
Abstract
Lung cancer remains a significant global health challenge, with advanced stages often limiting surgical options and necessitating systemic therapies, such as radiotherapy. Resistance to radiotherapy frequently undermines the treatment efficacy. This study explored the role of miR-7-5p in modulating the expression and radiosensitivity of plakophilin-2 (PKP2) in non-small cell lung cancer (NSCLC). Using clonogenic assays, CCK-8 assays, immunofluorescence staining, western blotting, and reporter gene assays, we assessed the effects of miR-7-5p overexpression and inhibition on A549 NSCLC cells. The results show that miR-7-5p overexpression enhanced radiosensitivity by increasing DNA damage (evidenced by higher γ-H2AX foci) and inhibiting non-homologous end joining (NHEJ) repair. Bioinformatic and experimental validation identified PKP2 as a direct target of miR-7-5p. PKP2 overexpression mitigated the radiosensitizing effects of miR-7-5p, confirming the miR-7-5p/PKP2 axis’s role in regulating radiosensitivity. This study highlights the potential of targeting the miR-7-5p/PKP2 pathway to overcome radiotherapy resistance in NSCLC and offers a promising therapeutic approach to enhance treatment outcomes.
Similar content being viewed by others


Introduction
Lung cancer is a significant and persistent threat to human health. In 2018, the estimated number of lung cancer-related deaths was 1,761,000, accounting for 18.4% of all cancer-related deaths. Additionally, in 2020, 229,000 new cases of lung cancer were diagnosed in the United States, representing 12.7% of all cancer diagnoses1. Non-small cell lung cancer (NSCLC) is the most common histological type of lung cancer, accounting for more than 80% of all lung cancer cases. Unfortunately, many patients are diagnosed with lung cancer at an advanced stage and thus lose the opportunity for surgical intervention. Consequently, systemic therapy has become the primary treatment for patients with advanced NSCLC. Radiotherapy, a key component of systemic therapy, often alleviates symptoms and mitigates tumor emergencies2. Radiotherapy is the gold-standard treatment for patients with stage III lung cancer. However, tumors are prone to developing resistance to radiotherapy during the treatment process, resulting in unsatisfactory treatment outcomes for many patients3.
Plakophilin-2 (PKP2) belongs to the plakophilin family and is a member of the Armadillo-like protein subfamily4. The literature reports that PKP2 plays an important role in the development of malignant tumors, including ovarian cancer and colon cancer5,6. Recent studies have shown that PKP2 is highly expressed in lung cancer tissues and promotes the occurrence and development of lung cancer7,8. In a previous study, we demonstrated the involvement of PKP2 in lung carcinogenesis and progression and found that PKP2 confers resistance to radiation therapy by promoting LIG4-dependent non-homologous end joining (NHEJ), a process associated with DNA damage repair9. Thus, the inhibition of PKP2 expression reduces radiotherapy resistance in patients. Many microRNAs (miRNAs) have been reported to be involved in the regulation of radiotherapy resistance in tumors10. miRNAs are non-coding RNAs (∼20–24 nucleotides) derived from longer stem-loop structures that function by binding to and inhibiting mRNAs11. Among the biomarkers valuable for assessing the effectiveness of radiotherapy in lung cancer, miRNAs appear to be the most appropriate12. For example, miRNA-218-5p increases the sensitivity of lung cancer cells to radiotherapy by inhibiting the activity of the anti-protein kinase, DNA-activated, catalytic polypeptide (PRKDC)13. Wei et al. found that miR-219a-5p enhances the sensitivity of non-small cell lung cancer cells to radiotherapy by targeting CD16414.
Defining the miRNAs that regulate PKP2 expression may enhance our understanding of the pathogenesis of radiotherapy resistance in lung cancer and provide new therapeutic approaches for these patients. This study aimed to investigate whether miR-7-5p can regulate PKP2 expression through 3′UTR binding and to explore the mechanism underlying miR-7-5p’s regulation of radiosensitivity in non-small cell lung cancer.
Materials and methods
Cell line
The A549 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Authentication was performed using short tandem repeat (STR) profiling, and the cells were tested for mycoplasma contamination. The cells were cultured for less than two months in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc.) and maintained at 37 °C in a humidified incubator with 5% CO2.
Antibodies, plasmid, and MiRNA
Human anti-PKP2 (HPA014314) was purchased from Sigma-Aldrich (St. Louis, MO, USA), and phospho-γH2AX (cat. no. 9718 S) was obtained from Cell Signaling Technology (Danvers, MA, USA). The miR-7-5p mimic, inhibitor, and negative control (NC) inhibitor were synthesized by RiboBio Co., Ltd. (Guangzhou, China). Transfection was performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA). The complete coding sequence (CDS) of human PKP2 was cloned into the pSin-puro vector, and the recombinant plasmid was verified by DNA sequencing (Ruiboxingke Biotech Co., Ltd., Beijing, China).
Irradiation treatment
Cells were irradiated using a Varian Trilogy linear accelerator (Varian Medical Systems, Palo Alto, CA, USA) at doses ranging from 0 to 6 Gy at a dose rate of 250 MU/min.
Luciferase assay
The wild-type (WT) PKP2 3′UTR sequence contained the predicted miR-7-5p binding site (5′-AGUCUUCC-3′, position 161–168). A mutant (MUT) version of PKP2 3′UTR was generated by site-directed mutagenesis, in which the seed sequence was altered (5′-AGUCUUCC-3′ → 5′-UCAUGAGG-3′), disrupting complementarity with miR-7-5p. HEK293 cells were co-transfected with 100 nM miR-7-5p inhibitor, miR-7-5p mimic, or miR-NC, along with the pGL3 reporter vector containing the PKP2 3′UTR (wild-type or mutant miR-7-5p binding site) and the control Renilla luciferase pRL-TK vector in 24-well plates. Firefly luciferase activity was measured 24 h post-transfection using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). Firefly luciferase activity was normalized to Renilla luciferase for relative reporter activity. Each experiment was performed in three independent trials, with each trial conducted in triplicate to ensure statistical robustness and reproducibility.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from A549 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA concentration and purity were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized from 1 µg of total RNA using the PrimeScript RT reagent Kit (Takara, Dalian, China). qRT-PCR was performed using SYBR Green Master Mix (Roche, Basel, Switzerland) on a LightCycler 480 II system (Roche). The relative expression levels of PKP2 mRNA were normalized to GAPDH and calculated using the 2 − ΔΔCt method. Primer sequences for PKP2 were as follows: forward, 5′-AGGACCTGGAGGTGCTGAAT-3′; reverse, 5′-TGGTGGTGGTGGTGGTGGTA-3′. Primer sequences for GAPDH were as follows: forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′; reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′.
Cell viability assays
Cells were seeded into 96-well plates at a density of 5000 cells per well and either subjected to irradiation treatment or left untreated. After treatment, 10 µL of CCK-8 reagent was added to each well, followed by incubation for 1.5 h. Optical density was measured at 450 nm using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA).
Clonogenic survival assay
Cells were seeded into six-well plates at a density of 5 × 103 cells per well and irradiated with the specified doses. Each experimental group consisted of three replicates. After 15 days of incubation at 37 °C, cells were washed with PBS, fixed in methanol for 15 min, and stained with 0.1% crystal violet for 60 min. Colonies containing at least 50 cells were counted under a light microscope.
NHEJ reporter assays
A total of 1 × 106 EJ5-GFP cells were electroporated with 12 µg of the I-SceI plasmid and the designated plasmids using a Bio-Rad GenePulser Xcell™ (Bio-Rad Laboratories, Hercules, CA, USA) at 250 V and 950 µF. GFP expression was measured 48 h post-transfection using flow cytometry to assess the efficiency of NHEJ.
Western blotting
Cells were lysed in RIPA buffer (150 mM NaCl, 0.5% EDTA, 50 mM Tris, and 0.5% NP-40) on ice for 30 min and centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatant was collected, and protein concentrations were quantified using a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were separated via 10% SDS-PAGE, transferred onto PVDF membranes (Millipore, Billerica, MA, USA), and blocked with 5% non-fat milk for 1 h. Membranes were incubated with primary antibodies, followed by peroxidase-conjugated secondary antibodies, and detected using an ECL chemiluminescence system (Beyotime, Shanghai, China). The bands were visualized using radiographic film (Carestream Health, Inc., Rochester, NY, USA).
Immunofluorescence staining
Cells were cultured in glass-bottom dishes (NEST, China), fixed with 4% paraformaldehyde (Thermo Fisher Scientific, Waltham, MA, USA) for 15 min at room temperature, and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA). Cells were incubated with primary antibodies against phospho-γH2AX (1:250 dilution, Cell Signaling Technology, Danvers, MA, USA) and secondary fluorescence-conjugated antibodies, followed by staining with Hoechst 33,342 (Thermo Fisher Scientific). Fluorescence images were captured using an Olympus IX81 microscope (Olympus Corporation, Tokyo, Japan), and double-strand break (DSB) foci were quantified using AxioVision software (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
All experiments were performed in triplicate. Data are presented as the mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). Statistical significance between groups was determined using a two-tailed Student’s t-test in SPSS version 18.0 (IBM Analytics, USA). A p-value < 0.05 was considered statistically significant.
Software and tools
Target gene prediction was performed using TargetScan 8.0 (http://www.targetscan.org/), miRBase (https://www.mirbase.org/), and miRDB (http://mirdb.org/). Statistical analysis and data visualization were conducted using GraphPad Prism (v9.5.1, https://www.graphpad.com/) and SPSS (v18.0, https://www.ibm.com/products/spss-statistics). γH2AX foci quantification was performed using ImageJ (Fiji, v1.53t, https://imagej.net/software/fiji/), and flow cytometry data were analyzed with FlowJo (v10.8.1, https://www.flowjo.com/).
Results
Result 1: miR-7-5p promotes sensitization of A549 cells to radiotherapy
To investigate the role of miR-7-5p in the radiosensitization of A549 lung cancer cells, A549 cells were treated with different doses of RT. Clone formation was significantly reduced in miR-7-5p overexpressing A549 cells and increased in the miR-7-5p knockdown group compared with that in the control group (Fig. 1A). The surviving fraction of miR-7-5p overexpressing A549 cells was significantly reduced following irradiation. Conversely, the viability of the miR-7-5p knockdown group increased (Fig. 1B). The CCK-8 assay (Fig. 1C) demonstrated decreased viability of miR-7-5p overexpressing cells and increased viability of the miR-7-5p knockdown group after irradiation compared to control cells (Fig. 1D). These findings suggested that miR-7-5p enhances the sensitivity of NSCLC cells to radiation.

miR-7-5p promotes A549 cell sensitization to radiotherapy. (A) Representative images showing clone formation in A549 cells after exposure to 0, 2, 4, and 6 Gy IR. (B) Clone formation survival of A549 cells after exposure to 0, 2, 4, and 6 Gy IR. Data are shown as mean ± SD from a 2-way ANOVA. (C,D) A549 cells were treated with 0–6 Gy of γ-irradiation for 1 to 5 days, and cell viability was measured daily using the CCK8 assay (relative percentage change compared to control). Data are shown as mean ± SD from a 2-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
Result 2: miR-7-5p enhances radiotherapy sensitivity by inhibiting NHEJ repair of DNA
Ionizing radiation induces cellular damage, particularly DNA damage. H2AX, known as H2A histone family member X (H2A), is a variant of chromosomal histone H2A. The level of γ-H2AX, produced by phosphorylation of H2AX, clearly reflects the degree of DNA damage and repair15. Immunofluorescence staining revealed that overexpression of miR-7-5p in A549 cells significantly increased the number of γ-H2AX foci following ionizing radiation (IR) (Fig. 2A,B), while inhibition of miR-7-5p significantly reduced the number of γ-H2AX foci (Fig. 2C,D). Additionally, the protein blotting results showed that γ-H2AX protein levels were elevated after the overexpression of miR-7-5p (Fig. 2E) and decreased after its inhibition (Fig. 2F). These results suggested that miR-7-5p may regulate DNA damage repair in lung cancer cells. NHEJ is a major pathway for DNA damage repair, in which the ends are joined without extensive homology. Recent studies have shown that miRNAs can control tumor radiosensitivity by affecting the DNA damage repair pathways16. We investigated the effect of miR-7-5p on radioresistance in lung cancer cells by evaluating its effect on NHEJ-mediated double-strand break (DSB) repair. Overexpression of miR-7-5p decreased the frequency of NHEJ-mediated repair in lung cancer cells (Fig. 2G), whereas miR-7-5p inhibition increased DSB repair (Fig. 2H). We used an EJ5-GFP-based chromosomal reporter gene to measure NHEJ (Fig. 2I).

miR-7-5p enhances radiotherapy sensitivity by inhibiting non-homologous end joining (NHEJ) repair of DNA. (A,C) A549 cells were collected for IF staining of γ-H2AX after 0 and 6 Gy IR exposure. Overexpression of miR-7-5p increased the number of γ-H2AX foci, while inhibition of miR-7-5p decreased γ-H2AX foci. Cell nuclei were stained with Hoechst33342 (blue) and γ-H2AX foci were stained green. Scale bar = 20 μm. (B,D) Quantitative analysis of DNA damage foci. Data are shown as mean ± SD from a Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001. (E,F) γ-H2AX protein levels in A549 cells after 6 Gy IR as detected by immunoblotting. Actin was used as a loading control. Full-length blots are shown in Supplementary Fig. S1. (G–I) Cells were analyzed and quantified by flow cytometry after co-transfection of I-SceI and PKP2 or control vector plasmids using electroporation, followed by 72 h of culturing of EJ5-GFP cells. Overexpression of miR-7-5p decreased NHEJ activity (G), while inhibition of miR-7-5p increased NHEJ activity (H). Chromosomal reporter gene pattern of J5-GFP (I). Data are shown as mean ± SEM from three independent experiments.
Result 3: miR-7-5p directly targets PKP2 to suppress radioresistance in NSCLC
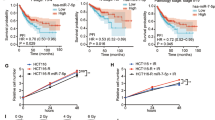
PKP2 is highly expressed in lung cancer tissues, and its elevated expression is associated with reduced survival. To further investigate the role of PKP2 in lung cancer, we analyzed its expression and clinical relevance using three tumor databases: GEPIA, UALCAN, and Kaplan–Meier Plotter. The results indicated that PKP2 expression was significantly higher in NSCLC tumor tissues than in normal lung tissues. Furthermore, patients with high PKP2 expression had lower survival rates, with a significant reduction in survival among those who underwent radiotherapy (Fig. 3A). Consistent with these findings, our previous study demonstrated that PKP2 plays a critical role in radiotherapy resistance in lung cancer9.

miR-7-5p directly targets PKP2 to suppress radioresistance in NSCLC. (A) PKP2 is highly expressed in lung cancer tissues, and patients with high PKP2 expression have lower survival rates, with or without radiotherapy. (B) Bioinformatic prediction of the binding sites between PKP2 and miR-7-5p. (C) Luciferase reporter gene assay showing that PKP2 is a target of miR-7-5p. Data are shown as mean ± SEM from three independent trials, each done in triplicate. (D) PKP2 expression in A549 cells was detected by overexpression and inhibition of miR-7-5p. Full-length blots are shown in Supplementary Figure S2 *P < 0.05, **P < 0.01, ***P < 0.001. (E) qRT-PCR analysis showing the effect of miR-7-5p overexpression and inhibition on endogenous PKP2 mRNA levels in A549 cells. GAPDH was used as an internal control. Data are shown as mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
To identify potential targets of miR-7-5p, we performed bioinformatics analysis using multiple databases, including TargetScan 8.0, miRbase, and miRDB. PKP2 was consistently identified as a target gene of miR-7-5p across all databases, underscoring its potential biological relevance. The interaction between miR-7-5p and PKP2 was supported by strong miRNA-target binding affinity, as predicted by TargetScan.
The predicted consequential pairing of miR-7-5p with the target region of PKP2 is illustrated in Fig. 3B. The seed sequence of miR-7-5p (positions 2–8 at the 5′ end) is 3′-CAGAAGG-5′, which aligns perfectly with the complementary sequence 5′-GUCUUCC-3′ in the wild-type PKP2 3′UTR (positions 161–167). This region is critical for miRNA binding and is highlighted with a bracket. The binding site is classified as an 8mer, with a context score of − 0.27 (96th percentile), a weighted context score of − 0.27, and a conserved branch length of 2.378. The PCT (probability of conserved targeting) is < 0.1, and the predicted relative KD (binding affinity) is − 5.445. These parameters indicate a strong and highly conserved interaction between miR-7-5p and PKP2, further supporting the biological relevance of this target.
To experimentally validate the interaction between miR-7-5p and PKP2, we performed a dual-luciferase reporter gene assay. The results showed that miR-7-5p significantly inhibited the luciferase activity of the PKP2 wild-type (WT) 3′UTR construct, whereas no inhibition was observed for the PKP2 mutant (MUT) 3′UTR (Fig. 3C). To further confirm the interaction between miR-7-5p and PKP2, we examined the effect of miR-7-5p on PKP2 protein expression in A549 cells. Western blot analysis revealed that overexpression of miR-7-5p via transfection with an miR-7-5p mimic significantly reduced PKP2 expression levels compared to the control. Conversely, inhibition of miR-7-5p expression via transfection with an miR-7-5p inhibitor significantly increased PKP2 expression levels (Fig. 3D). To further investigate the mechanism by which miR-7-5p regulates PKP2, we examined the effect of miR-7-5p overexpression on the endogenous PKP2 mRNA transcript levels in A549 cells. qRT-PCR analysis revealed that overexpression of miR-7-5p significantly reduced the levels of endogenous PKP2 mRNA compared to the control (Fig. 3E). Conversely, inhibition of miR-7-5p expression via transfection with an miR-7-5p inhibitor significantly increased PKP2 mRNA levels (Fig. 3E). These results indicate that miR-7-5p regulates PKP2 through transcriptional degradation rather than translational repression alone. Collectively, these findings demonstrate that PKP2 is a direct and functionally relevant target of miR-7-5p.
Result 4: miR-7-5p/PKP2 axis regulates radiosensitivity via DNA damage and NHEJ repair
To investigate whether the miR-7-5p/PKP2 axis regulates NSCLC radiotherapy sensitivity, A549 cells were transfected with an miR-7-5p mimic followed by PKP2 overexpression and then subjected to 6 Gy radiotherapy. The miR-7-5p mimic reduced clone formation in A549 cells after radiotherapy, whereas PKP2 overexpression reversed this effect (Fig. 4A,B). PKP2 overexpression inhibited miR-7-5p mimic-induced apoptosis (Fig. 4C). Furthermore, immunofluorescence staining showed that exogenous elevation of PKP2 rendered miR-7-5p overexpressing cells resistant to IR and reduced γ-H2AX foci formation in vitro (Fig. 4D,E). PKP2 upregulation reversed, at least partly, the miR-7-5p-induced reduction in NHEJ activity (Fig. 4F,G). These results suggested that the miR-7-5p/PKP2 axis modulates the radiosensitivity of NSCLC cells.

miR-7-5p/PKP2 axis regulates radiosensitivity via DNA damage and NHEJ repair. (A,B) Clonal survival of A549 cells after 6 Gy IR. Data are shown as mean ± SD from a Student’s t-test. (C) Cell viability after 6 Gy IR. Data are shown as mean ± SEM from three independent experiments. (D,E) IF staining of H2AX foci in the indicated cells after exposure to 6 Gy IR. Scale bar = 20 μm. Data are shown as mean ± SD from a Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001. (F,G) Detection of NHEJ activity in EJ5-GFP cells. Cells were transfected with I-SceI and miR-NC, miR-7-5p mimic, or wild-type PKP2 plasmids by electroporation, then analyzed by flow cytometry (F) and quantified (G). Data are shown as mean ± SEM from three independent trials.
Discussion
Lung cancer is a common malignant tumor, characterized by high morbidity and mortality rates. Clinically, it is managed with a combination of surgery, radiotherapy, and chemotherapy17. Radiotherapy is a crucial modality in the treatment of lung cancer. High-dose radiation can directly or indirectly kill cancer cells and inhibit their growth. Radiotherapy can enhance the effectiveness of surgery by reducing tumor size preoperatively or by eliminating residual cancer cells postoperatively. Despite advances in radiotherapy technology, some patients with lung cancer exhibit resistance to radiotherapy, leading to poor treatment outcomes and even disease progression18.
Therefore, understanding the mechanisms of radiotherapy resistance in lung cancer and developing strategies to overcome it are of great significance. Although the mechanisms of radioresistance in cancer cells remain unclear, they likely involve genetic and epigenetic abnormalities, including aberrant activation of oncogenes and growth factor signaling pathways, as well as disruptions in post-transcriptional gene regulation mechanisms19. Recent studies have elucidated the complex molecular mechanisms underlying this phenomenon and have identified miRNAs as emerging players in this intricate network.
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression post-transcriptionally. Extensive research supports the notion that miRNAs can mediate the radiosensitization of cancer cells by regulating target genes20,21. Among these, miR-7-5p, a significant miRNA, has garnered extensive attention from researchers in recent years. It plays a crucial regulatory role in various cellular processes. Studies have shown that miR-7-5p expression is often altered in various cancers, suggesting that it plays a significant role in cancer development and progression22,23. In addition, The role of miR-7-5p in lung cancer has also been confirmed, as studies have shown that propofol suppresses tumorigenesis by modulating the circ-ERBB2/miR-7-5p/FOXM1 axis24, while circATXN7 downregulation inhibits NSCLC growth by increasing miR-7-5p availability25, both highlighting its tumor-suppressive function. Several studies have reported the involvement of miR-7-5p in cancer chemotherapy resistance26. Ma et al. demonstrated that miR-7-5p inhibits cisplatin resistance in lung cancer27. Yang et al. found that the overexpression of miR-7 could reverse CDR1 silencing, enhancing the susceptibility of breast cancer cells to drug resistance28. In this study, we demonstrated that miR-7-5p enhances the sensitivity of lung cancer cells to radiotherapy. We found that the up-regulation of miR-7-5p significantly increased the radiosensitivity of NSCLC cells, whereas its down-regulation significantly decreased it. These results suggest that miR-7-5p plays a role in promoting radiosensitivity in NSCLC, in addition to its previously reported tumor-suppressive effects.
DNA damage can lead to cell death. DSBs are the most harmful type of DNA damage, and, if not repaired, can cause chromosomal aberrations, genomic instability, or cell death29. Recent studies have suggested that miR-7-5p modulates the efficacy of tumor therapies by altering DNA damage repair. Lai et al. found that miR-7-5p mediates PARP1 down-regulation, affecting DNA homologous recombination repair and adriamycin resistance in small-cell lung cancer30. Luo et al. reported that miR-7-5p overexpression inhibits the DNA repair abilities of PARP-1 and BRCA1, thereby inhibiting cell proliferation and promoting apoptosis31. Additionally, lncRNA ANRIL has been shown to promote homologous recombination (HR) repair by sponging miR-7-5p and upregulating PARP1 expression, further underscoring the complex regulatory network involving miR-7-5p in DNA repair mechanisms32. The effectiveness of radiation therapy relies on three pillars: tumor-targeting accuracy, dose optimization, and combination therapy. The biological processes underlying the latter two pillars are DNA damage and repair, which are intrinsic factors in the response to radiation exposure33. A key component of DNA repair is histone H2AX, which is rapidly phosphorylated on serine residues at the carboxyl terminus to form γ-H2AX at nascent DSB34. In mammalian cells, DSBs that occur throughout the cell cycle are primarily repaired via the NHEJ pathway35. We used an EJ5-GFP-based chromosomal reporter gene to measure NHEJ activity and found that upregulation of miR-7-5p reduced NHEJ-mediated repair in lung cancer cells. Therefore, we suggest that miR-7-5p may contribute to tumor resistance to radiotherapy by regulating NHEJ repair of DNA damage.
To further investigate the mechanism underlying the effect of miR-7-5p on cellular radiation resistance, we predicted downstream target genes of miR-7-5p and identified PKP2 as a potential target. miR-7-5p binds to the 3′-UTR of PKP2 mRNA. Using dual-luciferase and western blot experiments, we confirmed that PKP2 is a target of miR-7-5p. To clarify PKP2’s role in the miR-7-5p-mediated sensitization of lung cancer cells to radiotherapy, we overexpressed PKP2 in lung cancer cells transfected with the miR-7-5p mimic, followed by radiotherapy. Overexpression of PKP2 led to increased proliferation of A549 cells, and this increase in DSBs was reversed by miR-7-5p upregulation. miR-7-5p regulates the growth and apoptosis of A549 lung cancer cells after radiotherapy via PKP2. Additionally, PKP2 overexpression increases NHEJ repair in lung cancer cells after radiotherapy, suggesting that it promotes radioresistance by enhancing NHEJ repair. Our previous research demonstrated that PKP2 positively regulates LIG4 expression in a β-catenin-dependent manner and that PKP2 enhances radiotherapy resistance by promoting LIG4-mediated NHEJ repair9. These findings indicate that PKP2 plays a pivotal role in the DNA damage response (DDR), particularly in double-strand break (DSB) repair. Here, we primarily focused on demonstrating that miR-7-5p regulates radiosensitivity by targeting PKP2.
While our study demonstrates that miR-7-5p enhances radiosensitivity in NSCLC by targeting PKP2 and inhibiting NHEJ repair, it is important to note that the role of miR-7-5p in radiotherapy response can be context-dependent. For example, Tomita K et al. reported that miR-7-5p contributes to radioresistance in clinically relevant radioresistant cells by regulating intracellular Fe2 + content, which plays a critical role in radiation-induced cell death36. This finding contrasts with our results, where miR-7-5p overexpression enhanced radiosensitivity in NSCLC cells. The dual role of miR-7-5p in radiotherapy response highlights the complexity of miRNA-mediated regulation and underscores the importance of context-specific studies to fully understand the mechanisms underlying therapy resistance.
In this study, our results suggest that PKP2 is a novel target of miR-7-5p and that miR-7-5p increases the radiosensitivity of NSCLC by targeting PKP2. These results indicate that the miR-7-5p/PKP2 axis plays a key role in the radiosensitization of NSCLC cells. miRNA-targeting approaches may increase the sensitivity and efficacy of radiotherapy and potentially improve the prognosis of many cancer patients. However, further research is needed to improve cancer treatment and overcome radiation resistance and chemoresistance.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Ferlay, J. et al. Cancer statistics for the year 2020: an overview. Int. J. Cancer. 149 (4), 778–789 (2021).
Zhu, Z. et al. International consensus on radiotherapy in metastatic non-small cell lung cancer. Transl. Lung Cancer Res. 11 (9), 1763–1795 (2022).
Alhaddad, L., Osipov, A. N. & Leonov, S. The molecular and cellular strategies of glioblastoma and non-small-cell lung cancer cells conferring radioresistance. Int. J. Mol. Sci. 23 (21), 13577 (2022).
Hatzfeld, M. The armadillo family of structural proteins. Int. Rev. Cytol. 186, 179–224 (1999).
Gao, L. et al. Identification of PKP 2/3 as potential biomarkers of ovarian cancer based on bioinformatics and experiments. Cancer Cell. Int. 20, 509 (2020).
Niell, N. et al. The human PKP2/plakophilin-2 gene is induced by Wnt/beta-catenin in normal and colon cancer-associated fibroblasts. Int. J. Cancer. 142 (4), 792–804 (2018).
Wu, Y. et al. Plakophilin-2 promotes lung adenocarcinoma development via enhancing focal adhesion and epithelial-mesenchymal transition. Cancer Manag. Res. 13, 559–570 (2021).
Hao, X. L. et al. Plakophilin-2 accelerates cell proliferation and migration through activating EGFR signaling in lung adenocarcinoma. Pathol. Res. Pract. 215 (7), 152438 (2019).
Cheng, C. et al. CRISPR/Cas9 library screening uncovered methylated PKP2 as a critical driver of lung cancer radioresistance by stabilizing beta-catenin. Oncogene 40 (16), 2842–2857 (2021).
Chen, B. et al. Targeting non-coding RNAs to overcome cancer therapy resistance. Signal. Transduct. Target. Ther. 7 (1), 121 (2022).
Saliminejad, K. et al. An overview of MicroRNAs: biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 234 (5), 5451–5465 (2019).
Powrozek, T. & Malecka-Massalska, T. MiRNA and lung cancer radiosensitivity: A mini-review. Eur. Rev. Med. Pharmacol. Sci. 23 (19), 8422–8428 (2019).
Chen, X. et al. miRNA-218-5p increases cell sensitivity by inhibiting PRKDC activity in radiation-resistant lung carcinoma cells. Thorac. Cancer. 12 (10), 1549–1557 (2021).
Wei, T. et al. miR-219a-5p enhances the radiosensitivity of non-small cell lung cancer cells through targeting CD164. Biosci. Rep. 40 (7), 1 (2020).
Podhorecka, M., Skladanowski, A. & Bozko, P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J. Nucleic Acids. 2010, 1–15 (2010).
Chaudhry, M. A. Radiation-induced MicroRNA: discovery, functional analysis, and cancer radiotherapy. J. Cell. Biochem. 115 (3), 436–449 (2014).
Cabrera-Sanchez, J. Lung cancer occurrence after an episode of tuberculosis. Eur. Respir. Rev. 31 (165), 1–12 (2022).
Zhou, T. et al. Mechanisms and perspective treatment of radioresistance in non-small cell lung cancer. Front. Immunol. 14, 1133899 (2023).
Huang, R. & Zhou, P. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther. 5 (1), 60 (2020).
Liao, Y. et al. Non-coding RNAs in lung cancer: emerging regulators of angiogenesis. J. Transl. Med. 20 (1), 1–349 (2022).
Huang, X. et al. miRNA-95 mediates radioresistance in tumors by targeting the sphingolipid phosphatase SGPP1. Cancer Res. 73 (23), 6972–6986 (2013).
Korać, P., Antica, M. & Matulić, M. MiR-7 in cancer development. Biomedicines 9 (3), 325 (2021).
Haiping Xiao. MiR-7-5p suppresses tumor metastasis of non-small cell lung cancer by targeting NOVA2. Cell. Mol. Biol. Lett. 24 (1), 60 (2019).
Gao, J. et al. Propofol suppresses lung cancer tumorigenesis by modulating the circ-ERBB2/miR-7-5p/FOXM1 axis. Thorac. Cancer. 12 (6), 824–834 (2021).
Li, D., Fu, Z. & Donget, C. Downregulation of circATXN7 represses non-small cell lung cancer growth by releasing miR-7-5p. Thorac. Cancer. 13 (11), 1597–1610 (2022).
Gajda, E. et al. The role of miRNA-7 in the biology of cancer and modulation of drug resistance. Pharmaceuticals. 14 (2), 180 (2021).
Ma, P. et al. LINC02389/miR-7-5p regulated cisplatin resistance of non-small-cell lung cancer via promoting oxidative stress. Anal. Cell. Pathol. 2022, 6100176 (2022).
Yang, W. et al. Silencing CDR1as enhances the sensitivity of breast cancer cells to drug resistance by acting as a miR-7 sponge to down-regulate reggamma. J. Cell. Mol. Med. 23 (8), 4921–4932 (2019).
Li, L. Y. et al. DNA repair pathways in cancer therapy and resistance. Front. Pharmacol. 11, 629266 (2020).
Lai, J. et al. MiR-7-5p-mediated downregulation of PARP1 impacts DNA homologous recombination repair and resistance to doxorubicin in small cell lung cancer. BMC Cancer. 19 (1), 602 (2019).
Luo, H. et al. miR-7-5p overexpression suppresses cell proliferation and promotes apoptosis through inhibiting the ability of DNA damage repair of PARP-1 and BRCA1 in TK6 cells exposed to hydroquinone. Chem. Biol. Interact. 283, 84–90 (2018).
Du, Z., Zhang, F. & Liuet, L. LncRNA ANRIL promotes HR repair through regulating PARP1 expression by sponging miR-7-5p in lung cancer. BMC Cancer. 23 (1), 130 (2023).
Goldstein, M. & Kastan, M. B. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 66, 129–143 (2015).
Rogakou, E. P., Boon, C., Redon, C. & Bonner, W. M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell. Biol. 146 (5), 905–915 (1999).
Chang, H. H. Y. et al. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell. Biol. 18 (8), 495–506 (2017).
Tomita, K., Fukumoto, M. & Itohet, K. MiR-7-5p is a key factor that controls radioresistance via intracellular Fe(2+) content in clinically relevant radioresistant cells. Biochem. Biophys. Res. Commun. 518 (4), 712–718 (2019).
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Special Fund for the Natural Science Foundation of China (8216110654 to YS), Jiangxi Provincial Natural Science Foundation of China (20212ACB216013, 20204BCJL23052 to YS), Nanchang Natural Science Foundation of China (2021 -129 to YS), and Jiangxi Key Laboratory (2024SSY06042 to YS).
Author information
Authors and Affiliations
Contributions
F.Z. (The first author), J.Y. (co-author) and X.Q. (co-author) contributed equally to this work. Corresponding author Y.S. and Co-corresponding author C.C. conceived the project. F.Z., J.Y. and X.Q. designed the experiments. F.Z., J.Y., X.Q. and J.H. conducted all the experiments. J.Y. and X.Q. analyzed the data. F.Z. wrote the manuscript. Y.S. and C.C. reviewed and polished the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, F., Yang, J., Qiu, Xl. et al. Low expression of miR-7-5p promotes resistance to radiotherapy in lung cancer through direct upregulation of PKP2 expression. Sci Rep 15, 16847 (2025). https://doi.org/10.1038/s41598-025-01001-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01001-6
