
Abstract
Evidence indicates a connection between periodontitis (PD) and systemic lupus erythematosus (SLE), though the underlying co-morbid mechanisms remain unclear. This study sought to identify the genetic factors and potential therapeutic agents involved in the interaction between PD and SLE. We employed multi-omics methodologies, encompassing differential expression analysis, weighted gene co-expression network analysis (WGCNA), functional enrichment (GO/KEGG), LASSO regression, diagnostic model construction, protein-protein interaction (PPI) networks, immune infiltration profiling, computational drug prediction, molecular docking, and disease subtyping, to analyze PD and SLE expression datasets from the Gene Expression Omnibus (GEO) database (GSE10334, GSE16134, GSE50772, and GSE81622). Cross-analysis identified 32 crosstalk genes (CGs) common to both PD and SLE. LASSO analysis pinpointed three key diagnostic genes (TAGLN, MMP9, TNFAIP6) for both conditions. The resulting diagnostic models demonstrated robust efficacy in both training and validation datasets. Four topological algorithms in Cytoscape highlighted four central crosstalk genes (TAGLN, MMP9, TNFAIP6, IL1B). Additionally, hesperidin, doxycycline, and cytochalasin D emerged as potential therapeutic agents. Two subtypes (C1 and C2) of PD and SLE were delineated based on CG expression profiles. The development of diagnostic models, potential drug identification, and disease subtype classification are poised to enhance diagnosis and treatment. These findings aim to deepen the understanding of PD and SLE complexities.
Similar content being viewed by others
Introduction
Periodontitis (PD) is a common chronic inflammatory condition, affecting approximately half of all adults globally1, with severe periodontitis affecting 11.2% of the world’s population2. This presents a significant global health challenge. The main cause of PD is plaque accumulation, leading to gingival inflammation and irreversible damage to periodontal structures, resulting in tooth mobility and potential loss3. Recent research has highlighted the association between PD and systemic diseases, with proposed mechanisms including metastatic infections, bacterial toxin dissemination, and immune-mediated damage4.
Systemic lupus erythematosus (SLE) is a prevalent systemic autoimmune disease with a high mortality rate, particularly affecting women of childbearing age5,6. A prospective study has identified cardiovascular disease as the leading cause of death in SLE patients7. Emerging evidence indicates a potential connection between SLE and the human oral microbiota8. Research by Corrêa, Jôice Dias et al.9has shown that SLE can impact the composition of the subgingival microbiota, with periodontal disease closely linked to the intraoral microbiota10,11. These findings collectively suggest a plausible association between PD and SLE. Moreover, a Mendelian randomization (MR) study has indicated that SLE may elevate the risk of developing periodontitis12. Despite the growing body of evidence supporting a robust association between PD and SLE, there is a paucity of studies investigating their correlation at the gene expression level.
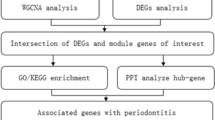
The rise of microarrays and high-throughput sequencing has popularized the use of bioinformatics in exploring disease associations. Transcriptomics analysis has become a favored method for detecting differences in gene expression across diseases due to its comprehensive evaluation of gene expression levels and the intricate relationships between genes and associated cell infiltration. In this research, we aimed to identify potential crosstalk genes (CGs) and key biological effects between PD and SLE using transcriptomics. We developed a disease diagnostic model employing machine learning and logistic regression algorithms, validated our findings across four datasets, and pinpointed immune-related hub genes through interaction network analysis. The correlation network of hub genes was confirmed using immune infiltration analysis. The study also unveiled three potential therapeutic agents, determined disease subtypes through consensus clustering analysis, and presented the study flowchart in Fig. 1. In conclusion, this study aims to offer novel insights for further exploration of the connection between PD and SLE, as well as advancements in diagnosis and treatment.

Flowchart of this study.
Materials and methods
Data sources
In this study, two PD datasets (GSE10334, GSE16134) and two SLE datasets (GSE50772, GSE81622) along with corresponding microarray platforms (GPL570, GPL10558) were sourced from the Gene Expression Omnibus (GEO) database13 (https://www.ncbi.nlm.nih.gov/geo/). GSE16163 and GSE81622 were utilized for disease validation. Detailed information on these datasets is presented in Table 1. Normalization of all expression matrices was performed using the “normalizeBetweenArrays” package in R software (version 4.4.2).
Identification of DEGs
GSE10334 and GSE50772 datasets underwent normalization and differential gene expression analysis between control and disease groups using the “limma” package14 in R (version 4.4.2). Criteria for differential expression were adjusted p-value < 0.05 and |logFC| ≥ 0.5. Volcano plots illustrating DEG distribution were generated using the “ggplot2” package. Heat maps displaying the expression patterns of the top 20 genes in both groups were created using the “(version 4.4.2)pheatmap” package in R.
WGCNA identification of key gene modules
Weighted gene co-expression network analysis (WGCNA) is a systems biology approach used to analyze gene expression data by clustering genes with similar expression patterns into modules. In this study, WGCNA in R was applied for gene clustering15. Initially, the gene expression matrix quality was assessed, and unsuitable genes and samples were excluded to ensure robust analysis. The pickSoftThreshold function was used to select an appropriate soft threshold for network construction. A topological overlap matrix was then constructed to identify gene modules using the dynamic tree-cutting algorithm. The correlation between modules and phenotypic data was calculated and visualized to identify key modules. Finally, genes within these key modules were extracted for further analysis.
Identification and functional enrichment of CGs
Differentially expressed genes (DGEs) from the GSE10334 and GSE50772 datasets were analyzed alongside genes from key WGCNA modules. The overlapping genes identified were termed crosstalk genes (CGs) for PD and SLE. Gene Ontology (GO) and KEGG16,17,18enrichment analyses of CGs were conducted using the DAVID19 bioinformatics resources (https://david.ncifcrf.gov/). Simultaneously, KEGG enrichment results were aligned with relevant genes, and the top 20 pathways were visualized using a Sankey bubble map via CNSknowall (https://www.cnsknowall.com/).
Determination of the best shared diagnostic genes
Expression matrices of CGs were extracted from the GSE10334 and GSE50772 datasets for LASSO regression analysis, a regularization technique that automates feature selection and reduces model complexity20. This method mitigates overfitting and enhances generalizability through regularization. The “glmnet” package21 and lambda.min were employed to identify feature genes in GSE10334 and GSE50772. Hierarchical 10-fold cross-validation determined the optimal regularization parameter (λ) for the PD (GSE10334) and SLE (GSE50772) training sets. λ was explored over a range from 10^−4 to 10^2 using coordinate descent. The PD dataset used a random seed of “12”, while the SLE dataset used “34”. A Wein plot intersected the LASSO analysis outcomes to identify shared diagnostic genes.
Diagnostic model construction and efficacy assessment
To evaluate the diagnostic efficacy of genes in PD and SLE, shared diagnostic genes were integrated into datasets GSE10334 and GSE50772. Logistic regression models for PD and SLE were developed using the “Irm” package22. Receiver Operating Characteristic (ROC) curves were generated with the “pROC” package23in R, with the area under the curve (AUC) serving as the metric for diagnostic efficacy. Bias-correction curves assessed prediction accuracy, while clinical decision curve analysis (DCA) evaluated the concordance between predicted and actual outcomes24. Datasets GSE16134 and GSE81622 served as validation sets, enhancing model validation through ROC analysis. Additionally, ROC curves for each diagnostic gene were plotted in both training and validation sets to demonstrate their predictive capabilities. Finally, column line graphs were created to depict the relationship between gene expression scores and disease risk in PD and SLE.
Screening for hub genes
To identify key interacting genes within CGs, a protein-protein interaction (PPI) network was constructed using the STRING database25. Cytoscape (3.10.2) facilitated the analysis of gene interactions within this network26. Four topological algorithms—MCC, EPC, Degree, and MNC—were employed to evaluate the pivotal genes in the CGs and their expression across various datasets. The screened genes were designated as hub genes for further analysis.
ssGSEA
Single-sample Gene Set Enrichment Analysis (ssGSEA) is an advanced analytical method that enables gene set enrichment analysis on individual samples, unlike traditional GSEA. ssGSEA is widely applicable in various biological studies, especially in oncology and immunology, allowing for the assessment of immune-related gene set activity in a single sample27. The “GSVA” package28 in R was employed to analyze the infiltration of 28 immune cells in both disease and control groups. Results are presented in box-and-line plots and correlation heatmaps.
Identification of drug candidates
To explore therapeutic agents for addressing PD and SLE co-morbidities, we employed the Enrichr29 platform (https://maayanlab.cloud/Enrichr/) to import diagnostic and key interacting genes identified as potential therapeutic targets. Subsequently, we screened drug candidates linked to these genes using the Drug Signature Database (DSigDB) within the same platform30.
Molecular docking is a computational technique used in biology and drug design to predict the binding modes and affinities between small molecules, such as drug active ingredients, and larger molecules like proteins or nucleic acids31. This method is crucial in drug discovery, enzyme engineering, and protein-ligand interaction studies. In this study, molecular docking was employed to assess the binding capabilities between drug molecules and proteins. Structure files for the drug molecules and proteins were sourced from PubChem32 (https://pubchem.ncbi.nlm.nih.gov/) and RCSB PDB33 (https://www.rcsb.org/), respectively. The molecules were then optimized using Chem3D (23.1.1) and PyMOL (4.6.0)34. AutoDockTools (1.5.7)35 facilitated the molecular docking process, with visualization conducted through PyMOL.
Consensus clustering analysis to identify disease subtypes
Consensus clustering is an ensemble learning technique employed to discern the underlying structure of data by synthesizing outcomes from multiple clustering analyses. This approach is particularly useful for delineating disease subtypes. To thoroughly assess the influence of CGs expression on PD and SLE, we extracted matrix data pertinent to CGs and applied the Pam algorithm within the “ConsensusClusterPlus” package36 to cluster and identify disease subtypes. We employed box-and-line plots to illustrate the differential expression levels of CGs across these subtypes, highlighting their distinctions. Additionally, Principal Component Analysis (PCA) was used to assess the effectiveness of consensus clustering.
Results
DEGs in PD and SLE datasets
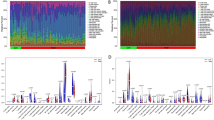
The datasets were standardized, and differentially expressed genes (DEGs) were identified using the limma package. The PD dataset (GSE10334) revealed 1,218 DEGs, comprising 691 up-regulated and 527 down-regulated genes. The SLE dataset (GSE50772) identified 3,326 DEGs, with 1,568 up-regulated and 1,758 down-regulated genes. Volcano plots depicted DEG expression patterns for both diseases (Fig. 2A and E), while heatmaps displayed the top 20 up- and down-regulated genes (Fig. 2B and F).

(A)-(D) DEGs and WGCNA results of PD; (E)-(H) DEGs and WGCNA results of SLE; (I) The highest scoring gene cluster in (D); (J)-(K) The highest scoring gene clusters in (H).
WGCNA network analysis
The sample clustering analysis retained all samples from GSE10334 and GSE50772. To ensure the network adhered to the scale-free topology, we applied soft thresholds of 6 and 7 for GSE10334 and GSE50772, respectively. Using a minimum module size of 50 and a cut height of 0.25, we visualized the relevant modules (Fig. 2C and G). For the PD and SLE samples, 23 and 15 modules were identified, respectively (Fig. 2D and H). To validate our findings, we selected modules with correlation coefficients exceeding 0.6 for further analysis (Fig. 2I-K). This included the blue module (|r| = 0.66, P < 0.001) in PD and the red (|r| = 0.64, P < 0.001) and pink (|r| = 0.69, P < 0.001) modules in SLE. We then integrated the genes from these key modules with the DEGs of both diseases and used Venn diagrams to visualize them (Fig. 3A), identifying 32 crosstalk genes (CGs).

(A) Cross-Wayne plots depicting differentially expressed genes (DEGs) and weighted gene co-expression network analysis (WGCNA); (B) Gene Ontology (GO) enrichment analysis of crosstalk genes; (C) Mulberry map illustrating the association between the top 20 KEGG pathways and the corresponding crosstalk genes.
GO and KEGG enrichment analysis
The 32 CGs underwent enrichment analysis using DAVID, with results prioritized by P-value. The biological process (BP) analysis suggested a strong association with inflammatory responses and immune factors. Cellular component (CC) and molecular function (MF) enrichment were primarily linked to extracellular pathways and protein binding (Fig. 3B). Further examination of the top 20 KEGG pathways and their genes highlighted a significant connection to immune-related pathways, such as the NF-kappa B signaling pathway, IL-17 signaling pathway, and cytokine interactions (Fig. 3C). This led to the hypothesis of a potential link between the pathogenesis of PD and SLE and immune system function.
Selection of shared diagnostic genes by LASSO analysis
To identify shared diagnostic genes for PD and SLE, we analyzed the CG matrices from GSE10334 and GSE50772 using the LASSO regression algorithm. This analysis screened 15 genes in GSE10334 and 6 in GSE50772 (Fig. 4A-D). Cross-referencing these results identified three promising shared diagnostic genes: TAGLN, MMP9, and TNFAIP6 (Fig. 4E). In the PD dataset, these genes showed elevated expression in the disease group (Fig. 4F). In contrast, within the SLE dataset, TAGLN was downregulated in the disease group, while MMP9 and TNFAIP6 were upregulated (Fig. 4G).

(A)-(D) Results of LASSO analysis for PD and SLE; (E) Intersection of PD and SLE after performing LASSO analysis; (F)-(G) Expression of three genes screened by LASSO analysis in the PD and SLE datasets. *P < 0.05; **P < 0.01; ***P < 0.001.
Model construction and assessment of diagnostic efficacy
To evaluate the predictive efficacy of the diagnostic genes, a prediction model was developed using three diagnostic genes and represented with column line graphs. The nomogram and decision curve analysis (DCA) confirmed the model’s satisfactory predictive performance (Fig. 5A and B). Additionally, receiver operating characteristic (ROC) curves were utilized to assess the combined diagnostic efficacy of these genes in both training and validation sets. The area under the curve (AUC) values surpassed 0.85 in both sets, indicating the model’s exceptional predictive capability (Fig. 5C and D).
To evaluate the predictive efficacy of individual diagnostic genes in PD and SLE, ROC curves for TAGLN, MMP9, and TNFAIP6 were generated across four datasets. In the PD dataset, all AUC values exceeded 0.75 (Fig. 6A and B), while in the SLE dataset, all AUC values surpassed 0.65 (Fig. 6C and D). These results indicate that the three genes exhibit satisfactory diagnostic efficacy and may serve as common diagnostic markers for PD and SLE co-morbidities.

(A) A diagnostic model for PD, featuring a column chart, calibration curve, and clinical decision curve; (B) A diagnostic model for SLE, also comprising a column chart, calibration curve, and clinical decision curve; (C) ROC curves for the PD diagnostic model in both training and validation sets; (D) ROC curves for the SLE diagnostic model in training and validation sets.

(A)-(B) ROC curves for diagnostic genes in PD datasets; (C)-(D) ROC curves for diagnostic genes in SLE datasets; (E) PPI network diagram of 32 crosstalk genes; (F) Central crosstalk genes identified by four topological algorithms in Cytoscape; (G)-(J) Expression profiles of central crosstalk genes in PD and SLE datasets.
Central genes in CGs
To identify potential interactions in CGs, we used the STRING database to construct a protein-protein interaction (PPI) network comprising 32 nodes and 53 edges (Fig. 6E). We applied four topology algorithms in Cytoscape to pinpoint four key interacting genes: MMP9, IL1B, CXCL8, and FCGR3B (Fig. 6F). These genes demonstrated elevated expression levels and p-values below 0.05 in both the PD and SLE datasets (Fig. 6G-J).
Analysis of immune infiltration
Previous enrichment analyses have established a strong link between the development of PD and SLE with immune factors. To explore this connection, we analyzed the infiltration of 28 immune cell types in both diseases using the ssGSEA method. In the PD dataset, infiltration levels of various B cells, T cells, and myeloid-derived suppressor cells (MDSCs) were significantly elevated in disease groups (Fig. 7A). In contrast, SLE showed significantly increased levels of activated dendritic cells, central memory T cells (CD8), eosinophils, and neutrophils, while effector memory T cells (CD4 and CD8) were notably reduced (Fig. 7B). Correlation analysis revealed a high concordance between immune cell infiltration and key interaction genes, highlighting a strong association between these genes and disease progression (Fig. 7C and D).

(A)-(B) Analysis of immune infiltration of 28 immune cells in PD and SLE datasets; (C) The connection between central crosstalk genes and immune cells in PD; (D) The connection between central crosstalk genes and immune cells in SLE. Red is a positive correlation and purple is a negative correlation. *P < 0.05; **P < 0.01; ***P < 0.001.
Identification of potential drugs and molecular docking
Diagnostic and key interaction genes were integrated into the Enrichr platform, and the top three drugs from the DSigDB library were identified based on P-value significance and gene composite scores. As shown in Table 2, the highest-scoring compounds are most pertinent to MMP9, CXCL8, and IL1B. These small molecules are potential therapeutic agents for PD and SLE co-morbidities. Molecular docking results (Fig. 8A-C) revealed that the binding energies of the nine drug-target pairs were all below − 5 kcal/mol, with MMP9 and IL1B displaying stronger affinities than CXCL8 (Fig. 8D). These results highlight the therapeutic potential of these small molecules for PD and SLE co-morbidities.

(A)-(C) Molecular docking results for nine gene-drug small molecule pairs generated by PyMol software (version 4.6.0); (D) Minimum binding energy between drug small molecules and target proteins calculated by AutoDockTools (version 1.5.7); (E)-(F) PD and SLE disease subtypes identified via CGs clustering analysis using the “ConsensusClusterPlus” package in R software (version 4.4.2); (G) CG expression differences between PD subtypes; (H) CG expression differences between SLE subtypes. *P < 0.05; **P < 0.01; ***P < 0.001.
Recognizing disease subtypes by CGs
Cluster analysis on the PD dataset GSE10334 and the SLE dataset GSE50772 identified two disease subtypes, C1 and C2 (Fig. 8E and F). Box-and-line plots depicted the expression of 32 CGs across these subtypes, highlighting the reliability of subtype classification through gene expression differences. In PD, most CGs showed higher expression in the C2 subtype compared to C1 (Fig. 8G). Conversely, in SLE, most CGs exhibited higher expression in the C1 subtype than in C2 (Fig. 8H). The results of PCA also showed significant differences between the subtypes (Supplementary material 1). These results underscore significant differentiation between PD and SLE subtypes, suggesting CGs are crucial in the pathogenesis of these diseases.
Discussions
PD and SLE are globally prevalent diseases whose rising incidence poses significant health challenges. Despite differing etiologies, research has identified notable associations between them. A recent systematic review and meta-analysis highlighted a bidirectional causal relationship, revealing that SLE patients face an increased risk of developing PD37. A Norwegian population-based study involving 1,990 SLE patients used logistic regression to analyze periodontitis risk, finding that SLE patients had nearly double the risk compared to controls, especially among younger individuals38. Additionally, a case-control study reported elevated cytokine levels in the saliva of PD and SLE patients39. Salivary IL-1B and IL-4 levels were positively correlated with periodontitis severity, suggesting their potential as biomarkers for periodontal damage in SLE patients. The study indicates that activation of auto-reactive B cells and an imbalance in immune cell populations, such as macrophages, neutrophils, CD4 + T cells, and dendritic cells, may underlie these mechanisms40. Research by C.P.C. Marques et al. further suggests that periodontitis might exacerbate SLE by sustaining elevated Toll-like receptor (TLR) expression, while periodontal therapy could mitigate SLE symptoms by reducing TLR expression41. Recognizing the link between these diseases and their inflammatory and immune responses is crucial, as shared mechanisms may connect their co-morbidities. Given the health risks and potential interactions between periodontitis and SLE, understanding the mechanisms underlying their co-morbidities is vital for effective prevention and treatment.
In this study, 32 common genes associated with both periodontitis (PD) and systemic lupus erythematosus (SLE) were identified. Enrichment analysis indicated that the disease group showed significantly elevated expression of pathways linked to inflammation and immune response compared to the control group. Previous research has suggested that innate immune dysregulation in SLE patients may elevate pro-inflammatory cytokines, such as IL-1B and IL-18, which are implicated in periodontitis pathogenesis42. Further investigation identified three diagnostic markers: TAGLN, MMP9, and TNFAIP6. Transgelin (TAGLN), an actin cross-linking protein prevalent in smooth muscle tissue, has been implicated in SLE development through its down-regulation43,44. TAGLN is also involved in angiogenesis, a process activated during tissue repair in PD, suggesting its potential role in the repair response of PD45. MMP9 is a matrix metalloproteinase (MMP) that has been shown to play a major role in myocardial infarction, heart failure, diabetes, hypertension and atherosclerosis46,47,48. Previous studies have linked increased MMP8 and MMP9 expression with periodontitis progression49,50. Liu et al.51demonstrated elevated MMP9 levels in gingival tissues of chronic periodontitis patients compared to healthy controls, regulated by extracellular matrix metalloproteinase inducer (EMMPRIN). MMP9 is also a sensitive marker for PD during orthodontic treatment, highlighting its diagnostic value52. Additionally, MMP9 is instrumental in immune complex regulation in systemic lupus erythematosus (SLE), suggesting its potential as an SLE biomarker53. Research indicates significant MMP9 overexpression in peripheral blood mononuclear cells of SLE patients versus healthy individuals54, and the combined effect of MMP9-C1562 T and MMP2-G1575 A alleles notably elevates SLE risk, underscoring MMP9’s significance in SLE55. TNFAIP6, also known as tumor necrosis factor α-stimulating gene 6 (TSG-6), exhibits anti-inflammatory properties56. Soyuan Um et al.57 found that TSG-6 secreted by human mesenchymal stem cells (hMSC) inhibits inflammatory responses via the p38 and ERK pathways in the mitogen-activated protein kinase cascade. Our study observed elevated TNFAIP6 expression in both PD and SLE, suggesting a potential protective response against inflammation, although further research is necessary to confirm this hypothesis. We subsequently developed disease risk prediction models for three diagnostic genes, evaluating their diagnostic efficacy with training and validation datasets. These models demonstrated satisfactory predictive performance, indicating that these biomarkers warrant further comprehensive investigation.
In a subsequent study, we employed a combination of four topological algorithms to identify four key genes from CGs: MMP9, IL1B, CXCL8, and FCGR3B. These genes demonstrated significantly elevated expression levels in the disease cohorts across the four datasets. IL1B, a well-known inflammatory cytokine, has been associated with variations that influence the susceptibility to PD and its progression58. Patients with chronic PD have been found to exhibit elevated serum levels of IL1B, leading to systemic effects59. IL1B has also been implicated in promoting T-cell Th17 differentiation, a process less prevalent in patients with SLE in the active phase compared to those in the moderate phase of the disease60. Additionally, studies have indicated that IL1B can enhance the expression of MMP, a collagenolytic enzyme that accelerates extracellular matrix degradation, ultimately leading to bone resorption and tissue destruction61. CXCL8, a prominent chemokine and potent angiogenic factor62, is commonly expressed in senescent cells and has the potential to induce endoplasmic reticulum stress and mitochondrial dysfunction. This, in turn, contributes to chronic inflammation, affecting peripheral cells and playing a role in age-related diseases63. Furthermore, research by Xin Chen et al.64suggests that CXCL8 may participate in inflammation-related pathways, such as the IL17 signaling pathway, thereby influencing the development of PD. FCGR3B is one of five genes encoding the Fcγ receptor, crucial for neutrophil recruitment to inflammatory sites and immune complex processing65. This gene is integral to inflammation and immune response. A study identified FCGR3B as a key gene linked to oxidative stress in PD66. Additionally, alterations in FCGR3B copy number predispose individuals to autoimmune responses, with reduced copy numbers associated with increased SLE risk67. Gene copy number indicates the gene’s presence in the genome, while gene expression refers to its transcription into mRNA and subsequent protein translation. Typically, increased gene copy number correlates with heightened gene expression. In this study, FCGR3B expression was elevated in SLE patients compared to healthy controls and a validation group. We hypothesize that increased FCGR3B expression may result from factors such as promoter activity, transcription factor binding, epigenetic modifications, mRNA stability, and translation efficiency. These genes are significantly enriched in immune-related pathways, notably IL-17 and NF-kappa B. Immune infiltration analysis underscores the association between key genes, such as MMP9, and immune cells. Investigating the connection between these pathways and specific immune cell types is crucial. Th17 cells primarily produce IL-17, which initiates and amplifies inflammation by releasing cytokines like IL-17 during immune responses. Activation of the IL-17 signaling pathway also stimulates macrophages to secrete tumor necrosis factor-alpha (TNF-a) and interleukin-6 (IL-6), further enhancing inflammation. Additionally, the IL-17 pathway interacts with immune cells such as neutrophils and dendritic cells to collectively modulate the immune response and inflammation68,69. When immune cells, including macrophages, T cells, and B cells, encounter inflammation, the NF-kappa B signaling pathway is activated, regulating gene expression (e.g., MMP9) and promoting the release of inflammatory mediators like chemokines. This attracts more immune cells to the inflammation site, intensifying the response. Moreover, NF-kappa B is involved in the negative feedback regulation of inflammation, preventing excessive reactions and maintaining immune homeostasis70,71. Overall, these four key genes are intricately linked to PD and SLE.
We selected the top three drugs—hesperidin, doxycycline, and cytochalasin D—from the DSigDB library in Enrichr for potential treatment of PD and SLE comorbidities, validating their binding efficacy through molecular docking. Hesperidin, a bioflavonoid abundant in citrus fruits, is absorbed orally via intestinal flora hydrolysis to glycosides, primarily distributing in the liver and intestines, with excretion through urine and bile. Its bioavailability is approximately 20%. Despite this, hesperidin is cost-effective to produce and can be enhanced with probiotics to improve efficacy in human studies72,73. Research indicates hesperidin reduces reactive oxygen species production and mitigates inflammation in PD, while also protecting epithelial barrier function74. Although less studied for SLE treatment, hesperidin has demonstrated antioxidant, photoprotective, anti-inflammatory, anticancer, and antimicrobial benefits in both healthy and diseased skin75,76. Doxycycline, a tetracycline antibiotic, is widely used in dermatology for its antimicrobial and anti-inflammatory effects77. Its bioavailability ranges from 73 to 95%, and it is fat-soluble, allowing for rapid absorption and extensive distribution in tissues such as gingival sulcus fluid, synovial fluid, and inflamed areas after oral administration78. Doxycycline is eliminated through bile secretion and glomerular filtration. It effectively inhibits MMP9 activity during traumatic injuries, like thermal injuries79. MMPs are extracellular matrix-degrading enzymes crucial for periodontal tissue remodeling80. Increasing evidence supports doxycycline’s efficacy in nonsurgical periodontal therapy81. Although its use in treating SLE is still being explored, doxycycline’s MMP9 inhibition indicates potential for SLE treatment. Cytochalasin D, a cytoskeleton inhibitor, effectively prevents rapid actin polymerization and promotes actin filament depolymerization in platelets82. Hiroaki Inaba et al. demonstrated its efficacy against Porphyromonas gulae, an animal periodontal pathogen, by inhibiting its adhesion and invasion of gingival epithelial cells83. Despite its potential, Cytochalasin D’s toxicity raises concerns about its pharmacokinetics and bioavailability. However, Huang et al.84showed that liposome-encapsulated Cytochalasin D can be dissolved in water for intravenous use, enhancing bioavailability and reducing side effects. Additionally, Cytochalasin D may inhibit NF-κB expression, modulating inflammation and cytokine expression85. Future research could focus on targeting lipid complexes or polymeric nanoparticles to inflammation sites, increasing local concentrations and reducing systemic toxicity. The molecular docking results demonstrated that the binding energies for all nine drug pairs were below−5 kcal/mol, indicating satisfactory binding efficacy. These findings suggest that the three drugs in question may have therapeutic potential for treating PD and SLE. While this study provides new insights into the treatment of both diseases, further clinical validation is necessary to confirm the efficacy of certain drugs.
Consensus cluster analysis of CGs identified two distinct subtypes in both PD and SLE, underscoring the pivotal role of CGs in the pathogenesis of these diseases. Immune infiltration analysis further emphasizes the significance of CGs in immune disorders. While this study elucidates the comorbidity between PD and SLE, it has limitations. The transcriptome data utilized, sourced from online databases, may lack comprehensiveness, particularly regarding regional and gender variations. Additionally, the predictive models and potential therapeutic drugs proposed require further clinical validation. Future research should focus on pathways that confirm the expression and impact of diagnostic genes in these diseases, and we encourage broader participation in this research.
Conclusions
Our research seeks to elucidate the genetic interactions between periodontitis (PD) and systemic lupus erythematosus (SLE) and to identify crosstalk genes (CGs) relevant to both conditions. We identified 32 CGs significantly associated with immune cells through immune infiltration analysis. Among these, TAGLN, MMP9, and TNFAIP6 emerged as potential diagnostic markers, facilitating the development of predictive disease models. Furthermore, MMP9, IL1B, CXCL8, and FCGR3B were pinpointed as pivotal crosstalk genes linking PD and SLE. Our findings also suggest potential therapeutic agents and disease classification strategies, offering new avenues for future PD and SLE research.
Data availability
The datasets generated and/or analysed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/repository. The datasets used and analyzed during the current study are available from the corresponding author on reasonable request. The accession number for GEO are GSE10334, GSE16134, GSE50772, and GSE81622.
Abbreviations
- PD:
-
Periodontitis
- SLE:
-
Systemic lupus erythematosus
- GEO:
-
Gene Expression Omnibus
- CGs:
-
Crosstalk genes
- DEGs:
-
Differentially expressed genes
- WGCNA:
-
Weighted gene co-expression network analysis
- ROC:
-
Receiver Operating Characteristic curve
- AUC:
-
Area under the curve
- DCA:
-
Decision curve analysis
- PPI:
-
Protein-protein interaction
- GSEA:
-
Single-sample Gene Set Enrichment Analysis
- DSigDB:
-
the Drug Signature Database
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- TLR:
-
Toll-like receptors
- TAGLN:
-
Transgelatin
- MMP:
-
Matrix metalloproteinase
- TSG − 6:
-
Tumor necrosis factor α-stimulating gene 6
- hMSC:
-
Human mesenchymal stem cells
References
Eke, P. I. et al. (2016). Periodontitis prevalence in adults≥ 65 years of age, in the USA. Periodontology 2000, 72(1), 76-95.
KassebaumN J et al. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J. Dent. Res. 93 (11), 1045–1053 (2014).
Kinane, D. F. et al. Jun. Periodontal diseases. Nature reviews. Disease primers vol. 3 17038. 22 (2017).
Van Dyke, T. E., Jan, A. & van Winkelhoff Infection and inflammatory mechanisms. J. Clin. Periodontol. 40, S1–7 (2013).
Yen, E. Y. & Singh, R. R. Brief report: lupus-an unrecognized leading cause of death in young females: a population-based study using nationwide death certificates, 2000–2015. Arthritis Rheumatol. 70, 1251–1255 (2018).
Yen, E. Y. et al. 46-Year trends in systemic lupus erythematosus mortality in the united States, 1968 to 2013: A nationwide Population-Based study. Annals Intern. Med. Vol. 167 (11), 777–785 (2017).
Papazoglou, N. et al. Atherosclerotic plaque progression and incident cardiovascular events in a 10-year prospective study of patients with systemic lupus erythematosus: the impact of persistent cardiovascular risk factor target attainment and sustained DORIS remission. Arthritis & rheumatology (Hoboken, N.J.), https://doi.org/10.1002/art.43097. (2024). 25 Dec.
Pessoa, L. et al. Host-microbial interactions in systemic lupus erythematosus and periodontitis. Front. Immunol. 10, 2602 (2019).
Corrêa, D. et al. Mar. Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome vol. 5,1 34. 20 (2017).
Dewhirst, F. E. et al. The human oral Microbiome. J. Bacteriol. Vol. 192, 5002–5017 (2010).
Griffen, A. L. et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6, 1176–1185 (2012).
Zhang, R. et al. Jun. Immune-mediated inflammatory diseases and periodontal disease: a bidirectional two-sample mendelian randomization study. BMC immunology vol. 25,1 39. 28 (2024).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. Vol. 41, D991–D995 (2013). Database issue.
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. Vol. 28 (1), 27–30 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Science: Publication Protein Soc. Vol. 28 (11), 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Sherman, Brad, T. et al. DAVID ortholog: an integrative tool to enhance functional analysis through orthologs. Bioinf. (Oxford England) Vol. 40, 10 (2024).
Xie, Y. et al. Bioinformatic analysis of underlying mechanisms of Kawasaki disease via weighted gene correlation network analysis (WGCNA) and the least absolute shrinkage and selection operator method (LASSO) regression model. BMC Pediatr. 23, 90 (2023).
Engebretsen, S. & Bohlin, J. Aug. Statistical predictions with glmnet. Clinical epigenetics vol. 11,1 123. 23 (2019).
Shariat, S. F. et al. Can nomograms be superior to other prediction tools? BJU international vol. 103,4 : 492-5; discussion 495-7. (2009).
Robin, X. et al. Mar. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC bioinformatics vol. 12 77. 17 (2011).
Vickers, A. J. & Holland, F. Decision curve analysis to evaluate the clinical benefit of prediction models. Spine Journal: Official J. North. Am. Spine Soc. Vol. 21 (10), 1643–1648 (2021).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. Vol. 51,D1, D638–D646 (2023).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Yi, M. et al. Jun. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Scientific reports vol. 10,1 10258. 24 (2020).
Hänzelmann, S. et al. Jan. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics vol. 14 7. 16 (2013).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. Bioinf. 31, 3069–3071 (2015).
Pinzi, L. & Rastelli, G. Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci. 20, 4331 (2019).
Kim, S. et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. Vol. 49,D1, D1388–D1395 (2021).
Burley, S. K. et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic acids research vol. 51,D1 : D488–D508. (2023).
Mooers, Blaine, H. M. Shortcuts for faster image creation in PyMOL. Protein Science: Publication Protein Soc. Vol. 29 (1), 268–276 (2020).
Morris, Garrett, M. et al. AutoDock4 and AutoDockTools4: automated Docking with selective receptor flexibility. J. Comput. Chem. Vol. 30 (16), 2785–2791 (2009).
Wilkerson, M. D. & Neil Hayes, D. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinf. (Oxford England) Vol. 26 (12), 1572–1573 (2010).
Hussain, S. et al. Periodontitis and systemic lupus erythematosus: A systematic review and meta-analysis. J. Periodontal Res. Vol. 57 (1), 1–10 (2022).
Bolstad, A. I. et al. Periodontitis in patients with systemic lupus erythematosus: A nationwide study of 1,990 patients. J. Periodontology Vol. 93 (3), 364–372 (2022).
Marques, C. P. et al. Salivary levels of inflammatory cytokines and their association to periodontal disease in systemic lupus erythematosus patients. A case-control study. Cytokine 85, 165–170 (2016).
Suárez, Lina, J. et al. Dec. Oral Dysbiosis and Autoimmunity: From Local Periodontal Responses to an Imbalanced Systemic Immunity. A Review. Frontiers in immunology vol. 11 591255. 8 (2020).
Marques, C. P. C. et al. Possible evidence of systemic lupus erythematosus and periodontal disease association mediated by toll-like receptors 2 and 4. Clin. Exp. Immunol. 183, 187–192 (2016).
Miranda, Leticia, A. et al. Increased interleukin-18 in patients with juvenile idiopathic arthritis and early attachment loss. J. Periodontology Vol. 76 (1), 75–82 (2005).
Dvorakova, M. et al. Transgelins, cytoskeletal proteins implicated in different aspects of cancer development. Expert Rev. Proteom. Vol. 11 (2), 149–165 (2014).
Tang, D. et al. Jul. Integrated analysis of mRNA, microRNA and protein in systemic lupus erythematosus-specific induced pluripotent stem cells from urine. BMC genomics vol. 17 488. 11 (2016).
Tsuji-Tamura, K. et al. The canonical smooth muscle cell marker TAGLN is present in endothelial cells and is involved in angiogenesis. J. Cell. Sci. Vol. 134 (15), jcs254920 (2021).
Ertl, G. & Frantz, S. Healing after myocardial infarction. Cardiovasc. Res. Vol. 66 (1), 22–32 (2005).
Jiang, B. et al. Salvianolic acid A, a novel matrix metalloproteinase-9 inhibitor, prevents cardiac remodeling in spontaneously hypertensive rats. PloS One Vol. 8 (3), e59621 (2013).
Kim, J. Y. et al. The stimulation of CD147 induces MMP-9 expression through ERK and NF-kappaB in macrophages: implication for atherosclerosis. Immune Netw. 9, 90–97 (2009).
Kinney, J. S. et al. Saliva/pathogen biomarker signatures and periodontal disease progression. J. Dent. Res. Vol. 90 (6), 752–758 (2011).
Nagarajan, R. et al. Biologic modelling of periodontal disease progression. J. Clin. Periodontology Vol. 46 (2), 160–169 (2019).
Liu, X. et al. Interaction between the Wnt/β-catenin signaling pathway and the EMMPRIN/MMP-2, 9 route in periodontitis. J. Periodontal Res. Vol. 53 (5), 842–852 (2018).
Luchian, I. et al. Feb. The Role of Matrix Metalloproteinases (MMP-8, MMP-9, MMP-13) in Periodontal and Peri-Implant Pathological Processes. International journal of molecular sciences vol. 23,3 1806. 4 (2022).
Ugarte-Berzal, E. et al. Mar. MMP-9/Gelatinase B Degrades Immune Complexes in Systemic Lupus Erythematosus. Frontiers in immunology vol. 10 538. 22 (2019).
Matache, C. et al. Matrix metalloproteinase-9 and its natural inhibitor TIMP-1 expressed or secreted by peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J. Autoimmun. Vol. 20 (4), 323–331 (2003).
Bahrehmand, F. et al. Matrix metalloproteinase 9 polymorphisms and systemic lupus erythematosus: correlation with systemic inflammatory markers and oxidative stress. Lupus 24 (6), 597–605 (2015).
Li, H. et al. Jun. Gut Subdoligranulum variabile ameliorates rheumatoid arthritis by promoting TSG-6 synthesis from joint cells. Frontiers in immunology vol. 15 1418717. 10 (2024).
Um, S. et al. TSG-6 secreted by mesenchymal stem cells suppresses immune reactions influenced by BMP-2 through p38 and MEK mitogen-activated protein kinase pathway. Cell. Tissue Res. Vol. 368 (3), 551–561 (2017).
Walther, K. A. et al. The role of polymorphisms at the Interleukin-1, Interleukin-4, GATA-3 and Cyclooxygenase-2 genes in Non-Surgical periodontal therapy. Int. J. Mol. Sci. 23, 7266 (2022).
De Alencar, J. et al. Jan. Influence of inflammasome NLRP3, and IL1B and IL2 gene polymorphisms in periodontitis susceptibility. PloS one vol. 15,1 e0227905. 24 (2020).
Tominaga, K. et al. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int. Immunol. Vol. 12 (2), 151–160 (2000).
Galozzi, P. et al. The revisited role of interleukin-1 alpha and beta in autoimmune and inflammatory disorders and in comorbidities. Autoimmun. Reviews Vol. 20 (4), 102785 (2021).
Heidemann, J. et al. Angiogenic effects of Interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J. Biol. Chem. 278, 8508–8515 (2003).
Di Micco, R. et al. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Reviews Mol. Cell. Biology Vol. 22 (2), 75–95 (2021).
Chen et al. CXCL8, MMP12, and MMP13 are common biomarkers of periodontitis and oral squamous cell carcinoma. Oral Dis. Vol. 30 (2), 390–407 (2024).
Lee, Young Ho et al. Association between FCGR3B copy number variations and susceptibility to autoimmune diseases: a meta-analysis. Inflammation research: official journal of the European Histamine Research Society… et al.] vol. 64,12 (2015): 983 – 91.
Zhang, Z. et al. Identification of Key Genes and Pathways Associated with Oxidative Stress in Periodontitis. Oxidative medicine and cellular longevity vol. 13 Sep. 2022. (2022) 9728172.
Mueller, M. et al. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am. J. Hum. Genet. Vol. 92 (1), 28–40 (2013).
Li, X. et al. IL-17 receptor-based signaling and implications for disease. Nat. Immunol. Vol. 20 (12), 1594–1602 (2019).
McGeachy, Mandy, J. et al. The IL-17 Family of Cytokines in Health and Disease. Immunity vol. 50,4 : 892–906. (2019).
Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspect. Biology Vol. 1 (6), a001651 (2009).
Guo, Q. et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct. Target. Ther. 9, 53 (2024).
Wdowiak, K. et al. Jun. Bioavailability of Hesperidin and Its Aglycone Hesperetin-Compounds Found in Citrus Fruits as a Parameter Conditioning the Pro-Health Potential (Neuroprotective and Antidiabetic Activity)-Mini-Review. Nutrients vol. 14,13 2647. 26 (2022).
Song, B. et al. Comprehensive review of Hesperetin: advancements in pharmacokinetics, Pharmacological effects, and novel formulations. Fitoterapia 179, 106206 (2024).
Chaves, Jaisa, O. et al. Integration of pressurized liquid extraction and in-line solid-phase extraction to simultaneously extract and concentrate phenolic compounds from lemon peel (Citrus limon L.). Food research international (Ottawa, Ont.) vol. 157 : 111252. (2022).
Abou Baker, Doha, H. et al. Jul. Biochemical and pharmacological prospects of Citrus sinensis peel. Heliyon vol. 8,8 e09979. 21 (2022).
Maquera-Huacho, P. et al. Effect of hesperidin on barrier function and reactive oxygen species production in an oral epithelial cell model, and on secretion of macrophage-derived inflammatory mediators during porphyromonas gingivalis infection. Int. J. Mol. Sci. 24, 10389 (2023).
Monk, E. et al. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol. Res. Vol. 63 (2), 130–145 (2011).
Thompson, E. J. et al. Population pharmacokinetics of Doxycycline in children. Antimicrob. Agents Chemother. Vol. 63 (12), e01508–e01519 (2019).
Stagg, H. et al. Doxycycline attenuates burn-induced microvascular hyperpermeability. J. Trauma. Acute Care Surg. Vol. 75 (6), 1040–1046 (2013). discussion 1046.
Sternlicht, M. D. & Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17, 463–516 (2001).
Zhang, Z. et al. Jan. Systemic doxycycline as an adjunct to nonsurgical periodontal therapy in diabetic patients with periodontitis: a systematic review and meta-analysis. Frontiers in physiology vol. 15 1479152. 22 (2025).
Shoji, K. et al. Cytochalasin D acts as an inhibitor of the actin-cofilin interaction. Biochem. Biophys. Res. Commun. Vol. 424 (1), 52–57 (2012).
Inaba, H. et al. Mar. Adhesion and invasion of gingival epithelial cells by Porphyromonas gulae. PloS one vol. 14,3 e0213309. 14 (2019).
Huang, F. et al. The antitumour activities induced by pegylated liposomal cytochalasin D in murine models. Eur. J. cancer (Oxford Engl. 48 (2012), 2260–2269 (1990).
Hassanein, Emad, H. M. et al. The role of inflammation in cadmium nephrotoxicity: NF-κB comes into view. Life Sci. 308, 120971 (2022).
Acknowledgements
Not applicable.
Funding
This work was supported by the Xinjiang Uygur Autonomous Region Natural Science Foundation Upper-level Program (Grant No. 2023D01 C116).
Author information
Authors and Affiliations
Contributions
L.Q conceptualized the idea and was responsible for the design of the study and the accuracy of the data analysis. JJ.W and QG.D performed the statistical analyses and wrote and revised the manuscript. All authors have read and approved the submitted version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Deng, Q. & Qi, L. Integrated bioinformatics, machine learning, and molecular docking reveal crosstalk genes and potential drugs between periodontitis and systemic lupus erythematosus. Sci Rep 15, 15787 (2025). https://doi.org/10.1038/s41598-025-00620-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00620-3
