteria at the genus level.
Discussion
As the high prevalence and lack of approved therapies for MASLD and CKD, these two diseases linked to cardiometabolic risk factors have gained increasing attention. The main findings of our well-characterized cohorts were as follows: (a) our results demonstrated that patients with MASLD and CKD had alterations of gut diversities and displayed distinct gut microbial compositions compared with healthy subjects; (b) patients with CKD displayed marked decreases in the relative abundance of SCFA-producing bacteria and fecal BCoAT level compared to the MASLD group. Moreover, the concentration of fecal BCoAT was inversely correlated with eGFR and I-FABP levels; (c) patients with CKD also had remarkably increased I-FABP levels, the biomarker of intestinal barrier dysfunction, than healthy controls and patients with MASLD; (d) Based on the ROC analysis, our data showed that the diagnostic value obtained from the different genera between groups exhibited a high diagnostic accuracy with an AUROC of 0.90, indicating distinct alterations of gut microbiota between MASLD and CKD. Finally, our data showed that subgroups of patients with coexisting both diseases exhibited mixed characteristics of MASLD and CKD regarding gut dysbiosis at the genus level. Nonetheless, the marked reduction in fecal BCoAT and barrier dysfunction in this disease entity was more likely similar to the CKD group.
Growing evidence has implicated the bidirectional relationship between gut microbiota and liver (gut-liver axis) and between gut microbiota and kidney (gut-kidney axis) in the development of MASLD and CKD, respectively8,11. Recent epidemiological data on MASLD and CKD have also been established and suggested that gut dysbiosis, metabolic dysfunction, unhealthy lifestyle, and dietary patterns could contribute to these two disorders’ pathogenetic mechanisms15. Most recent studies of gut microbiota in patients with CKD have focused on ESKD, but the data on less severe stages (e.g., CKD stage 3–4) are still lacking. This report characterized gut microbiota diversities and composition in patients with MASLD and CKD. Contrasted to healthy individuals, bacterial communities in patients with MASLD were less diverse and distinct from those of healthy controls. These data indicated that the richness, evenness, and variability in the clustering of gut microbiota were significant changes in patients with MASLD. Regarding the CKD group, a significant difference was found only in the beta diversities, whereas the alpha diversities were similar to healthy individuals. These results were in line with a recent systematic review demonstrating that alpha diversities are not consistently observed in patients with CKD, as only 40% of the reports displayed statistical significance in the indices16.
Current data reveal altered gut microbial composition at multiple taxonomic levels is demonstrated in patients with MASLD and CKD versus healthy subjects11,12. Generally, the bacterial phyla with a high abundance in the gut are Firmicutes, Bacteroidota, Actinomycetota, Proteobacteriota, Fusobacteria, and Verrucomicrobiota. Of these, Firmicutes and Bacteroidota are normally the two most common, representing approximately 90% of the whole population, and thus, the Firmicutes and Bacteroidota (F/B) ratio could reflect the homeostasis of the human gut17. In this study, we observed a trend of increased F/B ratio in patients with CKD versus healthy controls and other groups, although its difference was insignificant. Moreover, the enrichment of Proteobacteriota was observed in the CKD group, while Actinobacteria was found to be more abundant in healthy controls. These data suggest that the shifts of the F/B ratio at the phylum level might not be able to reveal gut dysbiosis associated with MASLD and CKD specifically. Thus, identifying lower taxonomies, such as the genus and species levels, is necessary.
Healthy individuals versus the patient groups in our study displayed a greater abundance of SCFA-producing bacteria at the genus level, including Bifidobacterium, Ruminococcus, Eubacterium, Coprococcus, and Erysipelotrichaceae. SCFAs, mainly acetate, propionate, and butyrate, are recognized as important gut metabolites that display several key physiological processes, such as maintaining intestinal barrier integrity and preventing endotoxins and bacterial translocation18. Moreover, SCFAs, particularly butyrate, have been shown to regulate host immunity and anti-inflammatory response at extra-intestinal sites and thus have been involved in various systemic disorders, including infections, cardio-metabolic diseases, and malignancies19. Among SCFA-producing bacteria enriched in our report, Bifidobacterium, a gram-positive, anaerobic bacterium belonging to the phylum Actinomycetota, is one of the most well-established probiotic strains that provide a positive health impact through its anti-inflammatory and mucosal-protective properties20. An animal model of MASLD showed that Bifidobacterium protected against liver injury; thus, a decrease in its abundance could lead to progressive liver inflammation21. Likewise, a supplement with probiotics containing Bifidobacterium might attenuate the deterioration of kidney function in an experimental mouse model and patients with CKD22.
Gut dysbiosis, as the reduction of beneficial bacteria and enriched potential pathogenic genera, is frequently described in cardio-metabolic disorders, including MASLD and CKD18. In this study, we detected significant changes in gut microbial composition between healthy controls and patients with MASLD. At the genus level, patients with MASLD exhibited increased Ruminococcus gnavus, Lachnoclostridium, and Phascolarctobacterium and the depletion of several SCFA-producing bacteria, including Coprococcus, Romboutsia, Ruminococcus, Erysipelotrichaceae_UCG-003, and Holdemanella when compared with healthy controls. Similar to our results, it was recently shown that Coprococcus might play an important role in decreasing visceral fat by producing SCFAs and vitamin B. In contrast, Lachnoclostridium could exhibit an opposing effect as being involved in the biosynthesis of harmful lipid compounds associated with MASLD23. Regarding Holdemanella, the bacterium was reported to modulate anti-inflammatory effects and metabolic functions, particularly in restoring glucagon-like peptide-1 (GLP-1) and improving insulin sensitivity in obese mice24. Our observations were also in line with a recent meta-analysis of MASLD, demonstrating the decrease of beneficial bacteria, including Faecalibacterium, Coprococcus, and Ruminococcus, as well as an enrichment in pathogenic bacteria such as Escherichia25.
Similarly, when compared with healthy subjects, patients with CKD displayed enriched harmful bacterial genera, including Ruminococcus gnavus, Intestinibacter, and Erysipelatoclostridium, whereas the reduction of SCFA-producing bacteria such as Bifidobacterium, Coprococcus, Erysipelotrichaceae_UCG-003, and Eubacterium. In this context, it has been shown that SCFA-producing bacteria, particularly butyrate-producing taxa, are progressively reduced in patients with advanced stages of CKD26,27. In our study, the relative abundance of Bifidobacterium was positively associated with improved kidney function as defined by eGFR, whereas Ruminococcus gnavus was inversely correlated with eGFR value. The enrichment of Ruminococcus gnavus, a gram-positive anaerobic bacterium, has been linked to intestinal inflammatory disorders, such as IBD and colon cancer, and chronic metabolic diseases, such as T2DM and obesity28. Moreover, recent findings in an animal model strongly indicated that oral administration of Ruminococcus gnavus could aggravate the pathological changes in the kidney29. Interestingly, the bacterium could also down-regulate the expression of intestinal tight junction proteins, generate uremic toxins, and promote inflammatory processes29. Thus, in the context of CKD, these data support the contributing role of gut dysbiosis through the gut-kidney axis in the development and progression of renal damage.
In addition to reducing SCFA-producing bacteria, our data revealed that the level of fecal BCoAT, reflecting butyrate production capacity by gut microbiota, significantly declined in patients with CKD compared with healthy controls and patients with MASLD. These key butyrate-producing taxa found in our study included Ruminococcus, Coprococcus, Eubacterium, and Erysipelotrichaceae_UCG-003. Moreover, fecal BCoAT level was positively correlated with eGFR, which was in agreement with recent publications demonstrating that fecal butyrate concentrations were linked to the decline in eGFR, particularly in advanced stages of CKD26,30. We further compared enriched bacterial genera between patients with CKD versus those with MASLD. Our results showed that the KCD group exhibited a higher relative abundance of genera that produce uremic toxins such as p-cresyl and indoxyl sulfate. For instance, Escherichia_Shigella, an important indole-producing taxa, has been consistently reported to be elevated in several studies of CKD and ESRD31. This bacterium also synthesizes ethanol and other endotoxins, leading to increased oxidative stress and gut permeability, and is considered a risk factor related to MASLD progression32,33. Besides Escherichia_Shigella, Collinsella, and Methanobacteria were also shown to be associated with the disease severity of CKD26. Notably, the genus Collinsella was reported to be associated with increased cholesterol levels, and its enrichment was related to symptomatic atherosclerosis and cerebrovascular events34.
To further understand the mechanisms by which SCFAs might be involved in the pathogenesis of CKD, we evaluated the alteration of circulating I-FABP, a reliable surrogate biomarker of intestinal barrier dysfunction. Our data showed that plasma I-FABP concentration was remarkably elevated in patients with CKD compared to healthy individuals and patients with MASLD. In this respect, a recent prospective study demonstrated that patients with early- and late-stage CKD were associated with decreased expression of intestinal tight junction components, indicating a potential mechanism of gut barrier disruption in CKD35. Interestingly, the increased plasma level of I-FABP was comparable between patients with CKD alone versus those with both diseases. Moreover, circulating I-FABP was negatively correlated with BCoAT level, suggesting a reverse relationship between butyrate production and the degree of intestinal permeability. Together, these data might indicate the complex interplay of host and gut dysbiosis in the development and progression of CKD. From one viewpoint, gut dysbiosis and impaired SCFA production trigger intestinal barrier permeability, allowing uremic toxin precursors into the circulation and thus promoting systemic inflammation and progressive kidney injury. On the other hand, the gradual decline in kidney function with accumulated uremic toxins per se can induce pathogenic bacterial overgrowth and declined saccharolytic bacteria that produce SCFAs, further compromising gut barrier integrity36,37. Thus, this vicious cycle between CKD and gut dysbiosis could have a detrimental effect on the functionality of the intestinal barrier integrity, possibly explaining a marked elevated plasma I-FABP concentration in patients with CKD observed in our study.
Regarding combined both diseases, this entity is considered more complex as gut microbiota and related metabolites could be altered by both MASLD and CKD and thus deserve further investigation. In the clinical aspect, patients who have simultaneous MASLD and CKD are associated with an increased risk of CVD, approximately two times higher than those with either MASLD or CKD alone38. In this study, patients with both diseases had a high relative abundance of Ruminococcus gnavus but reduced several SCFA-producing bacteria, including Bifidobacterium, Romboutsia, and Erysipelotrichaceae_UCG-003 compared to healthy individuals. Additionally, compared both diseases versus MASLD, bacterial genera previously reported in patients with CKD, such as Collinsella, Clostridium_su_stricto_1, and Catenibacterium39, were predominated in both diseases. Likewise, Collinsella, Phascolarctobacterium, Peptoclostridium, and Desulfovibrio were enriched in CKD versus both disease groups. Of note, the Desulfovibrio genus includes sulfate-metabolizing bacteria that generate cytotoxic effects on the progression of renal dysfunction in patients with CKD40. Additionally, fecal BCoAT levels in both disease groups were comparable to those in the CKD group but significantly lower than those in patients with MASLD. In contrast, plasma I-FABP levels were significantly elevated in CKD and both diseases compared to the MASLD group. Together, these data indicated that both diseases partially shared the characteristics of gut dysbiosis between MASLD and CKD in terms of their diversities and microbial communities. Nonetheless, this subgroup of patients had depleted fecal butyrate concentration and exhibited remarkable barrier dysfunction similar to CKD rather than those found in the MASLD group.
Despite the valuable insights provided by our study, several limitations should be acknowledged. First, our observational study was a cross-sectional cohort that could not provide a cause-and-effect relationship. Second, our analysis was based on 16 S rRNA gene sequencing, which offers limited resolution at the strain level and lacks comprehensive functional information about the gut microbiota. Whole genome or shotgun metagenomics would have provided more detailed insights into microbial communities’ functional potential and strain-level differences. Third, the relatively small sample size and absence of an independent validation cohort may limit the generalizability of our findings, particularly the robustness of the random forest classifier model used for distinguishing MASLD and CKD. Future studies with larger cohorts and external validation are warranted to confirm our results. Fourth, the complexity of gut microbiota is influenced by several factors, including dietary patterns, lifestyle, and environmental circumstances. Thus, these factors might limit the interpretation of microbial profiling, especially insufficient dietary information in our study. Finally, although efforts were made to control key confounders by matching study groups for age and sex, residual confounding from variables such as BMI, serum creatinine, and kidney function cannot be excluded. These factors could have influenced the observed gut microbial alterations and should be considered in future investigations.
In conclusion, this study explored the characteristics of gut microbiota and related biomarkers in MASLD- and CKD-alone, as well as the combined entity of MASLD and CKD. Our data emphasized the shared and distinct mechanisms of MASLD and CKD regarding the role of gut dysbiosis, impaired production of butyrate, and intestinal barrier dysfunction. Exploring the correlation between gut dysbiosis in patients with MASLD and CKD might provide clinical application prospects. Considering that no effective pharmacological therapies are the standard of care for MASLD and CKD, interventions to restore microbial balance and improve gut health might be an option. For instance, a recent meta-analysis of randomized controlled trials (RCTs) demonstrates that probiotics, prebiotics, or synbiotics effectively improve liver-related clinical outcomes in patients with MASLD41. Likewise, meta-analysis of RCTs in patients with CKD also supports the use of biotics as their supplement, which could alleviate metabolic indicators related to CVD risk42. Considering interventions beyond lifestyle and dietary modifications, these results could encourage the point that biotic treatment might also have clinical benefits for patients with MASLD and CKD.
Materials and methods
Participants and study design
A total of 180 patients with MASLD, CKD, and both diseases were prospectively recruited in the cross-sectional cohort between 2023 and 2024 in the outpatient liver and kidney clinics at the King Chulalongkorn Memorial Hospital, Thailand. To reduce the confounding effects, patients in each group were matched for age and gender. All participants provided written informed consent before collecting the sample specimens. The study was approved by the Institute Ethics Committee of the University (IRB No. 436-66) and was performed according to the Declaration of Helsinki and the principles of Good Clinical Practice.
The inclusion criteria for MASLD were Thai individuals aged ≥ 18 years with the diagnosis of liver steatosis using vibration-controlled transient elastography (VCTE) with controlled attenuation parameter (CAP, > 248 dB/m)43 and the combination of clinical phenotypes associated with metabolic dysfunction1. The diagnostic criteria of CKD with an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 for at least 3 months were defined following KDIGO’s 2024 Clinical Practice Guideline for Chronic Kidney Disease44. Exclusion criteria in this study were (1) presence of decompensated cirrhosis and complications including HCC, (2) CKD individuals who had eGFR < 15 mL/min/1.73 m2 (stage 5) or started long-term dialysis, (3) presence of significant medical conditions such as active malignancies and autoimmune disorders, (4) history of significant gastrointestinal tract disorders including previous intestinal surgery, inflammatory bowel disease (IBD), and pancreatic disease, and (5) excessive daily alcohol drinking of > 10 g in women and > 20 g in men. In addition to patients with MALSD and CKD, 22 healthy individuals with normal liver function tests, VCTE, and kidney function were recruited as the control group.
All participants in this study were recommended not to take nutritional or herbal supplements, antibiotics, proton pump inhibitors, probiotics, prebiotics, immunosuppressive drugs, or any medications that could potentially impact the gut microbiota analysis at least four weeks before the recruitment. The patient’s clinical information, including demographic data, anthropometric results, and laboratory assessments, was recorded at the enrollment.
The sample size in this study was calculated based on data from a previous report examining gut microbiota metabolites in patients with MASLD and CKD45. The number of participants in each group was calculated by G*Power version 3.1.9.646, and the estimated sample size in each group was approximately 60.
Vibration-controlled transient elastography (VCTE)
As appropriate, VCTE equipment (Echosens, Paris, France) with M-probe and XL-probe was applied to obtain CAP and liver stiffness measurement (LSM), indicating liver steatosis and fibrosis, respectively47. The accurate values were based on at least 10 validated assessments with a success rate of over 60% and an interquartile range of less than 30%.
Fecal collection, DNA extraction and sequencing
Fecal sample specimens were collected from the participants in the DNA/RNA Shield™-Fecal Collection tubes (Zymo Research Corp, Irvine, CA, USA). Then, the samples were vigorously shaken and stored at − 80 °C until analysis. According to the manufacturer’s instructions, the samples were extracted for DNA by the ZymoBIOMICS™ DNA Miniprep Kit (Zymo Research Corp, Irvine, CA, USA). The quality, including purity and concentration of total DNA, were then measured by DeNovix™ UV-Vis spectrophotometer and was preserved at − 20 °C until further testing. Afterward, amplicon-based 16 S rRNA gene sequencing in the V4 hypervariable regions was amplified using the forward primer 515 F (5′-GTGCCAGCMGCCGCGGTAA-3′) and the reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′). This process was followed by paired-end sequencing on the Illumina MiSeq 2 × 300 bp platform (San Diego, CA, USA) conducted at Mod Gut Co., Ltd. (Bangkok, Thailand).
Gut microbiota data processing and analysis
The microbiome analysis was performed using QIIME 248 (version 2024.2). The raw sequence data was demultiplexed using the q2-demux plugin. Then, reads with expected errors (maxEE) higher than 3.0 were discarded using denoising software DADA2 (via q2-dada2). After that, mitochondria and chloroplast 16 S sequences were eliminated. A phylogeny was created from representative sequences using the align_to_tree_mafft_fasttree command of the q2-phylogeny plugin. Alpha-diversity indexes and beta-diversity metrics were estimated using the q2-diversity plugin after rarefying the samples to 12,000 reads. Then, Principal Coordinate Analysis (PCoA) was performed on the beta-diversity distance metrics. The amplicon sequence variants (ASVs) were taxonomically assigned using the classify-sklearn naive Bayes taxonomy classifier against the Silva (version 138) 99% reference sequences. The Spearman Rank Coefficient was used to calculate the correlation between clinical parameters and microbial genera. The clustermap function from the seaborn library in Python version 3.10.13 was utilized to generate a correlation graph representing clusters. The diagnostic performance of microbial genera in distinguishing between groups was evaluated using the area under the receiver operating characteristic (ROC) curve, employing the Random Forest machine learning algorithm with default parameters from the RandomForestClassifier function in the sklearn.ensemble library. The original dataset was partitioned using the train_test_split function from the sklearn.model_selection library in Python version 3.10.13, with the parameters test_size = 0.33 and random_state = 42. Subsequently, the feature scores derived from the Random Forest were utilized to visualize the important features.
Quantification of fecal BCoAT gene
The quantification of the butyryl-CoA: acetate CoA transferase (BCoAT) gene in fecal samples, which reflects the ability of gut microbiota to produce butyrate, was performed by qPCR using 4× CAPITAL™ qPCR Green Master Mix (Biotech Rabbit) with the following degenerate primers: (forward primer) 5′-GCIGAICATTTCACITGGAAYWSITGGCAYATG-3′ and (reverse primer) 5′-CCTGCCTTTGCAATRTCIACRAANGC-3′ as described previously49. Briefly, the qPCR conditions started with a DNA-denaturation step at 95 °C for 15 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at a primer-specific temperature for 20 s, and extension at 72 °C for 30 s. The quantification of the BCoAT gene in each fecal sample was calculated and normalized using the V3–V4 gene as the representative of total bacteria.
Intestinal barrier dysfunction assessment
To quantify intestinal fatty acid binding protein (I-FABP) level, plasma extracted from peripheral blood samples and stored at − 80 °C was evaluated using commercially available enzyme-linked immunosorbent assay (ELISA) kits (Hycult Biotech enzyme-linked immunosorbent assay kit, Uden, Netherlands), following the manufacturer’s instructions. The plasma samples were diluted at a proportion of 1:2 before the measurement of I-FABP.
Statistical analysis
The patients’ characteristical information was performed using SPSS (version 22.0.0, SPSS Inc., Chicago, IL, USA) and GraphPad Prism Software (version 9.5.0, Boston, MA, USA). Chi-square was applied to analyze categorical variables. The independent samples t-test and one-way ANOVA were performed to compare the means between groups as appropriate. Statistical tests for alpha and beta diversities were conducted using Kruskal–Wallis and PERMANOVA (number of permutations = 999). A significant differential abundance analysis of microbiota was also carried out using linear discriminant analysis of effect size (LEfSe)50. Nonparametric factorial Kruskal–Wallis sum-rank tests were initially applied to select differentially distributed features among classes (p < 0.05). Consequently, the linear discriminant analysis (LDA) model was used to estimate their effect sizes, supported by 30-fold bootstrapping (cutoff = logarithmic LDA score of ≥ 1.0). For the Random forest analysis, a p-value of less than 0.05 indicated the statistical significance of discriminating important features.
Data availability
All data analyzed in this published article and its supplementary materials are available in the National Center for Biotechnology Information Gene Expression Omnibus repository under the BioProject accession number PRJNA1152299. The datasets used and/or analysed in the present study are available from the corresponding author upon reasonable request.
References
Rinella, M. E. et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 79, 1542–1556. https://doi.org/10.1016/j.jhep.2023.06.003 (2023).
Younossi, Z. et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69, 2672–2682. https://doi.org/10.1002/hep.30251 (2019).
Lazarus, J. V. et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 19, 60–78. https://doi.org/10.1038/s41575-021-00523-4 (2022).
Kovesdy, C. P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 12, 7–11. https://doi.org/10.1016/j.kisu.2021.11.003 (2022).
Targher, G., Chonchol, M. B. & Byrne, C. D. CKD and nonalcoholic fatty liver disease. Am. J. Kidney Dis. 64, 638–652. https://doi.org/10.1053/j.ajkd.2014.05.019 (2014).
Mantovani, A. et al. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: An updated meta-analysis. Gut 71, 156–162. https://doi.org/10.1136/gutjnl-2020-323082 (2022).
Roderburg, C. et al. Non-alcoholic fatty liver disease (NAFLD) is associated with an increased incidence of chronic kidney disease (CKD). Eur. J. Med. Res. 28, 153. https://doi.org/10.1186/s40001-023-01114-6 (2023).
Tilg, H., Cani, P. D. & Mayer, E. A. Gut microbiome and liver diseases. Gut 65, 2035–2044. https://doi.org/10.1136/gutjnl-2016-312729 (2016).
Jayakumar, S. & Loomba, R. Review Article: Emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Aliment Pharmacol. Ther. 50, 144–158. https://doi.org/10.1111/apt.15314 (2019).
Amini Khiabani, S., Asgharzadeh, M. & Samadi Kafil, H. Chronic kidney disease and gut microbiota. Heliyon 9, e18991. https://doi.org/10.1016/j.heliyon.2023.e18991 (2023).
Krukowski, H. et al. Gut Microbiome studies in CKD: Opportunities, pitfalls and therapeutic potential. Nat. Rev. Nephrol. 19, 87–101. https://doi.org/10.1038/s41581-022-00647-z (2023).
Aron-Wisnewsky, J. et al. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 17, 279–297. https://doi.org/10.1038/s41575-020-0269-9 (2020).
Wang, X. et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 69, 2131–2142. https://doi.org/10.1136/gutjnl-2019-319766 (2020).
Zou, K. H., O’Malley, A. J. & Mauri, L. Receiver-operating characteristic analysis for evaluating diagnostic tests and predictive models. Circulation 115, 654–657. https://doi.org/10.1161/CIRCULATIONAHA.105.594929 (2007).
Byrne, C. D. & Targher, G. NAFLD as a driver of chronic kidney disease. J. Hepatol. 72, 785–801. https://doi.org/10.1016/j.jhep.2020.01.013 (2020).
Stanford, J., Charlton, K., Stefoska-Needham, A., Ibrahim, R. & Lambert, K. The gut microbiota profile of adults with kidney disease and kidney stones: A systematic review of the literature. BMC Nephrol. 21, 215. https://doi.org/10.1186/s12882-020-01805-w (2020).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. https://doi.org/10.1038/nature08821 (2010).
Nogal, A., Valdes, A. M. & Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes. 13, 1–24. https://doi.org/10.1080/19490976.2021.1897212 (2021).
Mann, E. R., Lam, Y. K. & Uhlig, H. H. Short-chain fatty acids: Linking diet, the microbiome and immunity. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-024-01014-8 (2024).
O’Callaghan, A. & van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 7, 925. https://doi.org/10.3389/fmicb.2016.00925 (2016).
Fang, D. et al. Bifidobacterium pseudocatenulatum LI09 and Bifidobacterium catenulatum LI10 attenuate D-galactosamine-induced liver injury by modifying the gut microbiota. Sci. Rep. 7, 8770. https://doi.org/10.1038/s41598-017-09395-8 (2017).
Wang, I. K. et al. Effect of a probiotic combination in an experimental mouse model and clinical patients with chronic kidney disease: A pilot study. Front. Nutr. 8, 661794. https://doi.org/10.3389/fnut.2021.661794 (2021).
Nogal, A. et al. Circulating levels of the short-chain fatty acid acetate mediate the effect of the gut microbiome on visceral fat. Front. Microbiol. 12, 711359. https://doi.org/10.3389/fmicb.2021.711359 (2021).
Romani-Perez, M. et al. Holdemanella biformis improves glucose tolerance and regulates GLP-1 signaling in obese mice. FASEB J. 35, e21734. https://doi.org/10.1096/fj.202100126R (2021).
Li, F., Ye, J., Shao, C. & Zhong, B. Compositional alterations of gut microbiota in nonalcoholic fatty liver disease patients: A systematic review and meta-analysis. Lipids Health Dis. 20, 22. https://doi.org/10.1186/s12944-021-01440-w (2021).
Gao, B. et al. Butyrate producing microbiota are reduced in chronic kidney diseases. Sci. Rep. 11, 23530. https://doi.org/10.1038/s41598-021-02865-0 (2021).
He, M. et al. Gut microbial metabolites SCFAs and chronic kidney disease. J. Transl. Med. 22, 172. https://doi.org/10.1186/s12967-024-04974-6 (2024).
Crost, E. H., Coletto, E., Bell, A. & Juge, N. Ruminococcus gnavus: Friend or foe for human health. FEMS Microbiol. Rev. https://doi.org/10.1093/femsre/fuad014 (2023).
Hong, J. et al. Specific alternation of gut microbiota and the role of Ruminococcus gnavus in the development of diabetic nephropathy. J. Microbiol. Biotechnol. 34, 547–561. https://doi.org/10.4014/jmb.2310.10028 (2024).
Steenbeke, M. et al. Gut microbiota and their derived metabolites, a search for potential targets to limit accumulation of protein-bound uremic toxins in chronic kidney disease. Toxins (Basel) 13. https://doi.org/10.3390/toxins13110809 (2021).
Voroneanu, L. et al. Gut microbiota in chronic kidney disease: From composition to modulation towards better outcomes—A systematic review. J. Clin. Med. https://doi.org/10.3390/jcm12051948 (2023).
Zhu, L. et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 57, 601–609. https://doi.org/10.1002/hep.26093 (2013).
Shen, F. et al. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobil. Pancreat. Dis. Int. 16, 375–381. https://doi.org/10.1016/S1499-3872(17)60019-5 (2017).
Karlsson, F. H. et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245. https://doi.org/10.1038/ncomms2266 (2012).
Georgopoulou, G. A. et al. Altered expression of intestinal tight junctions in patients with chronic kidney disease: A pathogenetic mechanism of intestinal hyperpermeability. Biomedicines. https://doi.org/10.3390/biomedicines12020368 (2024).
Lambert, K. et al. Targeting the gut microbiota in kidney disease: The future in renal nutrition and metabolism. J. Ren. Nutr. 33, S30–S39. https://doi.org/10.1053/j.jrn.2022.12.004 (2023).
Tang, Z., Yu, S. & Pan, Y. The gut microbiome tango in the progression of chronic kidney disease and potential therapeutic strategies. J. Transl Med. 21, 689. https://doi.org/10.1186/s12967-023-04455-2 (2023).
Chung, G. E. et al. Combined effects of chronic kidney disease and nonalcoholic fatty liver disease on the risk of cardiovascular disease in patients with diabetes. Biomedicines https://doi.org/10.3390/biomedicines10061245 (2022).
Cao, C., Zhu, H., Yao, Y. & Zeng, R. Gut dysbiosis and kidney diseases. Front. Med. (Lausanne). 9, 829349. https://doi.org/10.3389/fmed.2022.829349 (2022).
Mazidi, M., Shekoohi, N., Covic, A., Mikhailidis, D. P. & Banach, M. Adverse impact of Desulfovibrio spp. And beneficial role of Anaerostipes spp. On renal function: Insights from a Mendelian randomization analysis. Nutrients https://doi.org/10.3390/nu12082216 (2020).
Rong, L. et al. Use of probiotics, prebiotics, and synbiotics in non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 38, 1682–1694. https://doi.org/10.1111/jgh.16256 (2023).
Zheng, H. J. et al. Probiotics, prebiotics, and synbiotics for the improvement of metabolic profiles in patients with chronic kidney disease: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 61, 577–598. https://doi.org/10.1080/10408398.2020.1740645 (2021).
Karlas, T. et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J. Hepatol. 66, 1022–1030. https://doi.org/10.1016/j.jhep.2016.12.022 (2017).
Levin, A. et al. Executive summary of the KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease: Known knowns and known unknowns. Kidney Int. 105, 684–701. https://doi.org/10.1016/j.kint.2023.10.016 (2024).
Obeid, R. et al. Plasma concentrations of trimethylamine-N-oxide, choline, and betaine in patients with moderate to advanced chronic kidney disease and their relation to cardiovascular and renal outcomes. J. Ren. Nutr. 34, 530–538. https://doi.org/10.1053/j.jrn.2024.03.009 (2024).
Kang, H. Sample size determination and power analysis using the G*Power software. J. Educ. Eval. Health Prof. 18, 17. https://doi.org/10.3352/jeehp.2021.18.17 (2021).
Cao, Y. T. et al. Accuracy of controlled attenuation parameter (CAP) and liver stiffness measurement (LSM) for assessing steatosis and fibrosis in non-alcoholic fatty liver disease: A systematic review and meta-analysis. EClinicalMedicine 51, 101547. https://doi.org/10.1016/j.eclinm.2022.101547 (2022).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Louis, P. & Flint, H. J. Development of a semiquantitative degenerate real-time PCR-based assay for estimation of numbers of butyryl-coenzyme A (CoA) coa transferase genes in complex bacterial samples. Appl. Environ. Microbiol. 73, 2009–2012. https://doi.org/10.1128/aem.02561-06 (2007).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Acknowledgements
We would like to thank the staff of the Center of Excellence in Hepatitis and Liver Cancer, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand, and ModGut Co., Ltd., Bangkok, Thailand, for supporting the sequencing and analysis of gut microbial data in this study.
Funding
This work was supported by the NSRF via the Program Management Unit for Human Resources and Institutional Development, Research and Innovation (PMU-B, grant number B36G660010), Thailand Science Research and Innovation Fund Chulalongkorn University (Fundamental Fund 2567) and Center of Excellence in Hepatitis and Liver Cancer, Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand.
Author information
Authors and Affiliations
Contributions
TD and PT designed the concept of the study; PS, PC, and PP performed the experiment; SU, PK, and PT provided the specimens and clinical data; NC, PC, and PP provided the suggestions for data analysis; TD, PC, and PP performed the statistical analysis, BB completed the data visualization and interpretation; TD and NC drafted the manuscript, SU, PK, and PT provided revision to the scientific content; PT reviewed, edited and supervised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dissayabutra, T., Chuaypen, N., Somnark, P. et al. Characterization of gut dysbiosis and intestinal barrier dysfunction in patients with metabolic dysfunction-associated steatotic liver disease and chronic kidney disease: a comparative study. Sci Rep 15, 15481 (2025). https://doi.org/10.1038/s41598-025-00237-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-00237-6

{kind=link}