
Abstract
Colorectal cancer (CRC) presents significant challenges due to limited targeted therapeutic options. This study integrates multi-omics analysis and AI to identify tumor antigens and immune gene targets for personalized immunotherapy. Using TCGA, differential expression and mutation analysis, we identified overexpressed and mutated genes in CRC. Among these, 62 neoantigens were shortlisted as potential tumor antigens. Survival analysis highlighted prognostic antigens, while their correlation with immune gene expression suggested these antigens could trigger immune activation. Three key neoantigens (TTK, EZH2, and KIF4A) emerged as promising candidates for immunotherapy. Based on immune gene activity, patients were categorized into three Immune Subtypes (IS). IS groups 1 and 2, characterized by high immune gene expression and immune activation markers, exhibited better survival outcomes, while IS 3, with low immune gene expression, showed poor survival and immune unresponsiveness. Neoantigen-based vaccines could potentially boost tumor recognition and improve survival for patients in immune-cold subtypes. Machine learning models like LightGBM, XGBoost, and XGBRF predicted optimal immune targets for vaccine design, validated through SHAP analysis. This study provides a machine learning- driven framework to identify tumor antigens and immune targets, offering a promising strategy for CRC immunotherapy tailored to immune subtype-specific responses.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) accounts for approximately 8.5% of cancer-related deaths and is one of the most prevalent and lethal malignant type of cancer worldwide. It is the second leading cause of cancer-related mortality globally. Standard treatment options for CRC include surgical tumor removal, often combined with radiation therapy and chemotherapy, which remains the most traditional approach for managing tumor recurrence and metastasis1. However, due to its major side effects, there is a pressing need for other therapies like targeted and immunotherapies. Studies have shown as part of immune checkpoint blockade (ICB), Pembrolizumab and nivolumab drugs that target programmed death 1 (PD-1) have 15% survival rates in mismatch repair-deficient (dMMR), also called microsatellite instability-high (MSI-H) types of CRC, whereas the other 85% accounts for mismatch repair-proficient (pMMR) or microsatellite-stable (MSS) types of CRC, where they show the worst survival rates2,3.
Cancer vaccines have emerged as the most promising immunotherapy option to tackle this highly fatal and prevalent malignancy. Unlike other traditional therapies, this method utilizes the immune system to combat cancer4. Many clinical trials targeting cancer vaccines have also shown promising results. Sipuleucel-T is the first FDA-approved cancer vaccine designed to treat prostate cancer. Over the past decade, it has demonstrated very promising results by increasing the overall progression rate of metastatic cancer5, and research studies have shifted to using whole-cell, DNA, mRNA, and protein-based platforms for vaccine design6. Clinical studies on mRNA-4157 and mRNA-5671 showed improved overall survival (OS) results in CRC patients, along with the mRNA vaccine, OncoVax, and GVAX, which are whole-cell and dendritic cell (DC) vaccines, also showed an overall immune response rate7.
Many immune subtyping analyses on cancer patients, where they are grouped patients according to immune gene expression levels, have revealed that those with higher immune gene expression tend to have better survival rates, while those with low immune gene expression have worse survival outcomes, which explains the role of immune genes in overall survival rates8, The primary reason for poor survival in patients with low immune gene expression is immune escape, where cancer cells either downregulate or completely suppress tumor antigen expression to evade immune surveillance, exposer of these tumor antigens to the immune system can be increased with the use of epitopes that interact with antigen-presenting cells (APC), and T cells, and thereby promoting the immune activation9.
Many research studies are targeting to incorporate epitopes from both tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) for anti-tumor immunity, and formulating a vaccine with this strategy can promote a robust immune response. Moreover, this approach is highly beneficial for personalized vaccines, as it allows the incorporation of patient-specific tumor antigens. However, identifying the key immune genes to target is a crucial aspect of vaccine design10. This challenge can be achieved using survival analysis-driven weighted gene network analysis (WGCNA), where all the similar correlating genes will be clustered in each module11,12. However, identifying the best hub gene is challenging, which can be aided with the use of machine learning and explainable AI techniques like SHapley Additive exPlanations (SHAP). The integration of WGCNA with ML-based feature selection enhances the precision of immune target identification, facilitating the development of highly specific cancer vaccines13,14,15.
The present study aims to integrate multi-omics to identify the best neo-antigens with a very high survival rate and best correlating with the immune gene following the tumor antigen curation. Immune subtyping has been done to explore the immune activity in different subgroups of CRC to identify the group beneficial from vaccination. Additionally, hub gene identification was conducted using Supervised machine learning algorithms such as Light Gradient Boosting Machine (LightGBM), Extreme Gradient Boosting (XGBoost), and XGBoost Random Forest (XGBRF) combined with an explainable AI SHAP algorithm. The detailed workflow used for this study is provided in Fig. 1

A detailed workflow illustrating all the steps of this study.
Results
Tumor antigen screening with integrative over-expressed and mutated genes
Using the limma library from R, 573 tumor samples were compared with the 54 normal samples. This analysis revealed a significantly higher number of downregulated genes compared to upregulated genes, with 12,273 upregulated genes and 37,013 downregulated genes. The Volcano plot from Fig. 2A illustrates the ratio of upregulated and downregulated genes and the number of log fold changes between overexpressed and under-expressed genes. After considering p-value < 0.0005 and fold change value greater than one, a total of 693 genes remained for further analysis. In mutation analysis from the 573 MAF files, it was observed that out of 573 samples, mutations were observed in 541 samples, and the APC gene, which is a tumor suppressor gene, was mutated in 73% of samples, and this may be the reason for uncontrolled cancer growth. The top 20 mutations varied from 12 to 73% of samples, and the most frequent mutation was observed as a missense mutation and a frame insertion mutation. Oncoplot of 573 samples is represented in Fig. 2B. For integrative analysis, genes that were mutated in a minimum of 15 samples were considered, and a total of 3287 genes were observed to have mutated in the minimum of 15 samples, after integrating the overexpressed with the mutated genes, 62 sharing genes were observed as represented in the Venn diagram from Fig. 2C, this integration is done to make sure the mutated gene are still overexpressed in tumor samples which makes them a neo-antigens specific to cancer, complete list of all genes used to screen tumor antigens can be found in supplementary Table 1 of spreadsheet 1.

Tumor antigen screening from over-expressed genes and mutated genes, (A) Volcano plot where red color dots are the over-expressed, blue dots represent under-expressed genes, and block dots are the not significant genes, (B) Oncoplot of CRC samples, (C) Venn diagram showing the Interesting genes from over-expressed and mutated genes.
Overall survival rate analysis and correlation of tumor antigen with the immune genes
For the selected 62 tumor antigens, patients were grouped into high-expression and low-expression samples based on expression counts. Using the Cox regression model, the hazard ratio (HR) value of these 62 tumor antigens was calculated, and out of them, 12 genes showed an HR value less than one with p value less the 0.05, HR value less than 1 indicates the best survival rates with fewer recurrence rates, while HR value greater than one represents the worst progress feature with high recurrence rates, forest plot of all 62 genes have been shown in Fig. 3A and HR scores with their p value for all 62 genes are presented in supplementary Table 2 of spreadsheet 2, Kaplan–Meier (KM) plot was plotted for these 12 genes, it was observed that the high-expression group exhibited significantly better survival compared to the low-expression group, KM plots of all 12 genes with their HR values have been represented in Fig. 3B and considered for further correlation analysis.

Survival and correlation analysis of tumor antigens, (A) Forest plot of all 62 interesting tumor antigens, each red dot in the plot represents HR value, (B) KM plot of 12 tumor antigens with low HR values, (C) Correlation of 9 tumor antigens with immune cells.
To compare the correlation of these 12 best survival tumor antigens with APCs, we have correlated these 12 tumor antigens with the APCs cells using Spearman’s correlation method in the TIMER 2.0 web tool. From 12 genes, it was observed that 3 genes were not listed in the database. Out of nine tumor antigens, 3 tumor antigens (TTK, EZH2, and KIF4A) showed a positive correlation with the TIMER these genes were considered based on the number of positive correlations with Tumor Immune Estimation Resource (TIMER), Estimating the Proportion of Immune and Cancer cells (EPIC), Microenvironment Cell Populations counter (MCP-counts), Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT), Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts Absolute Mode (CIBERSORT-ABS), Quantification of the Tumor Immune Contexture (QUANTSAQ), and XCELL databases with Colon Adenocarcinoma (COAD) project from TCGA. The correlation of 9 genes in these databases is illustrated in Fig. 3C.
Immune subtyping (IS) of CRC patients
Using the Consensus-Cluster-Plus library from R, we have clustered the CRC patients into nine clusters, The Consensus-Cluster algorithm works in an unsupervised clustering manner, where its core algorithm was built to subsample the data matrix based on p-Item and p-Feature parameters, for this analysis bootstrap was set to 500, with 80% of the sample underwent for resampling and max k value was set to 9. Till k value 3 sample was uniformly distributed, and from k value 4–9, an uneven distribution of samples was found. The optimal k value was considered based on the Consensus Delta Area (CDA) and the Consensus Cumulative Distribution Function (CDF). The delta area curve with a large value indicates a significant improvement in cluster stability as new clusters are added, while a smaller Delta Area suggests no improvement despite the addition of new clusters. For our dataset, the optimal k-value was determined to be 3, as the CDF plot also demonstrated stability at k-value 3 (Fig. 4A,B). Thus, we selected k value 3 as the best clustering K value, leading to the identification of 3 Immune Subtypes (IS). The consensus matrix of k value 3 is illustrated in Fig. 4C, the consensus matrix of k value from 4 to 9 clusters is illustrated in Supplementary Fig. 1A–F. The distribution of samples across these subtypes is as follows, IS1 had 167 samples, IS2 had 182 samples, and IS3 had the highest number of samples, with 224.

Immune subtyping of 573 CRC samples, (A) Delta area of all K values, (B) CDF plot of all K values, (C) Consensus matrix from k value 3.
Mutational landscape and overall survival rate analysis of immune subtypes
A High mutational burden in tumor samples represents a higher chance of survival rate. To analyze the mutational burden in each IS, an oncoplot was generated for each immune subtype to examine the mutational landscape. APC and TP53 mutations, which are associated with tumor suppression, were observed at high frequencies across all samples, with mutation percentages ranging from 57 to 79%. The majority of mutations were missense mutations. The tumor mutation burden (TMB) per Megabase(Mb) was calculated for each IS. In IS3 mean TMB of 6.79 was observed, which is very low compared to other groups. In IS1, the highest observed mutation count was 200, with 10 samples showing 50 mutations, leading to a mean TMB of 7.16. IS2 exhibited the highest mutation frequency, with a mean TMB of 12.66. Based on this analysis. IS1 and IS2 had a higher tumor mutation burden compared to IS3, which had the lowest TMB, oncoplot and its mutation burden are illustrated in Fig. 5A–F.

Mutational landscape and survival analysis of three IS groups, (A–B) Oncoplot and mutation burden of IS 1, (C–D) Oncoplot and mutation burden of IS 2, (E–F) Oncoplot and mutation burden of IS 3, (G) KM plot of all three IS.
An OS analysis was conducted for each IS groups to determine which subtype has the highest chances of survival, based on the number of death cases and the number of days patients remained alive. It was observed that IS3 had the highest number of death cases, with a survival rate of 73.2%, indicating the worst prognosis. IS2 showed a survival rate of 79.7%, while IS1 had the best survival rate at 83.8%, making it the most favorable immune subtype. In the KM plot, IS1 displayed a higher and more constant survival rate throughout the timeline compared to the other two IS groups. On the other hand, IS3 had a faster drop in the curve, indicating a quicker decline in survival, with each drop corresponding to death. IS2 represented an intermediate survival profile. The combined KM plot of each IS group is illustrated in Fig. 5G.
The molecular landscape of immune subtype
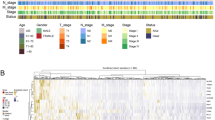
Expression levels of immune genes in tumor samples signify their immune status, and patients who are rich in immune gene expression levels have higher survival rates. To know the immune landscape of different IS, we have generated enrichment scores [ES] of 11 immune pathways in each IS. These scores were generated from the Gene Set Enrichment Analysis (GSEA) database, annotation was done using a C5 onco file from the GSEA database. Immune pathways across these IS were significantly differentially expressed. Activation of immune response and adaptive immune response, B cell-mediated immunity, humoral immune response, and humoral immune response mediated by circulating immunoglobulin were more significantly expressed at high levels in IS1 and IS2 when compared to IS3. In antigen processing and presentation of endogenous antigens, peptide antigens, and exogenous antigens, no significant differences were observed among three IS groups. Granulocyte activation and T cell-mediated cytotoxicity pathways were high in IS2 and IS3 compared to IS. Individual enrichment scores of each pathway of all samples in each IS can be found in supplementary Table 3 of spreadsheet 3. This revealed that overall, immune IS3 enrichment scores were very low when compared to IS1 and IS2, which indicates that IS3 belongs to an immune cold group (Fig. 6A).

Molecular landscape of three IS groups, (A) Heatmap of 11 immune pathways in three IS groups, (B) Heatmap of 22 immune gene scores in three IS groups, (C–E) Box plot of stromal, immune, and estimate scores of three IS groups, (F) Box plot of ICP genes in three IS groups, (G) Box plot of ICD genes in three IS groups.
Additionally, the CIBERSORT result was also similar to the GSEA immune pathway result, where a total of 22 immune-related gene expressions were compared between each IS. IS3 expression scores, especially in B cell memory, plasma cells, CD8, CD8 naive T cells, CD4 memory active, T cell-based delta, NK cells, monocytes, macrophages, dendritic active cells, and mast cell active cells, were very low compared to IS1 and IS2 groups. However, high expression was observed in B cell naive, T cell CD4 memory resting, T cell follicular, T cell follicular delta, macrophage M0, dendritic cells resting, eosinophils, and neutrophils of IS3 (Fig. 6B).
Furthermore, an estimate analysis was conducted to analyze stromal score, immune score, and estimate scores. Stromal scores help assess fibrotic and non-cancerous tissue in the tumor microenvironment, and the immune score indicates the levels of immune cell infiltration in the tumor. A high immune score is linked to better progress. Similarly, the estimate analysis integrates the stromal and immune scores to estimate tumor purity (lower purity = more non-cancerous cells). From our analysis, IS2 has low stromal, immune, and estimate scores. IS1 and IS2 have almost similar stromal scores, but the immune score and estimate scores in IS1 were high compared to IS3. This analysis revealed that IS2, with a low stromal score, has non-cancerous cells in its microenvironment. For IS3, although its stromal scores are similar to IS1, IS3 has a low immune score. Based on immune pathway analysis, CIBERSORT, and estimate analysis, it was observed that IS3 had very low immune cells. This is due to the underexposure of tumor antigens to the immune cells, which can be addressed by mRNA vaccination. mRNA vaccines help expose neoantigens externally, thereby increasing the immune response (Fig. 6C is IS1, Fig. 6D is IS2, Fig. 6E is IS3).
Apart from immune gene analysis in IS groups, expression levels of 24 immune checkpoint genes and 11 immune cell death genes in all IS groups were analyzed. Patients with high expression of Immune checkpoint genes (ICP) and immune cell death genes (ICD) are associated with better progress. For this analysis, since the expression levels vary significantly between genes, we applied a log10 scale to represent them in the box plot. IS1 and IS2 have very high expression levels compared to IS3 (Fig. 6F). Similar results were observed for ICD gene expression levels as well, gene levels were low in IS3 compared to IS1 and IS2 (Fig. 6G). From this analysis, it is clear that IS3 is best suited for vaccination since both ICP and ICD levels are low.
WGCNA analysis for correlation gene module generation and pathway analysis
From molecular and survival analysis, it is clear that patients from the IS3 group are best suited for vaccination. To select the best target immune gene that targets the vaccine to elicit an immune response, we clustered highly correlated immune genes. After running soft power from 1 to 20, soft power 5 achieved an R2 value of 0.9, indicating that the network follows a scale-free topology well. Additionally, mean connectivity helps assess how densely connected the network is at different soft-thresholding power values. At soft power 5, the mean connectivity is significantly reduced compared to lower power values, indicating that weak connections have been filtered out. This ensures that the network remains biologically meaningful by grouping genes into modules based on stronger co-expression relationships. Plots of soft-threshold and mean connectivity are illustrated in Fig. 7A.

WGCNA analysis to identify the best module, (A) Scale-free topology and mean connectivity to determine the best K value, (B) Dendrogram of clusters, (C) Bar plot of 17 different models identified and its gene count in each module, (D) Module-trail relationship between modules and IS group, (E) Forest plot of survival HR values of each module, (F) Box plot of low HR value modules with their KM plot, (G) Box plot of high HR value modules with its KM plots, (H) KEGG, molecular function and biological process of Turquoise module (I) KEGG, molecular function and biological process of brown module (J) KEGG, molecular function and biological process of Blue Module.
By keeping a minimum of 30 genes in each module, a dendrogram was constructed with a deep split of 3 and a height of 0.25. Module eigengenes (MEs) were calculated (Fig. 7B), and a total of 17 modules were obtained. A bar plot of the 17 modules and their gene counts is illustrated in Fig. 7C, the gene list in each module is tabulated in supplementary Table 4 of spreadsheet 4. When ME scores in each IS were compared, it was observed that IS3 had very low scores compared to the other two IS groups. The module-trait relationship also revealed that all modules show a positive correlation with IS1 and IS2 and a negative correlation with IS3 (Fig. 7D). Overall survival analysis for these 17 modules revealed that the best HR values were observed in the turquoise, brown, blue, light cyan, pink, salmon, magenta, yellow, midnight blue, black, and gray modules (Fig. 7E), ME scores in each IS, along with their Kaplan–Meier (K–M) plot for best HR value modules are illustrated in Fig. 7E and module with poor HR values in Fig. 7F. For the top three modules (turquoise, brown, and blue), KEGG, biological process, and molecular function analyses were performed. It was observed that the turquoise module was rich in immune-related pathways (Fig. 7H–J).
Machine learning and explainable AI for target gene identification for vaccines
For predicting the target genes that can recognize the vaccine with tumor antigens, we have selected the turquoise module along with its clinical data and gene expression counts. The count matrix was structured with the first column containing sample IDs, each subsequent column (except the last) representing gene expression counts, and the last column indicating the vital status of the patient (alive or deceased). Using Select-K-Best with ANOVA F-score and mutual information methods, we selected 20 features responsible for the alive status of patients. These selected features were then used to train the XGBoost, LightGBM, and XGBRF models. The performance of these models was evaluated using AUC, where LightGBM achieved the highest accuracy with an AUC of 93%, followed by XGBoost with 92%, and XGBRF with the lowest AUC at 86%.
Using SHAP AI, the top 5 hub genes were identified based on SHAP importance scores. These genes include IGLV7-43, IGKV2D-28, IGLV2-8, IGHV3-35, IGV3-74, and IGHV3-7. The confusion matrix, ROC curve, 20 selected features, and SHAP interpretation results are illustrated in Fig. 8A–D. These hub genes represent the best immune targets for vaccine development.

Machine learning and explainable AI for target identification, (A) Confusion matrix of Light GBM, XGB and XGBRF, (B) Features identified to train models of Light GBM, XGB and XGBRF (C) ROC curve of Light GBM, XGB and XGBRF, (D) SHAP result of Light GBM, XGB and XGBRF.
Discussion
CRC is considered the world’s third most diagnosed cancer, with the second-highest cancer-linked death rate. Despite advances in conventional therapies, the prognosis for advanced CRC remains poor, with very high rates of recurrence and metastasis. Additionally, due to the molecular heterogeneity in CRC patients, treatments are more associated with significant side effects and are not equally effective across all patients1. This explains the urgent need for advanced therapeutic strategies that can offer more personalized and effective treatment options. Immunotherapy, particularly cancer vaccines, has emerged as a promising alternative to traditional therapies. This approach is often associated with long-lasting immunity, fewer side effects, and the ability to target multiple tumor antigens simultaneously16. Although tumor antigens arise from our body, they are produced by cancer cells. The proteins that are produced by cancer are very specific to them and are not present in normal cells. Our immune cells recognize them as non-self and induce an immune response against these tumor antigens, which are raised from the mutations in normal genes. From our integrative differential and mutational analysis17, three tumor antigens, TTK, EZH2, and KIF4A, are identified, patients with high expression of these tumor antigens have better survival rates compared with the patients with low expression levels of these antigens, which interprets these tumor antigens are linked with the progress of patients18.
APCs play a pivotal role in both adaptive and innate immune responses. When they detect non-self-proteins, they process their components and initiate a specific immune response by activating T cells. Their ability to bridge innate immunity (rapid, non-specific defense) with adaptive immunity (targeted, memory-based defense) makes them essential for a well-coordinated immune response19,20. When the correlation of identified tumor antigens with macrophages, B cells, and dendritic cells was analyzed, a strong correlation was observed among them. This analysis revealed that these tumor antigens are actively recognized by APCs, which may explain the better prognosis in patients with overexpressed tumor antigen genes21. Designing an mRNA vaccine incorporating epitopes from these tumor antigens could elicit a strong and targeted immune response22.
Studies have shown that cancer patients with a high abundance of immune genes tend to have better prognoses due to the active recognition of tumor antigens by immune cells. However, patients whose tumor antigens are not exposed to immune cells have worse outcomes23. Vaccination with these antigen epitopes can externally present the antigens to immune cells, thereby activating them and enhancing the immune response in CRC patients24. To identify the patient group most likely to benefit from vaccination with these tumor antigens, we classified patients into three distinct groups based on the expression profile of immune genes. It was observed that the OS rates of IS3 were the worst compared to the other IS groups. Additionally, the mutational burden was generally lower in this group. Patients with a high mutation burden tend to have higher neoantigen expression, thereby encouraging active recognition of these neoantigens by immune cells25. The lower expression of neoantigens in IS3 may contribute to its poor survival rates. Furthermore, immune pathway activity and immune cell scores were lower in IS3, likely due to the limited exposure of neoantigens to immune cells26. The tumor microenvironment in IS3 contained a higher proportion of cancer cells. Additionally, patients with high expression of immune checkpoint genes and immune cell death-related genes tended to have better survival rates, whereas IS3 patients exhibited lower expression levels of these genes27,28. Based on this analysis, IS3 appears to be the most suitable group for vaccination, as vaccination could enhance the active immune response and improve patient outcomes in this subgroup. Additionally, to identify potential vaccine targets, we grouped the correlating immune gene sets using WGCNA analysis. For WGCNA analysis, we have included immune genes from all samples for correlation analysis, which is independent of the predetermined IS group, the inclusion of all samples could facilitate the comparative analysis of gene modules survival rates in each IS. Our WGCNA analysis resulted in approximately 17 modules, each containing correlated immune genes. After identifying the correlation between gene modules and immune subtypes, we further analyzed the overall survival (OS) of genes within each module to assess whether they were associated with high or low survival rates. The analysis revealed that the blue, brown, and turquoise modules were the most significant, with low hazard ratios and strong survival associations. In all those modules, the IS 3 group had very less counts compared to other groups, and29. This finding was important for selecting immune genes that might be most effective in targeting specific patient groups for vaccination.
Following the WGCNA analysis, we performed functional enrichment analysis, including Gene Ontology (GO) terms for biological processes (BP), molecular functions (MF), and KEGG pathway analysis. Among the 3 modules, the turquoise module exhibited the highest enrichment of immune-related pathways compared to the others. This module was significantly associated with key immune processes such as antigen processing and presentation, T cell receptor signaling, natural killer cell mediated cytotoxicity, cytokine -cytokine signaling pathways, Additionally, biological pathway analysis revealed a strong representation of immune-associated pathways, positive regulation of cytokine production, natural killer cell mediated immunity, regulation of lymphocyte and leukocyte mediated immunity further underscoring its relevance to the immune landscape. In contrast, other modules showed enrichment for pathways unrelated to immune activity, such as metabolic or structural processes, reinforcing the selection of the turquoise module as the most relevant for immune-related investigations. Given its predominant association with immune response mechanisms, this module was prioritized for further analysis to identify key immune hub genes and their potential implications in cancer immunotherapy. The turquoise module contained approximately 139 genes. We next performed hub gene identification within the turquoise module using machine-learning models, including LightGBM, XGBoost, and XGBRF. Almost all models identified similar top 20 genes, and their feature importance was explained using SHAP analysis, which provided insights into the contribution of each gene to the classification as a hub gene for the alive status of the patient. And this analysis revealed IGLV7-43, IGKV2D-28, IGLV2-8, IGHV3-35, IGV3-74, IGHV3-7 as targets for vaccinattion. All of these genes are related to immunoglobulin light and heavy variable-related genes, which are responsible for the active recognition of antigenic regions of non-self-proteins30,31,32,33.
Methods
Data retrieval and preprocessing
For this study, we have considered 573 CRC samples, which have both RNA seq data as well as mutation annotation files (MAF) data from TCGA. Each patient had a shared RNA-seq count file, MAF file, and linked clinical data. The clinical features considered for this study are tabulated in supplementary Table 5 in spreadsheet 5.
Tumor antigen screening with integrative overexpressed and mutated genes
Using the limma package34, differential gene analysis was performed on 573 CRC RNA-seq count files along with 54 normal RNA-seq count files. Similarly, for 573 MAF files, an oncoplot was generated using maftools35, and the list of mutated genes was saved in a CSV file for further screening. In the integration phase, overexpressed genes from the differential expression analysis were compared with the mutated gene list, and the shared genes were selected for downstream analysis.
Overall survival and correlation analysis of tumor antigens with immune genes
For the selected tumor antigen, overall survival analysis was carried out using the survival and survminer libraries in R36. Patients were classified into high-expression and low-expression groups based on tumor antigen expression levels. A Cox regression model37 was employed to calculate the hazard ratio (HR), and the best tumor antigen was further screened based on the highest survival rate. Tumor antigens with the best HR values were then screened to assess their correlation with immune genes. This correlation analysis was performed using the TIMER 2.0 web tool38, where associations were evaluated with B cells, dendritic cells, and macrophages. The tumor antigen showing the strongest correlation with immune cells was selected as a candidate tumor antigen.
Immune subtyping of CRC patients based on immune gene expression
Immune subtyping was performed to classify the patients of CRC according to their immune cell expression. For this analysis, only immune gene counts were considered, selecting 1,792 immune genes referenced from the ImmPort database. The corresponding expression levels were extracted to create an immune gene count matrix for subtyping. Immune subtyping was carried out using a clustering technique implemented in the Consensus-Cluster-Plus package in R39. The input was the expression levels of immune genes, the k-value was set to 9, and the best subtype was evaluated using the delta area plot and CDF plot.
Mutational landscape of immune subtypes
To analyze the mutational landscape of patients in different immune subtypes, the MAF files were merged with their corresponding immune subtype classifications, allowing for a comparative analysis of mutation landscapes across different immune subtypes. An oncoplot of different immune subtypes was generated to visualize their mutational landscape. Additionally, tumor mutation burden (TMB) was calculated for each tumor sample by determining the number of mutations per megabase (Mb) in each immune subtype, providing insights into the mutation load across different immune subtypes. Following the mutational landscape analysis, an overall survival analysis for each immune subtype was carried out to determine its survival rate40.
Immune landscape analysis of immune subtypes
After clustering the immune subtypes, their immune landscape was analyzed to characterize the immune profile of each subtype. To achieve this, the C5 ontology gene set was downloaded from the GSEA database41, which contains genes annotated by the same ontology terms. A total of 11 different immune pathways were considered to explore their activity in the different immune subtypes using enrichment scores. Similarly, using the CIBERSORT library, the expression levels of 22 different immune cells were analyzed in each immune subtype. Following this analysis42, the ESTIMATE library was used to explore the tumor stromal, immune, and estimate scores in each immune subtype43. Additionally, the expression levels of immune checkpoint genes and immune cell death genes were analyzed across different immune subtypes.
Grouping correlating immune genes using WGCNA and module selection
For WGCNA analysis, we first checked for missing values and filtered out low-quality genes to ensure data integrity. Genes with low variance were removed to enhance co-expression detection. The expression matrix was transposed to have genes as columns and samples as rows. To establish a scale-free network, we determined the optimal soft-thresholding power (β) by testing values between 1 and 20 using the pickSoftThreshold function, selecting the power where the scale-free topology model fit (R2) exceeded 0.85. Using this power, we computed the adjacency matrix to quantify gene co-expression relationships, then transformed it into a Topological Overlap Matrix (TOM) to incorporate network neighborhood information44. Hierarchical clustering was performed using TOM-based dissimilarity, and module detection was conducted using the blockwise modules approach, where the dynamic tree-cutting algorithm (cutreeDynamic) automatically identified gene modules with a minimum size of 30 genes. Modules with highly correlated eigengenes (correlation > 0.75) were merged using a threshold of 0.25. To identify biologically relevant modules, we calculated module eigengenes (MEs) and examined their correlation with immune subtypes. Significant module-trait relationships (p < 0.05) were visualized using heatmaps, and eigengene expression differences across immune subtypes were assessed using boxplots and scatter plots. Module sizes were visualized with bar plots.
Post-WGCNA analysis, survival analysis for all modules was performed to identify the module associated with the best survival outcome. For the best-performing module, KEGG pathway enrichment, biological process (BP), and molecular function (MF) analyses were conducted using ShinyGo45 to determine the presence of immune-related pathways, aiding in the identification of the most immunologically significant module.
Hub gene identification using machine learning and explainable AI
To identify key hub genes associated with patient survival, we employed machine learning models alongside the explainable AI algorithm SHAP. Feature selection was performed using the Select-K-Best method with ANOVA F-score and mutual information, ensuring that only the most relevant features were chosen. These selected features were then used to train XGBoost, LightGBM, and XGBRF models, chosen for their efficiency in handling high-dimensional genomic data, ability to capture complex feature interactions, and strong classification performance in biomedical applications. SHAP (SHapley Additive exPlanations) was applied to determine the most influential features for predicting patient survival. Unlike traditional feature importance methods, SHAP provides an interpretable and quantifiable measure of each feature’s contribution to the model’s predictions. By considering all possible feature combinations, SHAP ensures a fair distribution of feature influence, making it particularly valuable in biomedical research where understanding gene significance in survival prediction is crucial for targeted interventions such as vaccine development. Based on this analysis, the top five hub genes were identified as optimal targets for vaccine development46.
Conclusion
Colorectal cancer (CRC) remains a major global health challenge, with high mortality rates and limited treatment efficacy. Immunotherapy, particularly mRNA-based vaccines, offers a promising alternative by enhancing immune recognition of tumor antigens. This study identified TTK, EZH2, and KIF4A as key tumor-specific antigens linked to better survival. Immune subtyping revealed that IS3 had the worst prognosis with immune cold status, and can be immune hot upon vaccination, making it the most suitable group for vaccination. WGCNA analysis clustered immune-related genes into 17 modules, with the turquoise module being the most immune-enriched, and patients having these genes have better survival rates. Machine learning models identified IGLV7-43, IGKV2D-28, IGLV2-8, IGHV3-35, IGV3-74, and IGHV3-7 as top vaccine-recognizing targets from the turquoise module. These immunoglobulin-related genes play a key role in antigen recognition, making them ideal for immune response elicitors. This study provides a framework for personalized mRNA vaccines, particularly for immune-cold IS3 patients, offering a novel strategy to improve CRC treatment outcomes.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Abbreviations
- CRC:
-
Colorectal cancer
- ICB:
-
Immune checkpoint blockade
- PD-1:
-
Programmed death 1
- MSI-H:
-
Microsatellite instability-high
- MSS:
-
Microsatellite-stable
- APC:
-
Antigen-presenting cells
- TAAs:
-
Tumor-associated antigens
- TSAs:
-
Tumor-specific antigens
- WGCNA:
-
Weighted gene network analysis
- SHAP:
-
SHapley Additive exPlanations
- HR:
-
Hazard ratio
- K–M:
-
Kaplan–Meier
- TIMER:
-
Tumor immune estimation resource
- EPIC:
-
Estimating the proportion of immune and cancer cells
- MCP-counter:
-
Microenvironment cell populations counter
- CIBERSORT:
-
Cell-type identification by estimating relative subsets of RNA transcripts
- CIBERSORT-ABS:
-
Cell-type identification by estimating relative subsets of RNA transcripts absolute mode
- QUANTISEQ:
-
Quantification of the tumor immune contexture
- COAD:
-
Colon adenocarcinoma
- CDA:
-
Consensus delta area
- IS:
-
Immune subtypes
- TMB:
-
Tumor mutation burden
- OS:
-
Overall survival
- GSEA:
-
Gene set enrichment analysis
- ES:
-
Enrichment scores
- MEs:
-
Module eigengenes
- ANOVA:
-
Analysis of variance
- DC:
-
Dendritic cells
References
Arvelo, F., Sojo, F. & Cotte, C. Biology of colorectal cancer. Ecancermedicalscience https://doi.org/10.3332/ecancer.2015.520 (2015).
Adrianto, A. A., Riwanto, I., Sadhana, U., Paramita, D.K., Setyawan, H., Tjandra, K. C., Respati, D. R. P., Rampengan, D. D. C. H., Ramadhan, R. N., Jangkang, G. G. & Mahati, E. The efficacy and safety of pembrolizumab, Ipilimumab, and nivolumab monoteraphy and combination for colorectal cancer: A systematic review and meta-analysis. medRxiv, 2024–07 (2024).
Wang, C. et al. Regorafenib and nivolumab or pembrolizumab combination and circulating tumor DNA response assessment in refractory microsatellite stable colorectal cancer. Oncologist 25(8), e1188–e1194 (2020).
Sarvizadeh, M. et al. Vaccines for colorectal cancer: an update. J. Cell. Biochem. 120(6), 8815–8828 (2019).
Kantoff, P. W. et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363(5), 411–422 (2010).
Sobhani, N. et al. Therapeutic cancer vaccines: From biological mechanisms and engineering to ongoing clinical trials. Cancer Treat. Rev. 109, 102429 (2022).
Jia, W. et al. Colorectal cancer vaccines: The current scenario and future prospects. Front. Immunol. 13, 942235 (2022).
Perez Villamil, B. et al. Colon cancer molecular subtypes identified by expression profiling and associatedto stroma, mucinous type and different clinical behavior. BMC Cancer 12, 1–13 (2012).
Budhwani, M. et al. Immune-inhibitory gene expression is positively correlated with overall immune activity and predicts increased survival probability of cervical and head and neck cancer patients. Front. Mol. Biosci. 8, 622643 (2021).
Rejali, L. et al. Identification of antisense and sense RNAs of intracrine fibroblast growth factor components as novel biomarkers in colorectal cancer and in silico studies for drug and nanodrug repurposing. Environ. Res. 239, 117117 (2023).
Offringa, R., van der Burg, S. H., Ossendorp, F., Toes, R. E. & Melief, C. J. Design and evaluation of antigen-specific vaccination strategies against cancer. Curr. Opin. Immunol. 12(5), 576–582 (2000).
Tu, S., Zhang, H. & Qu, X. Screening of key methylation-driven genes CDO1 in breast cancer based on WGCNA. Cancer Biomark. 34(4), 571–582 (2022).
Norouzinasab, F. et al. Discovery of LINC01614 associated with the SPP1 gene in colorectal cancer. Pathol.-Res. Pract. 266, 155761 (2025).
Hasan, M. A. M., Maniruzzaman, M. & Shin, J. WGCNA and machine learning-based integrative bioinformatics analysis for identifying key genes of colorectal cancer. IEEE Access https://doi.org/10.1109/ACCESS.2024.3472688 (2024).
Iranmanesh, H. et al. The association of clinicopathological characterizations of colorectal cancer with membrane-bound mucins genes and LncRNAs. Pathol.-Res. Pract. 233, 153883 (2022).
Lynch, D. & Murphy, A. The emerging role of immunotherapy in colorectal cancer. Ann. Transl. Med. 4(16), 305 (2016).
Herberman, R. B. Immunogenicity of tumor antigens. Biochim. Biophys. Acta (BBA): Rev. Cancer 473(2), 93–119 (1977).
Rosenberg, S. A. et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 175(9), 6169–6176 (2005).
Suek, N., Campesato, L. F., Merghoub, T. & Khalil, D. N. Targeted APC activation in cancer immunotherapy to enhance the abscopal effect. Front. Immunol. 10, 604 (2019).
Habibzadeh, G. et al. Identification of lncRNA associated with the SERPINE1 gene in colorectal cancer through TGF-β pathway. Comput. Biol. Med. 190, 110037 (2025).
Ye, L. et al. Identification of tumor antigens and immune landscape in glioblastoma for mRNA vaccine development. Front. Genet. 12, 701065 (2021).
Fan, C., Qu, H., Wang, X., Sobhani, N., Wang, L., Liu, S. & Li, Y. Cancer/testis antigens: from serology to mRNA cancer vaccine. In Seminars in Cancer Biology, Vol. 76, 218–231 (Academic Press, 2021).
Chen, H. et al. An immune gene signature to predict prognosis and immunotherapeutic response in lung adenocarcinoma. Sci. Rep. 12(1), 8230 (2022).
Corulli, L. R. et al. Multi-epitope-based vaccines for colon cancer treatment and prevention. Front. Immunol. 12, 729809 (2021).
Yu, G. et al. Tumor mutation burden, expressed neoantigens and the immune microenvironment in diffuse gliomas. Cancers 13(23), 6092 (2021).
Chen, Y. et al. Immunological classification of pancreatic carcinomas to identify immune index and provide a strategy for patient stratification. Front. Immunol. 12, 719105 (2022).
Chen, Y. et al. A novel immune checkpoint score system for prognostic evaluation in pancreatic adenocarcinoma. BMC Gastroenterol. 23(1), 113 (2023).
Abutalebi, M. et al. Discovery of PELATON links to the INHBA gene in the TGF-β pathway in colorectal cancer using a combination of bioinformatics and experimental investigations. Int. J. Biol. Macromol. 270, 132239 (2024).
Liu, Z., Li, M., Hua, Q., Li, Y. & Wang, G. Identification of an eight-lncRNA prognostic model for breast cancer using WGCNA network analysis and a Cox-proportional hazards model based on L1-penalized estimation. Int. J. Mol. Med. 44(4), 1333–1343 (2019).
Cleary, M. L. et al. Clustering of extensive somatic mutations in the variable region of an immunoglobulin heavy chain gene from a human B cell lymphoma. Cell 44(1), 97–106 (1986).
Cui, M. et al. Immunoglobulin expression in cancer cells and its critical roles in tumorigenesis. Front. Immunol. 12, 613530 (2021).
Matsumoto, Y. & Ju, T. Aberrant glycosylation as immune therapeutic targets for solid tumors. Cancers 15(14), 3536 (2023).
Boncheva, V. B. et al. Identification of the antigens recognised by colorectal cancer patients using sera from patients who exhibit a Crohn’s-like lymphoid reaction. Biomolecules 12(8), 1058 (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47–e47 (2015).
Mayakonda, A. & Koeffler, H. P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRxiv, 052662 (2016).
Pawar, A., Chowdhury, O. R. & Salvi, O. A narrative review of survival analysis in oncology using R. Cancer Res. Stat. Treatm. 5(3), 554–561 (2022).
Benítez-Parejo, N., del Águila, M. R. & Pérez-Vicente, S. Survival analysis and Cox regression. Allergol. Immunopathol. 39(6), 362–373 (2011).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77(21), e108–e110 (2017).
Chen, S. et al. RNA adenosine modifications related to prognosis and immune infiltration in osteosarcoma. J. Transl. Med. 20(1), 228 (2022).
Khosroshahi, E. M. et al. Determining expression changes of ANO7 and SLC38A4 membrane transporters in colorectal cancer. Heliyon 10(14), e34464 (2024).
Canzler, S. & Hackermüller, J. multiGSEA: A GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinform. 21, 1–13 (2020).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Syst. Biol.: Methods Protocols https://doi.org/10.1007/978-1-4939-7493-1_12 (2018).
Stein, S. E. Estimating probabilities of correct identification from results of mass spectral library searches. J. Am. Soc. Mass Spectrom. 5(4), 316–323 (1994).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 1–13 (2008).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8), 2628–2629 (2020).
Chen, Y., Liao, R., Yao, Y., Wang, Q. & Fu, L. Machine learning to identify immune-related biomarkers of rheumatoid arthritis based on WGCNA network. Clin. Rheumatol. https://doi.org/10.1007/s10067-021-05960-9 (2022).
Acknowledgements
The authors extend their heartfelt gratitude to the management of REVA University, Manipal University, Institute of Bioinformatics, and AIMST University, Malaysia, for their unwavering support, valuable assistance, and consistent encouragement throughout this research endeavor.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
Karthick Vasudevan: Writing—review and editing, Writing—original draft, Conceptualization. Dhanushkumar T: Writing—review and editing, Writing—original draft, Visualization, Formal analysis, Data curation. Sripad Rama Hebbar: Writing—review and editing, Writing—original draft, Visualization, Validation, Formal analysis. Prasanna Kumar Selvam: Writing—review and editing, Writing—original draft, Formal analysis. Majji Rambabu: Writing—review and editing, Formal analysis. Anbarasu Krishnan: Writing—review and editing, Formal analysis. Rohini Karunakaran Writing—review and editing, Project administration, Conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vasudevan, K., T, D., Hebbar, S.R. et al. Multi-omics and AI-driven immune subtyping to optimize neoantigen-based vaccines for colorectal cancer. Sci Rep 15, 19333 (2025). https://doi.org/10.1038/s41598-025-01680-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01680-1
