
Abstract
The application of Machine Learning has become a revolutionary instrument in the domain of pharmaceutical research. Machine learning enables the modelling of Quantitative Structure Property Relationship, a crucial task in forecasting the physiochemical characteristics of drugs. In this study we utilized machine learning algorithms namely Artificial Neural Networks and Random Forest to predict physiochemical characteristics of Anti-malaria drugs. These models utilize several topological indices global variables quantifying the connectivity and geometric characteristics of molecules to estimate the ability of prospective antimalarial compounds to interact with the target enzyme and other physicochemical parameters. Molecular descriptors such as size, shape, and electronic structure indices are a way of mapping molecular properties into a set of quantitative data that can be analyzed by Machine Learning techniques. By carrying out regression analysis with the help of Artificial Neural Networks and Random Forest, the corresponding changes in the molecular structures and their effects on effectiveness and properties of the potential drugs can be predicted, thereby supporting the search for new therapeutic compounds. Machine learning not only observe the drug development process but also facilitates to look at chemical datasets with respect to high order non-linear relationship, which are essential to improve antimalarial drug candidates and pharmacokinetic properties.
Similar content being viewed by others
Introduction
Machine learning and graph theory are two fields with a lot of synergy and potential for high-level data analysis of system and network structures1. Graph theory, which is the branch of mathematics that deals with graphs as abstract data types that represent relations between objects, is rich in applications to representing data with relational structures such as use in chemistry and molecular biology, social networks and biological systems2. Machine learning techniques especially the graph based learning uses the structure of these graphs for the purpose of making predictions, data classification or pattern recognition3,4. For example, in the process of drug discovery, graphs can present molecules in which atoms are considered nodes and bonds as considered edges; this is used in the venture of determining or predicting chemical properties or biological activities dependent on certain topological features that can be learned by the machine learning algorithms5. In essence, this integration between machine learning and graph theory enhances efficiency in the prediction of elements in areas that depend on the interconnection of components such as computational biology6,7,8, social sciences9 and among others10,11,12. There are many molecular descriptors available but topological indices are most famous and are being widely used in studies due to their ability to find structural features of molecules in a numerical form13.
The topological indexs capacity to discriminate between similar molecules is crucial. Indices provide information about the topology of molecules which is helpful in prediction of physicochemical properties14. Topological indices are mathematical quantities calculated from the molecular graph of a given chemical compound and reflecting structural and connectivity characteristics of the compound. These indices quantify various characteristics of molecular structures such as the shape and size, and atom connectivity without requiring detailed quantum mechanical computations; thus they are effective statistics for investigating chemical properties and biological activities15,16. Some of the most known topological descriptors include the Wiener index which equal to the sum of the distance between all pairs of vertices in the graph and the Zagreb indices which emphasize the Vertex degree17. All these descriptors are extensively employed in Quantitative Structure-Activity Relationships (QSAR) and Quantitative Structure-Property Relationships (QSPR) where the molecular structure is related to solubility, boiling points or biological activity18,19. Topological indices reduce the complicated correlations between molecular characteristics and properties to a single number, which contains the indispensable structural parameters; these descriptive topological indices are particularly essential for predictive modeling in the drug development process, if a machine learning-based approach that can discover patterns and provide accurate predictions of new compounds is involved. Predictive power of QSPR analysis20 has been increased due to recent updates in machine learning models such as Artificial Neural Networks (ANN) and Random Forest (RF) such as antimalaria drugs were studied via QSPR analysis and python based approach for more accurate analysis. Non-linear and complex relationships between topological indices and properties can be handled by these models which is crucial for tasks related to prediction. The correlation between the elements’ functions and characteristics is established by the application of QSPR and QSAR21,22. Within the realms of theoretical and computational chemistry as well as other disciplines, a core idea is the correlation and prediction of physical, chemical, and biological activity based on the structure. Exclusive definitions of the fundamental topological indices are provided for both linked and undirected molecular graphs. Hydrogen atoms are excluded (referred to as “hydrogen suppressed”) and the analysis disregards the inclusion of double bonds and triple bonds. The topological index for a collection of molecular networks may be equal, suggesting that the index is unable to discriminate between the molecular networks.23.
Kulli24 introduced the concept of reverse vertex degree R(v), defined as
where \(\Delta (G)\) is the maximum degree of the graph G and d(v) is the degree of the vertex v.
Various researchers worked on the QSPR analysis of reverse degree based topological indices for some drugs used in the analysis of para-line graph of Remdesivir25, treatment of H1N126, COVID - 19 drugs27, prediction of physicochemical properties of benzene derivatives28, reverse degree based topological indices and its importance in chemical sciences29.
Various researchers worked on the computation of reverse degree based topological indices for general structures like Dendrimers30, Chain Hex-Derived Network31, hex-derived networks32, Silicate Network33, sodalite materials network34, fullerene cage networks35 and so on. Inspired by the aforesaid ideas, Vignesh et al.36 defined the reduced reverse vertex degree as
where \(\Delta (G)\) is the maximum degree of the graph G and d(v) is the degree of the vertex v.
The reverse degree-based versions of Zagreb indices, harmonic index, forgotten index, shilpa-shanmukha index, atom bond connectivity index, Randic index, sum connectivity index, geometric arithmetic index and the defined indices are dicussed in Table 1, and the reduced reverse degree-based versions of Zagreb indices, harmonic index, forgotten index, shilpa-shanmukha index, atom bond connectivity index, Randic index, sum connectivity index, geometric arithmetic index and the defined indices are dicussed in Table 2. These indices are critical for capturing features that influence physicochemical properties, as demonstrated in prior QSAR/QSPR studies.
In this study, the use of machine learning techniques such as ANN and RF with experimental data makes it easier for researchers to perform elaborate QSPR modeling. These models help to design drugs prospective to enhance their antiviral properties because these molecular models demonstrate how the structure of a molecule influences its biological activity. Therefore, embracing both the structural medicinal chemistry with computational method in relation to the design of other anti-malarial drugs is crucial this is more so with growing outbreaks whereby time is crucial. In conclusion, integrating machine learning to identify new anti-malarial treatment agents shortens the process of identifying better treatment options for the disorder indication that machine learning is a viable solution toward the health concern affecting the entire world. Here we envision to obtain correct predictions of these properties using Reverse and Reduce Reverse topological indices and to implement them in applied machine learning models. Thus, the proposed approach offers not only useful information regarding the molecular properties but also the prospects of applying machine learning to the advancement of drug discovery.
Motivation and significance
This situation is driven by the increasing demand for effective drugs in the global market, particularly for diseases like malaria. This demand puts pressure on the pharmaceutical value chains to discover faster and more efficient methods for producing these drugs. Conventional approaches to drug discovery are usually lengthy and expensive and therefore increased emphasis has to be placed in new technologies. Machine learning has become an invaluable tool in this field, dramatically improving a drug development pipeline through quick identification of the properties and possible effectiveness of a compound. More specifically the application of QSPR analysis in conjunction with Machine Learning yields a framework for approximating the biological activities of antimalarial drugs from their molecular characteristics. With the help of topological descriptors, QSPR models can identify calculations that forecast the pharmacological properties of a compound based on its structure, which will facilitate the identification of better drugs with a faster rate. This approach does not only fasten the identification of more potent antimalarial compounds but also decreases the number of hits that have to go through expensive experiments it therefore represents an important strategy in drug discovery at the present time.
A bibliometric analysis is shown in Fig. 1 (https://www.vosviewer.com version 1.6.20) clearly demonstrates the interest the world over in the analysis of degree-based topological indicators. This collection of national research investigations improves the field of graph theory through a enhance understanding of the topological indices and a multitude of uses for them. In the keywords bibliometric analysis of the degree-based topological indices in Fig. 2 (https://www.vosviewer.com version 1.6.20), we have presented information in several ways. The study findings show how extensively degree-based topological indices are talked about source (https://www.scopus.com).

Bibliometric analysis: different countries degree-based topological indices (https://www.scopus.com).

Bibliometric analysis: topological indices based on degree (https://www.scopus.com).

Antimalaria drugs.
Material and methods
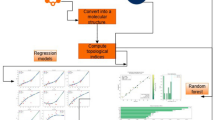
Fifteen antimalaria drugs were selected for this study. Figure 3a shows their molecular chemical structures of Atovaquone, Artemether and Artemoti and Fig. 3b shows their 2D structures, which include Artelinic acid, Atovaquone, Artemether, Artemotil, Quinine, Artemisinin, Triazole 1, Triazole 2, Triazole 3, Primaquine, Dihydroartemisic acid, Chloroquine, Mefloquine, Halofantrine and Lumefantrine denoted as “A1”, “A2”, “A3”, “A4”, “A5”, “A6”, “A7”, “A8”, “A9”, “A10”, “A11”, “A12”, “A13”, “A14” and “A15”48. To gather the physicochemical properties of the antimalaria drugs, the legacy ChemSpider (https://legacy.chemspider.com) database was utilised. This database is a reliable and lawful source of chemical information. The molecular graphs for each drug are constructed by converting their chemical structures into graph representations using specialised online available software Graph online(https://graphonline.ru/en/ Version V1.0) that records the interactions between atoms and bonds. Important molecular descriptors for the study, the Reverse and Reduced Reverse Topological Indices, were subsequently computed using an algorithm based on Python Programming (www.python.org Version 3.13.2). Using the appropriate Python libraries, two machine learning methods ANN and RF had their feature variables merged with these topological indices. The physicochemical properties of the antimalaria drugs were input into these models during training so that the accuracy and efficiency of each approach could be assessed. The accuracy of the models was evaluated by comparing the predicted values to the actual values using line graphs. This was done by integrating machine learning methodologies with molecular graph data. Tables 3 and 4 represents the values of Reverse topological indices and Reduce Reverse topological indices generated by Algorithm 1 and Algorithm 2, Table 5 represent the physicochemical properties.

Reverse topological indices and molecular descriptor calculation

Reduced reverse topological indices computation
Supervised machine learning techniques
In our study two Supervised Machine Learning based models have been employed for prediction. The first included method: ANN and second included method: RF. The general idea is to use these models to predict characteristics that can normally be tested in laboratories while reducing the time and the means that would normally take to do the testing. It is beneficial in handling analysis and computation of such small but large data sets in order to discover chemical behaviors in the shortest possible time, in contrast with the conventional approaches in computations.
Artificial neural network algorithm
Artificial Neural Network is an Machine learning model derived from the structure and function of biological neurons in the human brain. Consisting of interlinked layers of nodes (neurons), these ANN are supposed to make a distinction between given data and give outputs based on examples learnt. The structure of the network which is composed of an input layer; one or more hidden layers and output layer. The neurons personalize information by weighing inputs and applying a kernel activation formula so that the network learns non-linearity in data. This the network achieves by back propagating, a process by which errors in the output are taken as a basis for fine tuning the weights. Due to their learning mechanism, ANN are used for data analysis tasks that involve recognition of patterns such as image analyzers, speech or voice recognition systems and predictive modeling analytical systems. In the area of pharmaceutical science such as drug design, ANN have the capabilities to capture mapping between molecular structure and its corresponding, biological activity or physicochemical property and make prediction in QSPR and QSAR analysis. Due to the efficiency of handling and processing high dimensional data, ANNs are a robust tool for modeling the intricate mapping of input and output variables; hence playing a significant role in enhancing cheminformatics and pharmaceutical based fields. Table 6 represents the predicted values of Reverse indices indices generated by Algorithm-3 and Table 7 represents the error estimates for the predicted values.
Suppose we are given training data \((\textbf{a}_i, \textbf{b}_i)\) where \(\textbf{a}_i \in \mathbb {R}^M\) and \(\textbf{b}_i \in \mathbb {R}^N\). An ANN consists of multiple layers of units, where each unit computes a weighted sum of its inputs followed by an activation function. For the ANN model, we implemented a feedforward neural network using the TensorFlow/Keras library. The network architecture consists of:
-
Input layer The number of neurons is equal to the number of input features.
-
Hidden layers Two fully connected layers with 8 neurons each, using the ReLU activation function.
-
Output layer A single neuron with a linear activation function to predict continuous values.
We optimized the model using the Adam optimizer with a learning rate of \(\alpha = 0.001\). The loss function used was Mean Squared Error (MSE), which minimizes the difference between the predicted and actual values.
The key hyperparameters used in training are:
-
Learning rate: \(0.001\)
-
Batch size: \(32\)
-
Epochs: \(100\)
-
Activation function: ReLU (hidden layers), Linear (output layer)
-
Loss function: Mean Squared Error (MSE)
-
Optimizer: Adam
The pseudocode for training an ANN using backpropagation is as follows and their architecture is presented in Fig. 4 based on reverse and indices respectively:

Artificial neural network (ANN) training

ANN architecture for reverse topological indices.
Random forest algorithm
Random Forest is a generalize and strong supervise machine learning algorithm, which works by building a large number trees classifier during the training phase then using a majority voting to return a single prediction. With Random Forest each tree in the forest is created by a different random sample and set of features from training data, which helps avoid overfitting. In the case of classification, the output formed is the mean of the votes of all the trees for regression the output formed is the mean of all the responses. Such approach enhances the model stability its accuracy and its ability to perform well on unseen data. In fields like drug discovery the Random Forest is employed for QSPR and QSAR for estimating the biological activity or physicochemical characteristics of chemical compounds depending on structures. RF stands out for its applicability to datasets with numerous predictors and at the same time for the model interpretability; these properties make RF relevant for cheminformatics and pharmaceutical science tasks where the chance of obtaining accurate prediction with a primary focus on potential drug candidates is a priority. Table 8 represents the predicted values of Reverse indices generated by Algorithm 4 and Table 9 represents the error estimates for the predicted values.
Then, the pseudocode of the Random Forest algorithm is as follows:

Random forest regression
Computed vs. actual: physicochemical parameter analysis
In this section, we apply two typical machine learning methods Artificial Neural Networks and Random Forest to evaluate predicted and experimental physicochemical properties. Comparing the results generated by the models with the real data the accuracy of the predicted values of packages of many important parameters was estimated. The comparison was displayed and conducted with the help of a line graph that pointed out the relation between the actual data and the predicted numbers. In the line graph one gets quite clear impression of the performance of models, some of advantages and disadvantages of each kind of methods are shown. While artificial neural networks and random forests were used in different combinations, the accuracy of the qualities prediction varied depending on the case.
Assessing ANN and RF predictions and comparison of error metrics: reverse and reduced reverse indices
Using two different types of topological indices:In the case of the Reverse and Reduced Reverse topological indices we use two machine learning models to predict several physicochemical properties in order to evaluate the performance of both models. These indices are identified as topological descriptors that originate from the architecture of different molecules and are applied to enhance the accuracy of model projection for various chemical and physical events. By applying these models, line graphs display predicted or expected levels of several physicochemical properties such as Boiling Point (Fig. 5), ACD/LogP (Fig. 7), Polarizability (Fig. 9), Enthalpy of Vaporization (Fig. 11). To compare the performance of ANN and RF we employed Mean Squared Error (MSE), Mean Absolute Error (MAE), Root Mean Squared Error (RMSE) and the coefficient of determination (R-Squared) metrics. In all 3D bar graphs we employed (* and **) for Reverse and Reduce Reverse indices respectively. 3D bar graphs that show several error measures used to evaluate model performance are shown in Fig. 6 (Boiling point), Fig. 8 (ACD/LogP), Fig. 10 (Polarizability), Fig. 12 (Enthalpy of Vaporization)and other physiochemical properties displayed in Supplementary file. A portion of each figure indicates the comparison of Four important error metrics; MSE, MAE, RMSE and R-Squared of two different sets of conditions or models differentiated as (*), (**).In addition, the study aims to assess each machine learning models capacity in predicting these physicochemical characteristics together with the topological indices by comparing these predictions. This will provide light on the model’s performance with respect to various chemical descriptors.

Comparison for boiling point.

Comparison of boiling point for metrics.

Comparison for ACD/logP.

Comparison of ACD/logP for metrics.

Comparison for polarizability.

Comparison of polarizability for metrics.

Comparison for enthalpy of vaporization.

Comparison of enthalpy of vaporization for metrics.
Conclusion
In this study two different machine learning algorithms have been employed to predict wide range of physiochemical properties in malaria disease curing drugs. The main objective was to design a good and simple model for prediction that would greatly help the pharmaceutical companies, given that the drug discovery and development is a lengthy process. Using four measures Mean Squared Error , Mean Absolute Error , Root Mean Squared Error and \(R^2\) feasibility of models has been assessed. Among all models, the Reverse ANNs yielded the best results of the broad range of properties that categorized from Table 6 like Boiling Point for Reverse Figs. 5 and 6, ACD/LogP for Reverse Figs. 7 and 8, Polarizability for Reverse Figs. 9 and 10 and Enthalpy of Vaporization for Reverse Figs. 11 and 12 been evaluated. Thus, lowest MSE, MAE and RMSE, in addition to the highest \(R^2\) shown Metrics for Reverse indices of Neural Network with Table 7 depicted it has high predictive reliability and most accurate. On the other hand, Out of all models used in this work, Reduce Reverse ANNs demonstrated especially high performance in predicting the properties, including Molar Refractivity for Reduce Reverse as well as ACD/LogD for Reduce Reverse. The positive signs from preceding glimpses demonstrate its highest capability for which proofs are in the acts of visual comparisons illustrating its predictive accuracy and consistency from one evaluation to another Metrics for Reduce Reverse indices of Neural Network in Table 7. For Density of Reverse , Surface Tension of Reverse, the most accurate prediction was done by using Reverse RF Table 8. In other words, although every one of the algorithms presented has its benefits and pitfalls, ANNs demonstrated the highest level of success for the purpose of employing Reverse and Reduce Reverse topological indices in order to predict the properties of the examined antimalaria drugs. This finding does not only indicate that neural networks can be implemented in pharmaceutical research but also demonstrate that suitable machine learning approaches should be chosen according to the characteristics and needs of chemical datasets used for exploration. Compared with previous studies48, linear regression showed additionally a moderate predictive accuracy, but applying more complex algorithms such as ANN and RF increased the accuracy of the predictions. This shows that out of the three grouping techniques and considering the high non-linearity in the present data, complex models like ANN and RF give improved general predictive accuracy.
Data availability
All data generated or analysed during this study are included in this published article.
References
Balamurugan, M., Muthuraman, M. S., Porkodi, V., Sivaram, M., Sridharan, M., and Gangadevi, E. Combining Graph Theory and Machine Learning for Network-Based Medical Image Analysis. In 2024 International Conference on Trends in Quantum Computing and Emerging Business Technologies 1–5 (IEEE, 2024).
Diestel, R. Graph theory. Springer (print edition) Reinhard Diestel (eBooks) (2024).
Ahmed, W. et al. Exploring the role of topological descriptors to predict physicochemical properties of anti-HIV drugs by using supervised machine learning algorithms. BMC Chem. 18(1), 1–22 (2024).
Khan, A. et al. Computational and topological properties of neural networks by means of graph-theoretic parameters. Alex. Eng. J. 66, 957–977 (2023).
Gaudelet, T. et al. Utilizing graph machine learning within drug discovery and development. Brief. Bioinform. 22(6), bbab159 (2021).
Wu, Z., Shangguan, D., Huang, Q. & Wang, Y. K. Drug metabolism and transport mediated the hepatotoxicity of Pleuropterus multiflorus root: A review. Drug Metab. Rev. 56(4), 349–358 (2024).
Mahesh, B. Machine learning algorithms—a review. Int. J. Sci. Res. (IJSR) 9(1), 381–386 (2020).
Pei, J. et al. Quercetin-functionalized nanomaterials: Innovative therapeutic avenues for Alzheimer’s disease management. Ageing Res. Rev. 104, 102665 (2025).
Zhang, Q. Y. et al. Multi targeted therapy for Alzheimer’s disease by guanidinium-modified calixarene and cyclodextrin co-assembly loaded with insulin. ACS Nano 18(48), 33032–33041 (2024).
Zhou, Y. et al. Dermatophagoides pteronyssinus allergen Der p 22: Cloning, expression, IgE-binding in asthmatic children, and immunogenicity. Pediatr. Allergy Immunol. 33(8), e13835 (2022).
Wang, L. et al. An investigation into the correlation between intraperitoneal teicoplanin concentrations and treatment outcomes in peritoneal dialysis-associated peritonitis. Front. Pharmacol. 15, 1446774 (2024).
Ahmed, W., Ali, K., Zaman, S. & Agama, F. T. A python based algorithmic approach to optimize sulfonamide drugs via mathematical modeling. Sci. Rep. 14(1), 12264 (2024).
Ahmed, W., Zaman, S. & Ashraf, S. Topological characterisation of three classes of complex networks and their graphical representation and analysis. J. Micromech. Mol. Phys. 09(02n03), 77–89 (2024).
Arockiaraj, M., Paul, D., Rahul, M. P., Clement, J., Tigga, S., Balasubramanian, K. Topological and Entropy Indices in Qspr Studies of N-Carbophene Covalent Organic Frameworks. Available at SSRN 4790155.
Ahmed, W., Zaman, S. & Ashraf, S. Topological characterisation of three classes of complex networks and their graphical representation and analysis. J. Micromech. Mol. Phys. 09(02n03), 77–89 (2024).
Hayat, S. & Imran, M. On topological properties of nanocones CNCk [n]. Stud. Univ. Babes-Bolyai, Chem. 59(4), 113–128 (2014).
Hayat, S. & Asmat, F. Sharp bounds on the generalized multiplicative first Zagreb index of graphs with application to QSPR modeling. Mathematics 11(10), 2245 (2023).
Ahmed, W., Ali, K., Zaman, S. & Raza, A. Molecular insights into anti-Alzheimer’s drugs through predictive modeling using linear regression and QSPR analysis. Mod. Phys. Lett. B 38(27), 2450260 (2024).
Arockiaraj, M., Jeni Godlin, J. J., Radha, S., Aziz, T. & Al-Harbi, M. Comparative study of degree, neighborhood and reverse degree based indices for drugs used in lung cancer treatment through QSPR analysis. Sci. Rep. 15(1), 3639 (2025).
Ahmed, W., Ali, K., Zaman, S., Ahmad, F. & Ashebo, M. A. Molecular insights into anti-Alzheimer’s drugs through eccentricity-based predictive mathematical modeling using regression and QSPR analysis. J. Mol. Eng. Mater. 13, 1 (2024).
Zaman, S., Yaqoob, H., Ullah, A. & Sheikh, M. QSPR analysis of some novel drugs used in blood cancer treatment via degree based topological indices and regression models. Polycyclic Aromat. Compd. 44(4), 2458–2474 (2024).
Shanmukha, M. C., Basavarajappa, N. S., Shilpa, K. C. & Usha, A. Degree-based topological indices on anticancer drugs with QSPR analysis. Heliyon 6(6), e04235 (2020).
Katritzky, A. R. & Gordeeva, E. V. Traditional topological indexes vs electronic, geometrical, and combined molecular descriptors in QSAR/QSPR research. J. Chem. Inf. Comput. Sci. 33(6), 835–857 (1993).
Kulli, V. R. Reverse Zagreb and reverse hyper-Zagreb indices and their polynomials of rhombus silicate networks. Ann. Pure Appl. Math. 16(1), 47–51 (2018).
Liu, J. B. & Singaraj, R. M. Topological analysis of para-line graph of Remdesivir used in the prevention of corona virus. Int. J. Quantum Chem. 121(22), e26778 (2021).
Poojary, P. et al. Reverse topological indices of some molecules in drugs used in the treatment of H1N1. Biointerface Res. Appl. Chem 13(1), 71 (2022).
Arockiaraj, M., Greeni, A. B., Kalaam, A. A., Aziz, T. & Alharbi, M. Mathematical modeling for prediction of physicochemical characteristics of cardiovascular drugs via modified reverse degree topological indices. Eur. Phys. J. E 47(8), 53 (2024).
Naeem, M., Rauf, A., Akhtar, M. S. & Iqbal, Z. QSPR modeling with curvilinear regression on the reverse entropy indices for the prediction of physicochemical properties of benzene derivatives. Polycyclic Aromat. Compd. 44(3), 1452–1469 (2024).
Jayanna, G. K. A study of reverse topological indices and their importance in chemical sciences. Appl. Math. E-Notes 23(1), 175–186 (2023).
Gao, W., Younas, M., Farooq, A., Virk, A. & Nazeer, W. Some reverse degree-based topological indices and polynomials of dendrimers. Mathematics 6(10), 214 (2018).
Koam, A. N., Ahmad, A. & Qummer, A. A. On the Study of Reverse Degree-Based Topological Properties for the Third Type of p th Chain Hex-Derived Network. J. Math. 2021(1), 4540276 (2021).
Koam, A. N., Ahmad, A., Ahmad, Y. & Ahmad, Y. Computation of reverse degree-based topological indices of hex-derived networks. AIMS Math. 6(10), 11330–11345 (2021).
Jayanna, G. K. & Swamy, N. N. Multiplicative reverse geometric-arithmetic indices and arithmetic-geometric of silicate network. Biointerface Res. Appl. Chem. 12(3), 4192–4199 (2021).
Koam, A. N., Ansari, M. A., Haider, A., Ahmad, A. & Azeem, M. Topological properties of reverse-degree-based indices for sodalite materials network. Arab. J. Chem. 15(10), 104160 (2022).
Ahmad, A., Koam, A. N. & Azeem, M. Reverse-degree-based topological indices of fullerene cage networks. Mol. Phys. 121(14), e2212533 (2023).
Ravi, V., Siddiqui, M. K., Chidambaram, N. & Desikan, K. On topological descriptors and curvilinear regression analysis of antiviral drugs used in COVID-19 treatment. Polycyclic Aromat. Compd. 42(10), 6932–6945 (2022).
Gutman, I. & Polansky, O. E. Mathematical concepts in organic chemistry (Springer Science and Business Media, 2012).
Martınez-Martınez, C. T., Mendez-Bermudez, J. A., Rodrıguez, J. M. & Sigarreta, J. M. Computational and analytical studies of the harmonic index in Erdös-Rényi models. MATCH Commun. Math. Comput. Chem 85, 395–426 (2021).
Furtula, B. & Gutman, I. A forgotten topological index. J. Math. Chem. 53(4), 1184–1190 (2015).
Zhao, W., Shanmukha, M. C., Usha, A., Farahani, M. R. & Shilpa, K. C. Computing SS index of certain dendrimers. J. Math. 2021, 1–14 (2021).
Estrada, E., Torres, L., Rodriguez, L. & Gutman, I. An atom-bond connectivity index: Modeling the enthalpy of formation of alkanes. Indian J. Chem. 37A, 849–855 (1998).
Gao, W., Wang, W. F., Jamil, M. K., Farooq, R. & Farahani, M. R. Generalized atom-bond connectivity analysis of several chemical molecular graphs. Bul. Chem. Commun. 48(3), 543–549 (2016).
Randic, M. Characterization of molecular branching. J. Am. Chem. Soc. 97(23), 6609–6615 (1975).
Ali, A., Furtula, B., Redžepović, I. & Gutman, I. Atom-bond sum-connectivity index. J. Math. Chem. 60(10), 2081–2093 (2022).
Vujošević, S., Popivoda, G., Vukićević, ŽK., Furtula, B. & Škrekovski, R. Arithmetic-geometric index and its relations with geometric-arithmetic index. Appl. Math. Comput. 391, 125706 (2021).
Gutman, I. On hyper-Zagreb index and coindex. Bulletin (Académie serbe des sciences et des arts. Classe des sciences mathématiques et naturelles. Sciences mathématiques). 42, 1–8 (2017).
Ranjini, P. S., Lokesha, V. & Usha, A. Relation between phenylene and hexagonal squeeze using harmonic index. Int. J. Graph Theory 1(4), 116–121 (2013).
Awan, N. et al. QSPR analysis for physiochemical properties of new potential antimalarial compounds involving topological indices. Int. J. Quantum Chem. 124(11), e27391 (2024).
Acknowledgements
The authors extend their appreciation to Taif University, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-94).
Funding
This research was funded by Taif University, Saudi Arabia, Project No. (TU-DSPP-2024-94).
Author information
Authors and Affiliations
Contributions
W.A. led the research, including conceptualization, methodology, formal analysis, and drafting the original manuscript. T.A. contributed to data curation, visualization, and the review and editing of the manuscript. K.A. and E.E.M. was responsible for validation, resource acquisition, and manuscript revisions. M.T.S. and M.B.B. developed the software tools and conducted investigations. Shahid Zaman provided supervision and contributed to the critical review and final editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
The manuscript has been approved by all authors and consent for publication has been granted
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmed, W., Ashraf, T., Saleem, M.T. et al. Computational approaches in drug chemistry leveraging python powered QSPR study of antimalaria compounds by using artificial neural networks. Sci Rep 15, 19307 (2025). https://doi.org/10.1038/s41598-025-01594-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01594-y
Keywords
This article is cited by
-
A Soft Computing Strategy Combining Fuzzy Analytic Hierarchy and Fuzzy Artificial Neural Networks for Predictive Modeling and Therapeutic Ranking in Bowel Cancer Drug Development
International Journal of Computational Intelligence Systems (2026)
-
A hybrid computational framework for antidepressant drug design integrating machine learning algorithms and molecular modeling
Chemical Papers (2025)
-
Topological and statistical regression study of chemical structures using graph-theoretic descriptors: applications to cancer therapeutics
Chemical Papers (2025)
