
Abstract
The tumor microenvironment (TME) plays a critical role in ovarian cancer (OC) progression, yet the relationship between immune and stromal scores within the TME and prognostic outcomes remains poorly understood. Immune and stromal cell scores were computed using the “estimate” R package, which enabled the assessment of immune and stromal components in OC samples. We then performed univariate and multivariate Cox regression analyses to identify prognostic factors associated with these scores using data from The Cancer Genome Atlas (TCGA). Additionally, LASSO Cox regression were employed to identify key prognostic genes linked to immune infiltration. Our analysis of OC expression data identified 1,667 differentially expressed genes (DEGs) associated with immune and stromal scores. From these, we developed a 6-gene risk model, consisting of ALOX5AP, FCGR1C, GBP2, IL21R, KLRB1, and PIK3CG, which effectively stratified OC patients into high-risk and low-risk groups. Survival analysis and area under the curve (AUC) assessment confirmed the model’s strong predictive accuracy. Furthermore, drug sensitivity predictions indicated that sorafenib was particularly effective in high-risk patients, with this finding validated through in vitro experiments. The 6-gene TME-related risk model offers robust prognostic capabilities for OC and could serve as a valuable tool for clinical stratification and personalized treatment approaches.
Similar content being viewed by others


Introduction
Ovarian cancer (OC) stands as one of the deadliest cancers globally, characterized by a high mortality rate. By the year 2023, it is projected that over 300,000 new cases of OC will emerge worldwide, comprising 3.6% of all cancer diagnoses, with an anticipated death toll surpassing 190,000. Compounded by its subtle clinical presentation and the absence of effective early-stage screening methods, nearly 75% of cases are diagnosed at an advanced stage, leading to a dismal 5-year survival rate1. Despite advancements in combination chemotherapy, targeted therapy, and intraperitoneal chemotherapy, approximately 80% of OC patients initially respond to treatment, yet succumb to chemotherapy resistance and recurrent disease2. Thus, early detection and intervention are imperative for enhancing the quality of life and survival prospects for OC patients. The notable heterogeneity within OC has been shown to contribute to treatment failures and unfavorable prognoses3. Consequently, the urgent imperative lies in the exploration and establishment of a reliable prognostic model for OC, aiming to inform more tailored clinical interventions and ameliorate patient outcomes.
The advent of immunotherapy has transformed cancer treatment, with immune checkpoint inhibitors (ICIs) emerging as a cornerstone in managing various malignant tumors4. Ovarian cancer (OC) presents a notable challenge due to its profound immunosuppressive nature, underscoring the significance of ICIs in its therapeutic landscape. Despite promising indications that recurrent and metastatic OC patients may derive benefits from ICI immunotherapy, clinical outcomes have often fallen short, yielding an overall response rate ranging between 13 and 18%5. Understanding the intricate interplay of immune infiltration within the tumor microenvironment (TME) is pivotal in comprehending tumorigenesis and tumor progression, ultimately shaping the clinical prognosis for patients. Moreover, mounting evidence suggests a correlation between tumor mutation burden (TMB) and response to immunotherapy, further emphasizing the multifaceted nature of immune response modulation in cancer therapy6.
Genetic and epigenetic alterations within tumor cells are pivotal in tumor initiation and progression. However, emerging evidence also underscores the significance of interactions between tumor cells and their surrounding normal counterparts6. TME represents a complex milieu comprising cancer cells, extracellular matrix components, immune cells, and diverse molecular entities. Notably, immune infiltrating cells and stromal cells are garnering increased attention as principal cellular constituents of the TME7. Assessing the status of these cell types within the TME holds promise for refining diagnostic and prognostic evaluations in cancer patients. Immune dynamics constitute a critical facet of the TME, underscoring the importance of comprehending immune-related features in OC for risk stratification and targeted therapeutic interventions. The Estimation of Stromal and Immune cells in Malignant Tumor tissues using Expression data (ESTIMATE) method have proven effective in quantitatively characterizing the TME across various malignancies8. Despite previous studies investigating OC patients through the lens of immune cell infiltration, a comprehensive examination of the relationship between OC and immunity remains lacking, particularly regarding immune-related genes, immune cell infiltration, and transcription factors (TFs).
In this study, we conducted a thorough examination of the predictive capabilities of immune and stromal scores in OC patients. Subsequently, we developed a risk model utilizing TME parameters to enhance prognostic risk stratification, thereby enabling more informed treatment decisions for patients. The identification of DEGs in our analysis holds promise for advancing our comprehension of tumor progression and guiding immunotherapy strategies. Moreover, our findings may shed light on mechanisms underlying tumor evasion and lay the groundwork for the discovery of novel prognostic markers.
Materials and methods
Data download and processing
We retrieved mRNA expression, clinical information, and somatic mutation data of ovarian cancer (OC) samples from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Using perl (strawberry-perl-5.30.0.1-64bit) and R software, we processed the relevant data from both TCGA, GEO and ICGC (International Cancer Genome Consortium) datasets, facilitating comprehensible visualization. We utilized the entire TCGA cohort of 306 ovarian cancer patients as the experimental group, as all cases had both comprehensive gene expression data and complete clinical follow-up information required for survival analysis. To avoid overfitting concerns, we employed two independent GEO datasets, GSE17260 (this dataset included 110 ovarian cancer patients who received primary surgery and a platinum/taxane-based chemotherapy) and GSE14764 (a cohort with 80 ovarian cancer patients) as validation cohorts. The RNA-seq expression and clinical-pathological parameters of 85 OC patients were retrieved from ICGC. Based on median stromal and immune scores, we categorized the TCGA patients into high- and low-score groups. Differential expression analysis (DEGs) between these groups was conducted using the “limma” R package (v3.62.2), with stringent criteria set at a false discovery rate (FDR)-adjusted p-value < 0.05 and |log2 fold change (FC)| ≥ 1. To ensure the reliability of our findings, we performed cross-validation and have incorporated these methodological details in the revised manuscript9.
TME analysis
We utilized the “estimate” R package (v1.0.13) to gauge the infiltration levels of immune and stromal cells, resulting in immune scores and stromal scores. Concurrently, the ESTIMATE score encapsulated the combined sum of immune and stromal scores. Subsequently, we investigated the relationship between the expression levels of model genes and these scores using Spearman’s rank correlation coefficient test. Moreover, employing the CIBERSORT algorithm, we evaluated the presence of 22 types of infiltrating immune cells within each sample. For the CIBERSORT analysis, we utilized the LM22 gene signature as the reference matrix—a standardized signature widely adopted for broad immune cell deconvolution—which includes 22 immune cell types, to estimate the relative proportions of immune cell populations in the tumor microenvironment based on bulk RNA-seq data. The analysis was performed using the CIBERSORT R script, with 1,000 permutations to ensure robust statistical inference based on established best practices. We used quantile normalization as enabled by default, which is appropriate for RNA-seq data. Samples with a CIBERSORT P-value < 0.05 were considered to have reliable immune cell composition estimates, ensuring the accuracy and validity of the deconvolution results. These parameters were chosen based on their suitability for immune profiling in cancer studies and to maintain consistency with established best practices10.
WGNCA for the transcriptome of ovarian cancer
We employed WGCNA (R package WGCNA v1.73) to identify the association between co-expressed mRNA modules and either immune or stromal clusters11. Module eigengenes (MEs) were computed as the first principal component of each module, serving as a representative of all mRNAs within that module. Gene significance (GS) was determined as the intermediary p-value for each gene, reflecting the strength of the linear correlation between gene expression within the module and clinical features. Modules pertinent to the clusters were designated based on a significance threshold of FDR-adjusted p-values < 0.04, with the highest GS value selected for subsequent analysis. To construct a co-expression network, we selected a soft-thresholding power of 4 to achieve a scale-free topology, a key property of biological networks. This choice was guided by the criterion that the scale-free topology fit index (R²) exceeded 0.9, ensuring that the network approximates a scale-free distribution, where a few highly connected genes (hubs) play critical roles in network structure and function. This threshold was validated by inspecting the scale-free topology plot and mean connectivity, which confirmed that the chosen power balanced network connectivity and scale-free properties effectively. Gene modules were identified using hierarchical clustering, and modules with similar expression profiles were merged based on a module eigengene dissimilarity threshold (MEDissThres = 0.25) with verbose output (verbose = 3) to ensure clarity and reproducibility. Among the identified modules, the “turquoise module” was selected for further analysis because it demonstrated the highest correlation with stromal score, immune score, and ESTIMATE score, suggesting its strong relevance to the tumor microenvironment. This correlation was used as a biological criterion to prioritize modules likely associated with the study’s survival outcomes.
Validation of ESTIMATE-based immune grouping and HLA gene expression analysis
We employed the ESTIMATE algorithm to calculate Stromal Score, Immune Score, ESTIMATE Score, and Tumor Purity from ovarian cancer transcriptome profiles, providing quantitative measures of tumor microenvironment composition. These ESTIMATE-derived metrics were then used to validate and characterize the biological differences between molecular subgroups previously established through ssGSEA analysis (GSEABase v1.68 and GSVA v2.0.5), with results visualized through clustering heatmaps and statistical plots. To further investigate immunogenic differences between subgroups, we examined human leukocyte antigen (HLA) gene expression levels12. This integrated analytical approach allowed comprehensive evaluation of tumor microenvironment heterogeneity in relation to molecular subtypes, with important implications for understanding disease mechanisms and predicting treatment outcomes.
Comparative analysis of GO and KEGG pathways in key module
We utilized the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) functional analyses to investigate the roles of key mRNAs within the WGCNA module13,14. The mRNAs were categorized into three groups based on the outcomes of the GO analysis, including biological processes (BP), molecular functions (MF), and cellular components (CC). Several R packages, namely “limma”, “org.Hs.eg.db” (v3.20), “DOSE” (v4.0.0), “clusterprofiler” (v4.14.4), and “enrichplot” (v1.26.6) were employed for this analysis. mRNA set enrichment analysis (GSEA) was conducted to identify active pathways in both high- and low-risk groups, with significance determined by | NES |≥ 1 and FDR q < 0.0515.
Construction of the module related risk signature
Initially, we conducted unsupervised clustering analysis utilizing the “ConsensusClusterPlus” (v4.14.4) package. Our feature selection methodology involved several sequential steps to identify hub genes with prognostic significance. First, we preprocessed mRNA expression data by removing incomplete samples, normalizing the dataset, and eliminating batch effects to ensure data quality. WGCNA was then performed to identify modules of co-expressed genes correlated with clinical traits, focusing on key modules associated with survival outcomes. Principal Component Analysis (PCA) was applied to these modules to extract the first principal component (Module Eigengene) as a representative feature, reducing redundancy and summarizing expression patterns. To further screen potential biomarkers, we employed LASSO (Least Absolute Shrinkage and Selection Operator) Cox regression, using 10-fold cross-validation to optimize the regularization parameter (lambda) and select survival-associated genes while addressing multicollinearity (optimal λ = 3.08 determined by minimum partial likelihood deviance). The optimal lambda value was determined by minimizing the cross-validated partial likelihood deviance, which balances model complexity and predictive accuracy. The intersecting genes identified in both WGCNA and LASSO analyses were shortlisted as candidate biomarkers, ensuring robustness by integrating complementary methodologies. Finally, the prognostic significance of these genes was validated through Kaplan-Meier survival analysis, multivariate Cox regression, and evaluation of model performance using metrics such as the concordance index (C-index) and time-dependent ROC curves. This comprehensive approach allowed us to systematically narrow down and justify the selection of final hub genes for prognostic analysis. This procedure aimed to refine candidate mRNAs based on the minimum penalty parameter (λ = 3.08) criteria, ultimately generating a multiple-mRNA signature. The final risk score model, calculated as Σ (Coef mRNA × Expr mRNA), effectively stratified ovarian cancer patients into high- and low-risk groups (median cutoff) with significant survival differences Based on the median risk score, OC samples were stratified into high- and low-risk groups, and Kaplan-Meier analysis was performed to compare overall survival (OS) between these groups. Utilizing packages such as “timeROC” (v0.4), “survival” (v3.7), “survminer” (v0.5.5), and “survivalROC” (v1.0.3.1), we conducted receiver operating characteristic (ROC) analysis at 1-, 3-, and 5-year intervals to evaluate the prognostic signature’s performance. Additionally, we compared the area under the ROC curve (AUC) of this signature with other clinicopathological traits.
Evaluation of clinical independence and construction of the nomogram
Subsequently, we excluded EC patients with incomplete clinicopathological details, such as survival status and time, age, weight, clinical stage, tumor grade, and lymph node status. Univariate and multivariate Cox regression analyses were performed, incorporating both clinical indices and risk scores, to confirm the independence of the risk model. ROC curves were generated to evaluate the predictive performance of the model against other clinical features. Additionally, we examined the correlation between the risk signature derived from the risk model and clinical indices. Lastly, we utilized the “rms” R package (v7.0) to integrate the risk score and clinical characteristics, facilitating the construction of a nomogram16.
Wound healing and transwell assays
Cell migration was assessed using both wound healing and Transwell assays with the A2780 human ovarian cancer cell line (obtained from ATCC, authenticated by STR profiling). Initially, cells were seeded into 6-well plates and cultured until reaching 95% confluence. Subsequently, a sterile 10 µl pipette tip was utilized to create a scratch across each well, and wound closure was monitored and photographed under a microscope (Olympus, Tokyo, Japan) at 0 and 24 h. For the Transwell migration assays, the ovarian cancer cell line SKOV3 was added to the upper chambers in Dulbecco’s modified eagle medium (DMEM) supplemented with 1% fetal bovine serum (FBS), while the lower chambers were filled with DMEM containing 20% FBS. Following a 24-hour incubation period, cells that migrated through the membrane were fixed with methanol at room temperature for 30 min, followed by staining with crystal violet staining solution for an additional 30 min. The migrated cells were then counted under a Leica microscope (magnification, ×100). All experiments were conducted three biological replicates to ensure statistical significance of the study.
External validation of the risk signature
The predictive potential of the risk model was validated in the testing cohort of 49 surgically treated OC patients at the Department of Obstetrics and Gynecology, Cangzhou Central Hospital, in which RNA sequencing results and clinical data were available. All samples were from patients between January 2014 and December 2020. Total RNA isolation and reverse transcription-quantitative PCR procedures according to manufacturer’s instructions. This research was approved by the Institutional Ethics Committee (Human Research) of our hospital and informed consent was obtained from the patients.
Statistical analysis
Data calculations and statistical analyses were conducted using R programming (version 4.0.2, https://www.r-project.org/). We conducted our statistical analysis using a univariate and multivariate Cox proportional hazards regression model to evaluate the association between gene expression and overall survival, ensuring that the proportional hazards assumption was met by testing Schoenfeld residuals with no significant violations. Missing gene expression data were handled using multiple imputation based on predictive mean matching to preserve data variability, while cases with missing survival times were excluded to maintain model validity. For WGCNA, significant modules were identified using a module-trait correlation threshold of (p < 0.05), and multiple comparisons were controlled using False Discovery Rate (FDR) adjustment with a cutoff of FDR < 0.05. LASSO Cox regression used cross-validation to optimize the regularization parameter (lambda) for selecting survival-associated genes, balancing model parsimony and predictive accuracy. Kaplan-Meier analysis employed the log-rank test to compare survival curves with a significance threshold of (p < 0.05), while multivariate Cox regression results were deemed significant if the hazard ratio’s 95% confidence interval excluded 1 and the p-value was below 0.05. All analyses were performed using R with key packages including “survival” (v3.7-0), “glmnet” (v4.1–8), “WGCNA” (v1.73), and “survminer” (v0.5.0), ensuring a robust and reproducible workflow. All experiments were performed with three biological replicates, and statistical analyses were conducted to assess differences.
Results
Identification of immune- and stromal-related DEGs in TCGA dataset
In our quest to uncover novel genes within the OC microenvironment associated with both immune and stromal scores, we conducted RNA-Seq analysis on 306 OC cases sourced from the TCGA cohort. Initially, we stratified the samples according to the median immune and stromal scores derived from ESTIMATE, facilitating a comparative analysis between high and low-scoring groups. Differential expression analysis revealed a total of 864 significantly upregulated genes and 313 significantly downregulated genes in relation to high and low immune scores, depicted visually through heatmap and volcano plot representations (Fig. 1A-B). Similarly, comparison of samples with high versus low stromal scores identified 1,029 significantly upregulated genes and 219 significantly downregulated genes (Fig. 1C-D). These findings offer valuable insights into the genetic landscape of OC microenvironment, potentially paving the way for novel therapeutic targets and prognostic markers.

Differential expression analysis of immune- and stromal-related genes in ovarian cancer using the TCGA dataset. (A) Volcano plot showing the distribution of differentially expressed genes (DEGs) between high and low immune score groups, identified based on ESTIMATE analysis. Upregulated genes are highlighted in red, and downregulated genes are highlighted in turquoise. Non-significant genes are shown in black. (B) Heatmap displaying the expression profiles of immune score-related DEGs, with columns representing individual samples and rows representing genes. Samples are grouped by high and low immune scores. (C) Volcano plot illustrating the DEGs between high and low stromal score groups, with upregulated and downregulated genes similarly highlighted in red and turquoise, respectively. (D) Heatmap of stromal score-related DEGs, showing gene expression patterns across samples grouped by stromal score.
Function of immune and stromal-related DEGs
Overlap of DEGs derived from immune and stromal scores yielded 761 genes (595 upregulated and 166 downregulated) for subsequent functional enrichment analysis encompassing GO and KEGG pathways (Fig. 2A-B). The top 8 functional annotations from GO analysis, depicted in Fig. 2C, revealed enrichment in immune cell activation, extracellular structure organization, and cellular regulation. Furthermore, KEGG pathway analysis highlighted involvement in cytokine interaction, hematopoietic cell lineage, and cell adhesion molecules (Fig. 2D). These findings shows that ESTIMATE/Stromal scores correlate negatively with risk, but Immune score does not collectively underscoring the enrichment of common genes in immune and extracellular matrix functions.

Identification and functional enrichment analysis of intersecting immune- and stromal-related DEGs. (A) Venn diagram illustrating the overlap of upregulated genes between the immune and stromal score-related DEG sets, revealing 595 shared genes. (B) Venn diagram showing the overlap of downregulated genes between the two DEG sets, identifying 166 shared genes. (C) Gene Ontology (GO) enrichment analysis of the 761 intersecting genes, with the top eight biological processes displayed, including immune cell activation, extracellular structure organization, and cellular regulation. (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the intersecting genes, highlighting pathways such as cytokine-cytokine receptor interaction, hematopoietic cell lineage, and cell adhesion molecules. These results emphasize the involvement of intersecting genes in immune-related functions and extracellular matrix regulation.
Detection of immunity-related module and hub genes by WGANA
To pinpoint the genes with the most robust association with both sex and immune/stromal scores, we employed WGCNA. Leveraging the immune and stromal scores, we constructed a co-expression network using the R package “WGCNA” revealing modules and genes significantly linked to immune-related clusters. Optimal construction of a scale-free network was achieved with a soft threshold (β = 3), as depicted in Fig. 3A-B. Subsequently, the gene network was visualized alongside meta-modules (Fig. 3C), and after parameter adjustments, DEGs were categorized into 9 distinct modules (FigureS1 and Fig. 3D). Notably, the turquoise module (details are shown in Table S1) emerged as the most correlated with the three clusters (Fig. 3E). Hub genes within modules correlated with ESTIMATE score indices were identified using a threshold of MM score > 0.8 and GS score > 0.5, yielding 75 hub genes for the immune score (Fig. 3F), 96 for the stromal score (Fig. 3G), and 152 for the overall ESTIMATE score (Fig. 3H). Subsequently, a total of 175 genes were selected for further analysis. GO and KEGG pathway enrichment analyses were then conducted to elucidate biological processes, cellular components, and molecular functions of these genes, with a significance threshold of P < 0.05 and enrichment > 2.0. The top 10 results from both analyses were visualized in bubble diagrams. Among the findings, the top three enriched biological processes included T cell activation, regulation of lymphocyte activation, and neutrophil activation, while the top cellular components included the external side of the plasma membrane, secretory granule membrane, and tertiary granule. Moreover, the top molecular functions comprised immune receptor activity, cytokine receptor activity, and cytokine binding (Fig. 4A). Furthermore, KEGG pathway analysis highlighted the importance of the PI3 K-Akt signaling pathway in tumor progression (Fig. 4B).

Weighted gene co-expression network analysis (WGCNA) to identify immune- and stromal-related modules and hub genes. (A) Analysis of the scale-free fit index for various soft-thresholding powers, determining the optimal soft threshold (β = 3) for constructing a scale-free network. (B) Mean connectivity versus soft-thresholding powers, demonstrating the criteria for optimal network construction. (C) Heatmap showing meta-modules of eigengene correlations, revealing clusters of co-expressed genes. (D) Dendrogram of gene clustering based on topological overlap, where branches represent gene modules, dynamically cut and merged to form final modules. (E) Heatmap of correlations between modules and ESTIMATE scores, identifying the turquoise module as the most significantly associated with immune and stromal scores. (F-H) Scatter plots of module eigengene correlations with immune score, stromal score, and combined ESTIMATE score in the turquoise module. Genes with module membership (MM) > 0.8 and gene significance (GS) > 0.5 were defined as hub genes, identifying 75 immune-related, 96 stromal-related, and 152 ESTIMATE-related hub genes, respectively.

Enrichment analysis of hub genes associated with immune and stromal scores. (A) GO enrichment analysis of 175 hub genes, with the top 10 biological processes, cellular components, and molecular functions visualized. Key processes include T cell activation, regulation of lymphocyte activation, and neutrophil activation. (B) KEGG pathway enrichment analysis, highlighting the PI3 K-Akt signaling pathway as critical in tumor progression. The results provide insights into the roles of hub genes in the immune microenvironment and cancer biology.
Consensus cluster analysis for selected key gene expression profiles
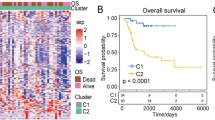
Subsequently, employing the 175 identified genes, consensus clustering analysis was conducted to explore their relationship with OC subtypes. Through analysis of the cumulative distribution function (CDF) value, the 306 OC patients were categorized into three clusters (k = 3, Fig. 5A-D). These clusters were denoted as cluster 1 (n = 117), cluster 2 (n = 130), and cluster 3 (n = 61). Principal component analysis (PCA) was employed to illustrate the variations in gene expression levels across these subgroups (Fig. 5E). Notably, patients within cluster 1 exhibited a tendency towards longer survival compared to those in clusters 2 and 3 (Fig. 5F), underscoring the significant prognostic relevance of these DEGs.

Consensus clustering of OC patients based on WGCNA-identified DEGs and their prognostic implications. (A) Consensus clustering matrix categorizing 306 ovarian cancer patients into three clusters (k = 3), based on the expression profiles of 175 hub genes. (B-D) Cumulative distribution function (CDF) plots and delta area plots showing the stability of clustering for k values from 2 to 9. The optimal k was determined to be 3. (E) Principal component analysis (PCA) of the three clusters, demonstrating distinct gene expression patterns among the subtypes. (F) Kaplan-Meier survival curves for the three clusters, revealing significant differences in overall survival, with cluster 1 showing a trend towards improved survival outcomes. These findings suggest the prognostic relevance of the identified molecular subtypes.
Functional analysis of the risk score model
We delved deeper into the association between the risk score and ESTIMATE-related scores, encompassing immune score, stromal score, and ESTIMATE score. Our analysis revealed a notably negative correlation between ESTIMATE score, stromal score, and risk score, with r values of −0.27 and − 0.28, respectively (both p < 0.01, Fig. 6A-B), indicating a lower stromal cell presence in the high-risk group. However, the correlation between immune score and risk score was found to be insignificant (Fig. 6C). These findings highlight the influence of tumor immune microenvironment variation on the unfavorable prognosis observed in the high-risk group of OC patients. To elucidate the key pathways associated with the risk signature, GSEA was conducted, revealing 68 significantly enriched pathways between low and high-risk groups based on criteria of FDR < 0.25 and P-value < 0.05. Notably, the top five signaling pathways identified in the high-risk group included the Notch signaling pathway, Wnt signaling pathway, tight junction, oocyte meiosis, and VEGF signaling pathway (Fig. 6D). Moreover, patients were stratified into four subgroups based on immune score and risk score, revealing distinct prognostic outcomes. Specifically, patients with high immune score and low risk score exhibited the most favorable prognosis, while those with low immune score and high-risk score demonstrated the poorest prognosis. Conversely, patients with high immune score and low risk score showed the best prognosis (Fig. 6E). These results underscore the significant relationship between ESTIMATE score and risk score and the consequential functional relevance of the risk signature.

Association between tumor microenvironment and the risk score. (A-B) Negative correlations between the risk score and ESTIMATE Score (r = −0.27) and Stromal Score (r = −0.28), indicating lower stromal content in high-risk patients. (C) Lack of significant correlation between the risk score and Immune Score, suggesting a differential relationship between immune and stromal components in the tumor microenvironment. (D) GSEA identifying the top five pathways significantly enriched in high-risk patients, including the Notch signaling pathway, Wnt signaling pathway, and VEGF signaling pathway, which are critical in tumor progression. (E) Kaplan-Meier survival analysis stratifying patients into four groups based on combined immune score and risk score, with the high immune score and low-risk group showing the best prognosis and the low immune score and high-risk group showing the worst prognosis.
Establishment and validation of the risk signature based on hub genes
Following the identification of the hub turquoise module, we proceeded to construct a risk model utilizing the mRNA expressions derived from this module. Initially, a subset of these genes was selected for further analysis through additional LASSO regression, involving 175 mRNAs (Fig. 7A-B). Subsequently, six hub genes were identified: ALOX5 AP, FCGR1 C, GBP2, IL21R, KLRB1, and PIK3 CG. Table 1 presents these genes along with their corresponding coefficients utilized for calculating each subject’s risk score. The risk score formula was derived as follows: Risk score = (ALOX5 AP* 0.0065) + (FCGR1 C* 0.0784) - (GBP2*0.0902) - (IL21R*0.0338) - (KLRB1*0.0102) + (PIK3 CG*0.0574). Subsequently, OC patients within the TCGA dataset were stratified into high-risk and low-risk groups based on the median risk score. Correlation analyses were conducted to assess the expression relationships among the hub genes (Fig. 7C). Heatmaps were utilized to depict the differential expression levels of the six mRNAs and clinicopathological features between the high- and low-risk groups. Notably, vital status, tumor residual disease, tumor status, recurrence, lymphatic invasion, and stage exhibited differential distribution across the two risk groups (Fig. 7D). Moreover, the high-risk group displayed a considerably higher mortality rate compared to the low-risk group (Fig. 7E-F). Evaluation of the 6-gene signature’s role in OC prognosis was performed through K-M curves for the TCGA-OC cohort, indicating significant differences in OS between the high- and low-risk groups (P < 0.01, Fig. 7G). Furthermore, a time-dependent ROC curve analysis demonstrated an AUC of 0.74 for the OS prediction model (Fig. 7H), underscoring the prognostic potential of the risk score derived from the immune and stromal signature as an independent factor in OC. We further conducted functional validation of ALOX5 AP, one of the six key genes identified, through in vitro experiments. First, we validated the knockdown efficiency of siALOX5 AP, as shown in Figure S2A. Using CCK8 and EdU staining assays, we assessed the proliferation of SKOV3 cells after knockdown. Figure S2B-D demonstrate that knockdown of ALOX5 AP significantly reduced the proliferation rate of the SKOV3 cell line. Additionally, we performed correlation analysis between the expression levels of the six key genes and 22 types of immune cells, as illustrated in Figure S2E. These results indicate that the expression of key genes is significantly correlated with the content of immune cells, especially CD8 + T cells and M1 macrophages.

Construction and validation of the immune- and stromal-related risk model in the TCGA cohort. (A) LASSO coefficient profiles of the 175 genes derived from the turquoise module, showing the shrinkage of coefficients as λ changes. (B) Ten-fold cross-validation for determining the optimal λ in the LASSO regression model, identifying six hub genes (ALOX5 AP, FCGR1 C, GBP2, IL21R, KLRB1, and PIK3 CG) as significant contributors to the risk model. (C) Correlation matrix showing the expression relationships among the six hub genes, with positive and negative correlations depicted by color intensity. (D) Heatmap illustrating the differential expression patterns of the six hub genes and their association with clinicopathological features (e.g., tumor residual disease, recurrence, and lymphatic invasion) and risk score groups. (E-F) Distribution of the risk scores and survival status among TCGA-OC patients, indicating higher mortality in the high-risk group. (G) Kaplan-Meier survival analysis comparing overall survival (OS) between high- and low-risk groups, revealing significantly worse outcomes in the high-risk group. (H) Time-dependent ROC curve analysis, showing the predictive performance of the risk model with an AUC of 0.744 for OS in the TCGA cohort.
Construction and validation of the prognostic-nomogram model
Subsequently, we conducted both univariate and multivariate Cox regression analyses in the TCGA-OC cohort to evaluate the independent prognostic significance of the 6-gene risk signature. Univariate analysis revealed significant correlations between prognosis and age, stage, tumor status, tumor residual, and risk score (Fig. 8A). Moreover, multivariate analysis identified grade, tumor residual, and the risk score as independent prognostic factors in TCGA-OC patients (Fig. 8B; all P < 0.05). To further assess the prognostic utility of the risk signature, a nomogram model based on these four independent risk factors was established, including grade, tumor residual, and risk score (Fig. 8C), with corresponding scores outlined in Table 2. Calibration curves demonstrated favorable consistency between expected and observed survival rates for 1-, 3-, and 5-year overall survival (Fig. 8D). Patients were subsequently stratified into three equally sized subgroups: low-, moderate-, and high-score groups—based on the total points from the nomogram. The overall survival curve depicted in Fig. 8E revealed that patients with high scores exhibited the worst prognosis. Furthermore, ROC analysis illustrated the nomogram’s ability to accurately predict survival outcomes, with AUC values of 0.755, 0.7963, and 0.846 for 1, 3, and 5 years, respectively (Fig. 8F). We applied the model to 49 ovarian cancer patients treated at our hospital, performing transcriptome sequencing and validation. As shown in Figure S2, the results confirmed that the model has high predictive accuracy (Figure S3). We also compared the impact of common prognostic factors, such as age and FIGO stage, with the predictive accuracy of the nomogram, as shown in Figure S4. The results indicate that the nomogram has the highest accuracy for prognostic prediction. Collectively, these findings suggest that the nomogram model provides enhanced reliability in predicting overall survival in OC patients.

Development and validation of a nomogram for predicting OC prognosis. (A) Univariate Cox regression analysis identifying factors significantly associated with OS in the TCGA cohort, including age, tumor stage, residual tumor status, and risk score. (B) Multivariate Cox regression analysis confirming that grade, residual tumor status, and the risk score are independent prognostic factors. (C) Nomogram integrating clinicopathological features (age, tumor residual status) and the 6-gene risk signature for predicting 1-, 3-, and 5-year OS. Each feature contributes to a total score for individualized survival prediction. (D) Calibration curves assessing the agreement between predicted and observed OS probabilities, indicating good calibration for the nomogram. (E) Kaplan-Meier survival analysis comparing OS among low-, moderate-, and high-score groups, revealing poorer prognosis in the high-score group. (F) ROC curves for the nomogram, showing AUC values of 0.755, 0.7963, and 0.846 for predicting 1-, 3-, and 5-year OS, respectively, demonstrating its robust predictive performance.
External validation of the 6-gene risk signature
To evaluate the prognostic efficacy of our risk model, we applied the risk score algorithm to independent validation GEO cohorts. Results from the validation cohort indicated that OC patients classified into the high-risk group exhibited significantly poorer OS and PFS rates in GSE17260 (Fig. 9A-B), as well as OS in GSE14764 (Fig. 9C) and ICGC database (Fig. 9D), compared to those in the low-risk group. The corresponding AUC values for survival prediction were 0.751, 0.741, 0.812 and 0.817, respectively (Fig. 9E-H). These findings underscored the robust predictive capability of the 6-gene risk model in accurately prognosticating outcomes for OC patients.

Validation of the 6-gene risk model in external datasets. (A-B) Kaplan-Meier survival analysis of the risk signature in the GSE17260 cohort, showing significant differences in OS (P < 0.01) and progression-free survival (PFS) (P < 0.01) between high- and low-risk groups. (C) Kaplan-Meier survival analysis in the GSE14764 cohort, revealing significantly poorer OS in the high-risk group. (D) Kaplan-Meier survival analysis in the ICGC cohort, revealing significantly poorer OS in the high-risk group. (E-H) Time-dependent ROC curves for OS and PFS in GSE17260, GSE14764 and ICGC cohorts, with AUC values ranging from 0.741 to 0.817, confirming the model’s strong prognostic performance across independent cohorts.
Chemotherapeutic drug sensitivity analysis and validation
To explore variations in drug sensitivity across different risk groups, we employed the “pRRophetic” package for chemotherapeutic agent analysis. Our observations revealed that patients categorized into the low-risk group exhibited heightened sensitivity to sorafenib (Fig. 10A). The concentration selection of Sorafenib is based on previous literature reports17. Sorafenib enhances the sensitivity of the SKOV3 cell line to platinum-based drugs (Figure S5). Subsequently, we conducted in vivo experiments utilizing the SKOV3 and A2780 cell lines to validate the efficacy of sorafenib. Transwell (Fig. 10B-C) and wound healing assays (Fig. 10D-E) demonstrated that sorafenib effectively inhibited invasion and metastasis in ovarian cancer cell lines in vitro.

Chemotherapeutic sensitivity and in vitro validation of sorafenib efficacy. (A) Box plot illustrating differences in sorafenib sensitivity between high- and low-risk groups, showing significantly increased sensitivity in the low-risk group. (B-C) Transwell invasion assays demonstrating reduced invasion capacity in SKOV3 and A2780 cells treated with 20 µM sorafenib. *, p < 0.05; **, p < 0.01; ***, p < 0.001. (D-E) Wound healing assays showing impaired migration in SKOV3 and A2780 cells treated with 20 µM sorafenib, highlighting its potential to suppress metastasis in vitro. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Ovarian cancer (OC) presents a grim prognosis, characterized by significant individual variability and a high degree of treatment resistance. Current therapies, including surgery and chemotherapy, often result in limited efficacy, contributing to high recurrence rates and an overall low 5-year survival rate for OC patients18. This situation underscores the urgent need to identify reliable tumor markers, develop precise prognostic strategies, and explore innovative therapies to enhance treatment outcomes. Importantly, the tumor microenvironment (TME) plays a pivotal role in cancer progression by facilitating immune evasion and promoting tumor survival, which profoundly impacts patient prognosis19. Immune and stromal cell infiltration within the TME can either support or hinder anti-tumor responses, highlighting the need for prognostic markers that can reflect this complex interplay. This study addresses these challenges by introducing an immune- and stromal-related prognostic model, offering valuable insights into OC management and therapeutic innovation.
The study identified six differentially expressed genes (DEGs) – ALOX5 AP, FCGR1 C, GBP2, IL21R, KLRB1, and PIK3 CG – closely associated with OC prognosis. These genes represent diverse biological pathways and mechanisms critical to cancer progression and immune regulation. ALOX5 AP (Arachidonate 5-Lipoxygenase-Activating Protein) plays a central role in the arachidonic acid metabolism pathway, which is pivotal in regulating inflammatory processes and leukotriene synthesis. Dysregulation of ALOX5 AP has been linked to chronic inflammation and cancer progression, including OC, through its influence on immune cell infiltration and tumor growth20. Similarly, FCGR1 C (Fc Gamma Receptor Ia), a member of the Fc receptor family, is instrumental in mediating antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis. This gene’s activity within the TME is crucial for modulating immune activation and influencing OC outcomes21,22.
GBP2 (Guanylate-Binding Protein 2), although primarily recognized for antiviral defense, has emerged as a significant player in cancer progression. Its ability to regulate cellular processes essential for tumor growth and invasion positions it as a promising therapeutic target23. IL21R (Interleukin-21 Receptor), a key regulator of immune responses, impacts TME dynamics by influencing immune cell infiltration and anti-tumor immunity. Dysregulation of IL21R has been implicated in reduced immune surveillance, emphasizing its potential as a prognostic marker and therapeutic target24. KLRB1 (Killer Cell Lectin-Like Receptor Subfamily B Member 1), associated with natural killer (NK) cell activity, underscores the importance of innate immunity in cancer defense. Enhancing KLRB1 expression could strengthen NK cell-mediated anti-tumor responses, providing a valuable avenue for therapeutic development25. Lastly, PIK3 CG (Phosphoinositide-3-Kinase Gamma), a critical component of the PI3 K signaling pathway, is integral to tumor cell proliferation, survival, and immune evasion. Targeting PIK3 CG pathways offers a promising strategy for disrupting tumor progression and improving therapeutic outcomes26.
The immune- and stromal-related prognostic model developed in this study holds significant potential for guiding immunotherapy decisions in ovarian cancer. By evaluating the expression levels of key genes such as ALOX5 AP, IL21R, and KLRB1, clinicians could identify patients likely to benefit from immune checkpoint inhibitors, including PD-1/PD-L1 inhibitors. For instance, high expression of IL21R and KLRB1 may indicate enhanced immune cell infiltration and activity, suggesting a favorable response to immunotherapy. Conversely, dysregulation of ALOX5 AP and PIK3 CG could signal immune evasion mechanisms, potentially predicting resistance to PD-1 inhibitors27,28. This model could thus serve as a complementary tool to existing biomarkers, such as PD-L1 expression and tumor mutational burden, to refine patient stratification and optimize immunotherapy eligibility. Further validation in clinical trials is warranted to establish its utility in real-world settings.
The six-gene model demonstrated robust predictive accuracy and was validated using external GEO datasets. By integrating these genes, the model provides a comprehensive tool for predicting OC prognosis and guiding clinical decision-making. Unlike traditional models focused on tumor proliferation or apoptosis, this study’s approach emphasizes the interplay between immune-related biomarkers and the TME, addressing a critical gap in OC research.
A notable contribution of this study is the drug sensitivity analysis, which identified sorafenib as a potential therapeutic agent for OC. Sorafenib, a multikinase inhibitor, effectively suppressed OC metastasis in our analysis, highlighting its therapeutic relevance. Its ability to target key pathways implicated in tumor progression and metastasis aligns with the biological mechanisms underlying the six-gene model. This finding underscores the potential of integrating immune-related biomarkers with targeted therapies to achieve superior treatment outcomes. Additionally, sorafenib’s efficacy in inhibiting metastatic processes suggests its utility in combination regimens with immunotherapies or other targeted treatments. Such combinations could enhance treatment efficacy, mitigate resistance mechanisms, and ultimately improve patient survival29.
By incorporating drug sensitivity analysis, this study extends beyond prognostic modeling to offer actionable therapeutic insights. The identification of sorafenib as a promising agent for OC not only validates the relevance of the six-gene model but also provides a foundation for future research into personalized therapeutic strategies. The study’s approach highlights the value of integrating molecular profiling with drug development to optimize cancer treatment paradigms.
Despite its significant contributions, this study has limitations that warrant further exploration. As a retrospective study utilizing publicly available data, our findings are subject to inherent biases associated with retrospective analyses and the limitations of data collection within TCGA and GEO. Another limitation is the lack of direct experimental validation of the identified genes and their roles in therapy resistance. While the six-gene model provides robust prognostic insights, in vitro and in vivo studies are necessary to elucidate the functional mechanisms of these genes within the TME. Furthermore, external validation of the model in diverse populations and independent datasets is essential to establish its generalizability and robustness30. Expanding the study to include longitudinal follow-up data could also enhance our understanding of the model’s predictive capabilities over time. Efforts to integrate multi-omics data, such as proteomics and metabolomics, could further refine the model by capturing additional layers of biological complexity. Finally, the use of external validation datasets may introduce variability due to differences in patient demographics, sample processing, and sequencing platforms, as well as limited sample sizes and incomplete clinical information.
Lastly, while sorafenib’s potential as an anti-metastatic agent is promising, its efficacy must be validated through rigorous clinical trials involving OC patients. These trials should evaluate sorafenib’s performance in combination with other therapies, such as immune checkpoint inhibitors or targeted agents, to determine optimal treatment regimens. Understanding the molecular underpinnings of sorafenib’s activity within the TME could also reveal new therapeutic opportunities and biomarkers for patient stratification31,32. While our in vitro experiments demonstrated sorafenib’s anti-metastatic effects on OC cell lines, we acknowledge that without verifying their alignment with the 6-gene risk profiles, these results reflect general drug activity rather than direct validation of the predicted risk-dependent sensitivity. This represents a limitation for clinical translation.
In conclusion, this study presents a novel immune- and stromal-related prognostic model that integrates key biomarkers with clinical insights. By addressing the limitations and expanding upon the findings, future research can pave the way for more effective and personalized approaches to OC management, ultimately improving prognosis and quality of life for patients33.
Conclusion
In conclusion, our study underscores the clinical significance of a six-gene signature in enabling early risk stratification and prognostic monitoring of OC. This signature holds promise for predicting prognosis in OC patients, with higher risk scores indicative of poorer outcomes. Further exploration of the underlying mechanisms governed by these genes could offer insights into personalized treatment approaches for OC, potentially shaping the landscape of OC management in the future.
Data availability
The datasets analyzed during the current study are available in the Cancer Genome Atlas repository, [https://portal.gdc.cancer.gov/].
References
Webb, P. M. & Jordan, S. J. Global epidemiology of epithelial ovarian cancer. Nat. Rev. Clin. Oncol. https://doi.org/10.1038/s41571-024-00881-3 (2024).
Kuroki, L. & Guntupalli, S. R. Treatment of epithelial ovarian cancer. BMJ 371, m3773. https://doi.org/10.1136/bmj.m3773 (2020).
Lheureux, S., Braunstein, M. & Oza, A. M. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J. Clin. 69, 280–304. https://doi.org/10.3322/caac.21559 (2019).
Geisler, A. N. et al. Immune checkpoint inhibitor-related dermatologic adverse events. J. Am. Acad. Dermatol. 83, 1255–1268. https://doi.org/10.1016/j.jaad.2020.03.132 (2020).
Vogel, A., Meyer, T., Sapisochin, G., Salem, R. & Saborowski, A. Hepatocellular carcinoma. Lancet 400, 1345–1362. https://doi.org/10.1016/S0140-6736(22)01200-4 (2022).
Zhang, Z. et al. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 14, 45. https://doi.org/10.1186/s13073-022-01050-w (2022).
Du, W., Xia, X., Hu, F. & Yu, J. Extracellular matrix remodeling in the tumor immunity. Front. Immunol. 14, 1340634. https://doi.org/10.3389/fimmu.2023.1340634 (2023).
Scire, J. et al. EstimateR: an R package to estimate and monitor the effective reproductive number. BMC Bioinform. 24, 310. https://doi.org/10.1186/s12859-023-05428-4 (2023).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Garcia-Rio, F. et al. Biological biomarkers in respiratory diseases. Arch. Bronconeumol. 58, 323–333. https://doi.org/10.1016/j.arbres.2022.01.003 (2022).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Yang, Q. et al. Single-Cell RNA sequencing reveals the heterogeneity of Tumor-Associated macrophage in Non-Small cell lung Cancer and differences between sexes. Front. Immunol. 12, 756722. https://doi.org/10.3389/fimmu.2021.756722 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Snipen, L., Angell, I. L., Rognes, T. & Rudi, K. Reduced metagenome sequencing for strain-resolution taxonomic profiles. Microbiome 9, 79. https://doi.org/10.1186/s40168-021-01019-8 (2021).
Poddar, M. S., Chu, Y. D., Pendharkar, G., Liu, C. H. & Yeh, C. T. Exploring cancer-associated fibroblast-induced resistance to tyrosine kinase inhibitors in hepatoma cells using a liver-on-a-chip model. Lab. Chip. 24, 5043–5054. https://doi.org/10.1039/d4lc00624k (2024).
Miyakawa, R. et al. SPON1 is an independent prognostic biomarker for ovarian cancer. J. Ovarian Res. 16, 95. https://doi.org/10.1186/s13048-023-01180-8 (2023).
Geistlinger, L. et al. Multiomic analysis of subtype evolution and heterogeneity in High-Grade serous ovarian carcinoma. Cancer Res. 80, 4335–4345. https://doi.org/10.1158/0008-5472.CAN-20-0521 (2020).
Kattimani, V. S. & Lingamaneni, K. P. Natural bioceramics: our experience with changing perspectives in the reconstruction of maxillofacial skeleton. J. Korean Assoc. Oral Maxillofac. Surg. 45, 34–42. https://doi.org/10.5125/jkaoms.2019.45.1.34 (2019).
Drozdowicz, L. et al. Sexual health in child and adolescent psychiatry: Multi-Site implementation through synchronized videoconferencing of an educational resource using standardized patients. Front. Psychiatry. 11, 593101. https://doi.org/10.3389/fpsyt.2020.593101 (2020).
Nimmerjahn, F. & Ravetch, J. V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47. https://doi.org/10.1038/nri2206 (2008).
Rahvar, F., Salimi, M. & Mozdarani, H. Plasma GBP2 promoter methylation is associated with advanced stages in breast cancer. Genet. Mol. Biol. 43, e20190230. https://doi.org/10.1590/1678-4685-GMB-2019-0230 (2020).
Yang, R. et al. Human T-bet governs the generation of a distinct subset of CD11c(high)CD21(low) B cells. Sci. Immunol. 7, eabq3277. https://doi.org/10.1126/sciimmunol.abq3277 (2022).
Lu, H. et al. Rapamycin prevents spontaneous abortion by triggering decidual stromal cell autophagy-mediated NK cell residence. Autophagy 17, 2511–2527. https://doi.org/10.1080/15548627.2020.1833515 (2021).
Ghigo, A. et al. A PI3Kgamma mimetic peptide triggers CFTR Gating, bronchodilation, and reduced inflammation in obstructive airway diseases. Sci. Transl Med. 14, eabl6328. https://doi.org/10.1126/scitranslmed.abl6328 (2022).
Han, M. G., Jang, B. S., Kang, M. H., Na, D. & Kim, I. A. PI3Kgammadelta inhibitor plus radiation enhances the antitumour immune effect of PD-1 Blockade in syngenic murine breast cancer and humanised patient-derived xenograft model. Eur. J. Cancer. 157, 450–463. https://doi.org/10.1016/j.ejca.2021.08.029 (2021).
Ji, H. et al. Integrated genomic, transcriptomic, and epigenetic analyses identify a leukotriene synthesis-related M2 macrophage gene signature that predicts prognosis and treatment vulnerability in gliomas. Front. Immunol. 13, 970702. https://doi.org/10.3389/fimmu.2022.970702 (2022).
Bongetta, D., Calloni, T., Colombo, E. V., Versace, A. & Assietti, R. Do matrix metalloproteases mediate the SARS-CoV-2-related damage to the central nervous system? Brain Behav. Immun. 88, 35. https://doi.org/10.1016/j.bbi.2020.05.050 (2020).
Buckeridge, J. Taxonomy: swallow the costly medicine. Nature 546, 600. https://doi.org/10.1038/546600c (2017).
Wang, Y. et al. Anti-PD-1 antibody armored gammadelta T cells enhance anti-tumor efficacy in ovarian cancer. Signal. Transduct. Target. Ther. 8, 399. https://doi.org/10.1038/s41392-023-01646-7 (2023).
Wong, J. L. et al. IL-15 synergizes with CD40 agonist antibodies to induce durable immunity against bladder cancer. Proc. Natl. Acad. Sci. U S A. 120, e2306782120. https://doi.org/10.1073/pnas.2306782120 (2023).
Sanderson, M. et al. Predicting 30-day mortality in patients with sepsis: an exploratory analysis of process of care and patient characteristics. J. Intensive Care Soc. 19, 299–304. https://doi.org/10.1177/1751143718758975 (2018).
Author information
Authors and Affiliations
Contributions
Conceptualization: Yiping Yu and; Experiment: Yiping Yu and Wen Yin; Jing Feng; Data curation: Jing Feng; Formal analysis: Wen Yin and Sumin Qian; Investigation: Jing Feng; Methodology: Sumin Qian; Writing – original draft: Jing Feng; Writing – review & editing: Sumin Qian, Yiping Yu, and Jing Feng.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The data used in this study were obtained from The Cancer Genome Atlas (TCGA), a publicly available database containing de-identified patient information. As such, no additional ethical approval was required for the use of these data. All TCGA data were collected and shared in accordance with ethical guidelines and institutional review board (IRB) approvals at the contributing institutions.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Y., Yin, W., Feng, J. et al. Development and validation of a risk model for effective immune and stromal related signature predicting prognosis of patients with ovarian cancer. Sci Rep 15, 16556 (2025). https://doi.org/10.1038/s41598-025-01212-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01212-x
