
Abstract
Staufen double-stranded RNA-binding protein 1 (STAU1) plays a significant role in cancer development and is associated with survival outcomes in patients with lung cancer. However, its specific functions and molecular mechanisms in lung adenocarcinoma (LUAD) remain underexplored. We conducted a comprehensive analysis of the role and mechanism of STAU1 in A549 cells via RNA sequencing (RNA-seq) and in vitro experiments. STAU1 is highly expressed in A549 cells, and the proliferation, invasion, and migration capabilities of siSTAU1 cells are markedly inhibited, while the level of apoptosis is increased. Through RNA-seq analysis, we identified 197 differentially expressed genes (DEGs) and 1,362 STAU1-regulated alternative splicing events (ASEs). The DEGs were specifically enriched in cell adhesion pathways, whereas the ASE genes were predominantly associated with cell division and the cell cycle. Furthermore, we validated the expression of several genes related to proliferation, invasion, and migration, as well as the AS patterns. Specifically, the expression levels of CFHR1, KLF2, and RHOB were upregulated in the siSTAU1 samples, whereas the expression of MASTL and STC2 was downregulated. Additionally, the AS patterns of BIN1 and SNHG17 were abnormal, which was corroborated by PCR experiments. Our study suggests that STAU1 has oncogenic characteristics and may modulate these genes to influence the proliferation, invasion, and migration of lung adenocarcinoma cells. This research offers new insights that may contribute to the diagnosis and treatment of LUAD.
Similar content being viewed by others


Introduction
Lung cancer (LC) represents one of the leading causes of cancer-related mortality globally1, with five-year survival rates ranging from 4 to 17%, contingent upon the stage of the disease at the time of diagnosis. Approximately 3 million individuals are projected to succumb to lung cancer by 20352. Lung cancer is categorized into two principal histological types: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC)3. NSCLC accounts for approximately 80–85% of all primary lung cancers and can be further subdivided into four subtypes: lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), large cell carcinoma, and bronchial carcinoid. Among these subtypes, LUAD is the most prevalent subtype of NSCLC and represents the most common primary lung tumor. Currently, the five-year survival rate for patients diagnosed with lung adenocarcinoma (LUAD) is less than 50%, whereas the survival rate for patients with distant metastasis is approximately 7%4. In the past decade, substantial advancements have been made in the diagnosis and treatment of NSCLC. The utilization of autoantibodies, microRNAs (miRNAs), circulating tumor DNA, exosomes, and other potential biomarkers has enhanced the determination of the diagnosis and prognosis of lung cancer5. Additionally, various novel therapeutic strategies, including molecular profiling and immunogenicity testing, are progressively improving the long-term survival rates of patients by increasing both their efficacy and safety5. Nevertheless, clinical outcomes frequently diverge from anticipated results. Therefore, it is imperative to develop and identify innovative therapeutic strategies or biomarkers aimed at increasing the survival rates of lung cancer patients.
Staufen double-stranded RNA binding protein 1 (STAU1) is a crucial double-stranded RNA binding protein that has been demonstrated to play a significant role in multiple stages of RNA metabolism, including alternative splicing6 (AS), transport7, stability8, translation and degradation9. Through these molecular mechanisms, STAU1 influences several physiological pathways associated with cellular decision-making processes, such as differentiation, proliferation, migration, apoptosis, and autophagy10,11,12. Furthermore, research has indicated that STAU1 is involved in the progression of various malignancies13. Large-scale comparative analyses of tumor and normal tissues have shown that the expression levels of STAU1 are increased in the majority of cancers, including prostate cancer14, high-grade glioma15, and stage IA and IB lung squamous cell carcinoma16. Consequently, researchers propose that STAU1 may function as an oncogene that enhances cancer cell proliferation and contributes to malignant phenotypes16,17. The mechanism of action of STAU1 varies across different cancer types. For example, LncCCLM inhibits lymph angiogenesis related to cervical cancer in vitro and lymph node metastasis in vivo by promoting the STAU1-mediated degradation of IGF-1 mRNA18. In LNCap prostate cancer cells, STAU1 activates the mTOR pathway to regulate cell proliferation; conversely, in PC3 prostate cancer cells, STAU1 influences cell migration and invasion through the activation of focal adhesion kinase14. These findings indicate that STAU1 has specific regulatory effects on the proliferation, migration, and invasion of different types of cancer cells.
Researchers have reported that STAU1 expression is significantly elevated in both lung adenocarcinoma and squamous cell carcinoma patients compared with normal controls. Specifically, 85.7% of adenocarcinomas and 86.7% of squamous cell carcinomas displayed strong to very strong staining for STAU119. Furthermore, patients with high STAU1 expression in lung cancer patients have a longer recurrence-free survival period19. However, a study conducted by K Wang16, which analyzed 602 lung squamous cell carcinoma samples, identified STAU1 as a potential risk factor for poor patient prognosis. The reasons underlying these contradictory findings remain unclear at present. In conclusion, the role of STAU1 in lung cancer—whether it exerts a positive or negative influence—suggests its potential as a novel biomarker and molecular target for targeted therapy. Unfortunately, the precise function and molecular mechanisms of STAU1 in lung cancer have not been sufficiently investigated.
In this study, we hypothesized that STAU1 may contribute to the development of lung cancer by affecting the proliferation, invasion, and migration of A549 cells, particularly in light of its elevated expression in lung adenocarcinoma tissues and its strong association with the progression of lung cancer. To test this hypothesis, we knocked down STAU1 in human lung adenocarcinoma cells (A549) and performed a series of experiments to assess cell proliferation, apoptosis, invasion, and migration while also acquiring transcriptomic data (RNA-seq) regulated by STAU1 through high-throughput transcriptomic sequencing. Next, we analyzed the impact of STAU1 knockdown on transcriptional regulation and alternative splicing events in A549 cells to preliminarily explore the molecular mechanisms that underlie the involvement of STAU1 in lung adenocarcinoma. The molecular targets identified in this study may represent potential therapeutic targets for future treatment strategies aimed at treating lung adenocarcinoma.
Methods
SiRNA information
All siRNA duplexes were purchased from Gemma (Suzhou, China). We designed three siRNAs targeting STAU1, designated as si-1843, si-1419, and si-1488. The sequence information for these siRNAs is provided in Supplementary Table S2. Following an initial screening, we selected si-1488, which exhibited the highest knockdown efficiency, for use in subsequent RNA-seq analysis. The knockdown efficiency results of the siRNAs are presented in Supplementary Fig.5.
Cell culture and transfections
A549 cells (CL-0481, Procell Life Science & Technology Co., Ltd., China) and PC-9 cells (CL-0668, Procell Life Science & Technology Co., Ltd., China) were cultured at 37 °C with 5% CO2 in basal medium supplemented with 10% fetal bovine serum (FBS) (10091148, Gibco, USA), 100 µg/mL streptomycin, and 100 U/mL penicillin (SV30010, HyClone, USA). For siRNAtransfection, Lipofectamine was utilized following the manufacturer’s instructions. The transfected cells were harvested after 48 h for RT‒qPCR and Western blotting analysis.
Assessment of gene expression
cDNA synthesis was performed via a reverse transcription kit (R323-01, Vazyme, China) at 42 °C for 5 min, 37 °C for 15 min, and 85 °C for 5 s on a mastercycler (T100, Bio-Rad, USA). qPCR was carried out on an ABI QuantStudio 5, followed by denaturation at 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 1 min. Each sample was analyzed in triplicate with technical replicates. The concentration of each transcript was then normalized to that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and the mRNA level was analyzed via the 2−ΔΔCT method. Comparisons were performed with two-way ANOVA by using GraphPad Prism software (Version number 8.0, San Diego, CA). The primers used for quantitative (q)PCR analysis are presented in Table SI.
Western blot
A549 cells were lysed in ice-cold RIPA buffer (PR20001, Proteintech, China) containing a protease inhibitor cocktail (4693116001, Sigma, USA) and incubated on ice for 30 min. The samples were then heated for 10 min in boiling water with protein loading buffer (P1040, Solarbio, China), loaded onto a 10% SDS‒PAGE gel and transferred to 0.45 mm PVDF membranes (ISEQ00010, Millipore, USA). The PVDF membranes were then blocked for 1 h at room temperature and incubated overnight at 4 °C with primary antibodies against STAU1 (anti-STAU1, 1:1,000; antibody produced in rabbits; 14225-1-AP; Proteintech, China) and GAPDH (1:10,000; antibody produced in mice; 60004-1-Ig; Proteintech, China), followed by incubation with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit, 1:10,000; SA00001-2; Proteintech, China; or anti-mouse, 1:10,000; AS003; ABclonal, China) for 45 min at room temperature. Finally, the membranes were visualized via chemiluminescence detection via enhanced ECL reagent (P0018 FM, Beyotime, China).
RNA extraction and sequencing
Total RNA was extracted via TRIzol (15596-018; Ambion, USA). The RNA was further purified through two rounds of phenol‒chloroform extraction and subsequently treated with RQ1 DNase (M6101; Promega, Madison, WI, USA) to eliminate any DNA contamination. The quality and concentration of the purified RNA were assessed by measuring the absorbance at 260 nm and 280 nm (A260/A280) via an ultrafine spectrophotometer (N50 touch, IMPLEN, Germany). Additionally, the integrity of the RNA was confirmed via 1.0% agarose gel electrophoresis.
RNA-seq assays were conducted by Wuhan Ruixing Biotechnology Co., Ltd. (http://www.rxbio.cc). For each sample, 1 µg of total RNA was treated with RQ1 DNase (M6101, Promega, USA) to eliminate DNA prior to the preparation of a directional RNA-seq library via the VAHTS® Universal V8 RNA-seq Library Prep Kit for Illumina (NR605, Vazyme, China). mRNAs were isolated via VAHTS mRNA capture beads (N401, Vazyme, China). The fragmented RNA was then converted into double-stranded cDNA. After the end repair and A-tailing steps, the DNAs were ligated to VAHTS RNA Multiplex Oligos Set 1 for Illumina (N323, Vazyme, China), followed by amplification of the ligated products. These products were purified and quantified before being stored at −80 °C until sequencing. The strand marked with dUTP (the 2nd cDNA strand) is not amplified, allowing strand-specific sequencing. For high-throughput sequencing applications, libraries were prepared according to the manufacturer’s guidelines and sequenced on an Illumina NovaSeq Xplus system utilizing 150 nt paired-end reads.
Cell proliferation assay
Cell proliferation was assessed via the BeyoClick™ EdU-488 cell proliferation assay kit. The cells from both the control and experimental groups were treated as required and incubated at 37 °C with 5% CO2 for 48 h prior to conducting the EdU experiments.
Flow cytometric analysis of cell apoptosis
An Annexin V-APC/7-ADD apoptosis assay kit (40304 ES 60; Yeasen, Shanghai, China) was used for the apoptosis assay. The treated and control cells were incubated with 5 µL of Annexin V-APC for 15 min and then with 10 µL of PI reagent for another 15 min in the dark. Apoptosis levels were subsequently assessed via flow cytometry (FACSCanto, BD, USA).
Cell invasion and migration assays
In vitro migration and invasion assays were performed using transwell chambers (3422, Corning, USA). A549 cells in 0.2 mL of serum-free medium were added to the transwell chambers with an 8 μm filter, and then, 600 µL of 10% FBS (10091148, Gibco, China) was added to the chambers, which served as a chemoattractant in the lower chamber. For the cell invasion assays, the cells were incubated for 48 h at 37 °C and 5% CO2, and for the cell migration assays, the cells were incubated for 24 h under the same conditions. The cells remaining on the upper membrane surface of the inserts were then removed with a cotton swab, and the total number of cells that migrated into the lower chamber was fixed with 4% paraformaldehyde (P0099, Beyotime, China) for 20 min and then stained with 0.1% crystal violet (C0121, Beyotime, China). The migrating cells were observed and counted under an inverted microscope (MF52-N, Mshot, China) at 200× magnification.
RNA-Seq Raw data clean and alignment
Initially, raw reads with more than 2 N bases were removed. Next, adaptors and low-quality bases were trimmed from the raw sequencing data via the FASTX-Toolkit (version 0.0.13). Reads shorter than 16 nt were also excluded. The cleaned reads were subsequently aligned to the GRCh38 genome via HISAT2, allowing for up to 4 mismatches20. Only uniquely mapped reads were utilized for counting gene read numbers and calculating fragments per kilobase of transcript per million fragments mapped (FPKM)21.
Differentially expressed gene (DEG) analysis
The R Bioconductor package DESeq2 was used to screen out the differentially expressed genes (DEGs). An adjusted P value < 0.01 and a fold change ≥ 2 or ≤ 0.5 were set as the cutoff criteria for identifying DEGs.
Alternative splicing analysis
The alternative splicing events (ASEs) and regulated alternative splicing events (RASEs) among the samples were identified and quantified via the ABLas pipeline. In summary, ABLs detect ten types of ASEs on the basis of splice junction reads, which include exon skipping (ES), alternative 5’ splice site (A5SS), alternative 3’ splice site (A3SS), mutually exclusive exons (MXE), intron retention (IR), mutually exclusive 5’ UTRs (5pMXE), mutually exclusive 3’ UTRs (3pMXE), cassette exons, A3SS & ES, and A5SS & ES. To evaluate RBP-regulated ASEs, Student’s t test was performed to evaluate the significance of the ratio alteration of AS events. Events that reached significance at a P value cutoff corresponding to a false discovery rate cutoff of 5% were classified as RBP-regulated ASEs.
Functional enrichment analysis
To determine the functional categories of the DEGs, Gene Ontology (GO) terms and KEGG pathways were identified via the KOBAS 2.0 server. Hypergeometric tests and Benjamini‒Hochberg false discovery rate (FDR) control procedures were used to define the enrichment of each term.
Analysis of gene expression in LUAD
The expression of STAU1 in LUAD patients was analyzed via the TCGA & UALCAN databases. The prognostic significance of gene expression levels was determined via Kaplan–Meier survival curve analysis.
Analysis of important gene-related expression information via a TNM plot
TNMplot (http://www.tnmplot.com/) was used to display important gene expression information in tumors and normal tissues. The dataset includes 56,938 unique samples from the GEO, GTEx, TCGA, and TARGET databases.
Immunohistochemical analysis of LUAD via the HPA
The Human Protein Atlas (HPA) program (https://www.proteinatlas.org/) offers detailed information regarding the tissue and cellular distributions of 26,000 human proteins. In this database, the researchers used highly specific antibodies and immunoassays (western blotting, immunofluorescence, and immunohistochemistry) to measure in detail the expression of each protein in 64 cell lines, 48 normal human tissues, and 20 tumor tissues. The HPA was utilized to visualize the immunohistochemical findings of STAU1 in both normal tissue and lung adenocarcinoma samples.
GSEA
GSEA is an analytical method for analyzing genome-wide expression profile microarray data. It can identify functional enrichment by comparing genes with predefined gene sets. We utilized GSEA-based enriched Gene Ontology (GO) biological process analyses.
Statistical analyses
The data from at least three independent experiments are presented as the mean ± standard deviation (SD). The data were analyzed via either Student’s t test (two-group comparisons) or two-way ANOVA (more than two groups). A p value of < 0.05 was considered to indicate statistical significance (*p 0.05; **p < 0.01; ***p < 0.001). All the graphs were generated via GraphPad Prism software (version 8.0, San Diego, CA).
Results
STAU1 is significantly upregulated in lung adenocarcinoma (LUAD)
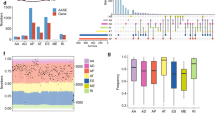
To further investigate the role of STAU1 in LUAD, we employed online tools such as TNMplot and UALCAN to analyze the expression of STAU1 in tumor tissues obtained from LUAD patients. The results demonstrated that STAU1 was significantly overexpressed in LUAD (Fig. 1A), with protein levels increasing in correlation with the progression of individual patient grades (Fig. 1B). Among the samples from the four stages of LUAD, the STAU1 levels were markedly higher in stages 1–3 samples than in normal samples; however, stage 4 samples had a smaller sample size (Fig. 1C). Additionally, with the exception of lepidic adenocarcinoma, squamous cell carcinoma, and colloid adenocarcinoma—which had smaller sample sizes—STAU1 protein levels were significantly elevated across other pathological types of lung cancer (Fig. 1D). This finding was further supported by immunohistochemical analysis of LUAD data sourced from the Human Protein Atlas (HPA) database (Fig. 1E).

Expression of STAU1 in clinical patients. (A) Boxplot generated to show the expression of STAU1 in LUAD samples on the basis of sample type via the TNMplot database. (B) Boxplot of the protein expression of STAU1 in LUAD according to tumor grade generated via the UALCAN database. (C) Boxplot of the protein expression of STAU1 in LUAD according to tumor stage generated via the UALCAN database. (D) Boxplot showing the protein expression of STAU1 in LUAD patients on the basis of tumor classification via the UALCAN database. (E) HPA was used to analyze the results of immunohistochemical analysis of STAU1 in LUAD.
The silencing of STAU1 results in the inhibition of cell proliferation, invasion, and migration while simultaneously promoting apoptosis
To investigate the biological role of STAU1 in LUAD, we chose two human lung adenocarcinoma cell lines, A549 and PC-9, and carried out a series of experiments. We transiently knocked down STAU1 in these two cell lines via transient transfection and then assessed cell proliferation, apoptosis, invasion, and migration capabilities. In the experiment, we employed three siRNAs targeting STAU1, namely si-1419, si-1843, and si-1488, to reduce STAU1 expression in the two cell lines. Compared with the NC group, the mRNA and protein levels of STAU1 in the si-STAU1 group were significantly lower, indicating that all three siRNAs could effectively knock down STAU1 (Fig. 2A-B, Supplementary Fig. 1 A-B, Supplementary Fig. 2 A-B). In A549 cells, knocking down STAU1 with si-1488 and si-1843 led to notable changes. Specifically, A549 cell proliferation was significantly suppressed, and apoptosis increased (Fig. 2C-D; Supplementary Fig. 1 C-D). Moreover, cell invasion and migration abilities were also markedly reduced (Fig. 2E-F, Supplementary Fig. 1E-F). To further confirm the function of STAU1 in LUAD, we repeated the same procedure in the PC-9 cell line and obtained similar results (Supplementary Fig. 2). Collectively, these results show that knocking down STAU1 can inhibit the proliferation, invasion, and migration of lung adenocarcinoma cells and promote apoptosis. This suggests that normally expressed STAU1 may enhance the pro-tumor properties of lung adenocarcinoma cells.

Knockdown of STAU1 inhibits the proliferation, metastasis and invasion and promotes the apoptosis of A549 cells. (A) The histogram shows the RT‒qPCR results for the NC and si_STAU1-1488 samples. The error bars represent the means ± SEMs. **** P value < 0.0001. (B) The results of the western blot experiments revealed that the STAU1 knockdown was successful. (C) The results of the cell proliferation experiments revealed that the knockdown of STAU1 inhibited the proliferation of A549 cells. The error bars represent the means ± SEMs. * P value < 0.05. (D) The results of the cell apoptosis experiments revealed that the knockdown of STAU1 promoted the apoptosis of A549 cells. The error bars represent the means ± SEMs. ** P value < 0.01. (E) The results of the cell invasion experiments revealed that the knockdown of STAU1 inhibited the invasion of A549 cells. The error bars represent the means ± SEMs. *** P value < 0.001. (F) The results of the cell migration experiments revealed that the knockdown of STAU1 inhibited the migration of A549 cells. The error bars represent the means ± SEMs. * P value < 0.05.

Stau1 regulates gene expression in human A549 cells. (A) PCA based on the FPKM values of all the detected Stau1 knockdown genes. The ellipse for each group is the confidence ellipse. (B) Volcano plot showing all differentially expressed genes (DEGs) between Stau1 knockdown (siStau1) and NC samples. (C) Hierarchical clustering heatmap displaying the expression of DEGs. (D) Scatter plot showing the most enriched GO pathway results of the upregulated DEGs. (E) The GSEA enrichment analysis results revealed the hallmark epithelial mesenchymal transition pathways. (F) Bar plot showing the expression patterns and significant differences in DEGs among several important genes. The error bars represent the means ± SEMs. ***P value < 0.001. The histogram shows the RT‒qPCR results for the Stau1-knockdown (siStau1) and NC samples. The error bars represent the means ± SEMs. **** P value < 0.0001.

Expression of STAU1 targets in clinical patients. (A) Boxplot generated to show the expression of STAU1 target genes in LUAD on the basis of sample type via the TNMplot database. (B) Boxplot generated by the UALCAN database showing the protein expression of STAU1 targets in LUAD on the basis of tumor grade. (C) Boxplot showing the protein expression of STAU1 targets in LUAD patients on the basis of tumor classification via the UALCAN database.

Correlation of STAU1 target genes with STAU1 expression and prognostic analysis of target genes. (A) Prognostic analysis of the STAU1 target genes. Comparison of overall survival according to the risk score calculated from the STAU1 target genes. (B) Scatter plots showing the correlation between the expression of STAU1 target genes and the expression of STAU1 in lung adenocarcinoma.

STAU1 regulates alternative gene splicing in A549 cells. (A) Bar plot showing the percentage of alternative splicing events (ASEs). X-axis: ASE percentage. Y-axis: the different types of AS events. (B) Overlap analysis among different types of alternative splicing events. The orange dots indicate the gene groups used for overlap analysis. The bar with the number indicates the number of genes in each group or two overlapping groups. (C) Bar plot showing the number of ASEs per gene; the median number is marked with a dashed line. X and Y are marked in parentheses. Y-axis: number of genes. X-axis: number of ASEs. (D) Boxplot showing the standard deviation of the ratio of each type of alternative splicing event (ASE) in all samples. (E) Boxplot showing the difference in the ratio of each type of alternative splicing event (ASE) across all samples. (F) Pie chart showing the proportions of RASG types. (G) Scatter plot showing the most enriched GO biological process results of the regulated alternative splicing genes (RASGs). (H) Bar plots showing the ratio change in RASEs in two genes (blue: alternative splicing exon; white: exon). The error bars represent the means ± SEMs. ** P value < 0.01, * P value < 0.05. RT‒PCR showing the significantly differentially expressed genes (AS events) between the STAU1-knockdown (siSTAU1) and NC samples. The right panel shows the quantitative results. The error bars represent the means ± SEMs. ***p value < 0.001, * P value < 0.05.
STAU1 modulates the transcriptomic profile of A549 cells
To further elucidate the role of STAU1 in regulating the oncogenic characteristics of A549 cells, we conducted RNA sequencing (RNA-seq) on samples treated with si-STAU1 and a negative control (NC). Principal component analysis (PCA) revealed a clear distinction between the si-STAU1 and NC samples (Fig. 3A), indicating that the knockdown of STAU1 significantly altered the transcriptomic profile of A549 cells. A total of 197 differentially expressed genes (DEGs), including 143 upregulated genes and 54 downregulated genes, were identified following the silencing of STAU1 in A549 cells (Fig. 3B). The number of upregulated DEGs in the si-STAU1 samples was significantly greater than that of downregulated DEGs, suggesting that STAU1 may exert transcriptional inhibitory effects in A549 cells. The expression patterns of all DEGs were visualized via a hierarchical clustering heatmap, which revealed largely inconsistent distribution patterns between the si-STAU1 and NC groups (Fig. 3C). Gene Ontology (GO) pathway enrichment analysis revealed that the DEGs resulting from STAU1 knockdown were enriched predominantly in pathways associated with cell adhesion, homophilic cell adhesion via plasma membrane adhesion molecules, and cell proliferation (Fig. 3D). To further investigate the specific biological significance of the gene sets closely associated with those regulated by STAU1, we performed gene set enrichment analysis (GSEA) on all the genes and found that the knockdown of STAU1 may influence the epithelial‒mesenchymal transition (EMT) pathway (Fig. 3E). Additionally, we identified 13 genes that are closely linked to cancer proliferation, invasion, and migration. As illustrated in Fig. 3F, the genes HAS2, RHOB, MESD, SNAP25, BTG2, HABP4, SFRP4, BBC3, CFHR1, KLF2, and MYL9 were upregulated, whereas MASTL and STC2 were downregulated (Fig. 3F and Supplementary Fig. 3). We validated the expression of CFHR1, KLF2, RHOB, and MASTL via RT‒qPCR, and the results were corroborated by sequencing data (Fig. 3F).
The expression of STAU1 targets in clinical patients
On the basis of these findings, we further investigated the role of target genes regulated by STAU1 in the context of lung cancer. The results demonstrated that these target genes presented significant differences in expression levels between lung cancer samples and normal controls. Specifically, data obtained from the TNM plot database indicated that MASTL and STC2 were upregulated, whereas KLF2, RHOB, HAS2, MYL9, and BTG2 were downregulated in LUAD (Fig. 4A and Supplementary Fig. 3). Additionally, analysis of the UALCAN database revealed that the expression of CFHR1, RHOB, and MYL9 was lower in LUAD tissues than in normal lung tissues (Fig. 4B and Supplementary Fig. 3). Furthermore, tumor classification data from the UALCAN database supported these findings, indicating that CFHR1, RHOB, and MYL9 were consistently downregulated across various subtypes of adenocarcinoma, including papillary adenocarcinoma, acinar adenocarcinoma and solid adenocarcinoma (Fig. 4C and Supplementary Fig. 3). Taken together, these findings indicate that STAU1 may play a substantial role in the progression of lung cancer because of its regulatory influence on these genes.
Correlation of STAU1 target genes with STAU1 expression and prognostic analysis of target genes
To further elucidate the role of target genes regulated by STAU1 in lung cancer, we performed survival analysis on these genes, which revealed that MASTL, HABP4, and BTG2 are significantly associated with lung cancer prognosis. Specifically, low expression levels of HABP4 and BTG2 were correlated with poor prognostic outcomes, whereas elevated MASTL levels were associated with reduced survival time among lung cancer patients (Fig. 5A and Supplementary Fig. 3). Furthermore, to assess the regulatory impact of STAU1 on DEGs, we utilized the Lung Adenocarcinoma dataset from The Cancer Genome Atlas (TCGA) to examine the correlation between STAU1 and the identified target genes. This analysis revealed a positive correlation between STAU1 and both MASTL and STC2, as well as a negative correlation with KLF2 and BTG2 (Fig. 5B and Supplementary Fig. 3), which is consistent with the results obtained from RNA-seq. These findings indicate that the genes whose expression is modulated by STAU1 in A549 cells are significantly associated with increased oncogenic characteristics.
STAU1 modulates alternative splicing events in A549 cells
STAU1 is an RNA-binding protein (RBP) that plays a crucial role in regulating alternative splicing (AS) patterns of its target genes. In this study, we examined the effects of STAU1 on AS in A549 cells. Analysis of RNA-seq data revealed a significant number of alternative splicing events (ASEs), identifying a total of 1,362 differential ASEs in A549 cells, with a selection criterion of a p value ≤ 0.05. This analysis identified a total of 619 upregulated and 743 downregulated ASEs, with the number of downregulated events exceeding that of the upregulated events (Fig. 6A). The predominant types of variable splicing included intron retention (IntronR), alternate 3’ splice site usage (A3SS), and alternate 5’ splice site usage (A5SS), followed by cassette exon and exon skipping (ES) events (Fig. 6A). Notably, there was minimal overlap among the genes regulated by STAU1 across the various types of splicing events, with the majority of individual genes experiencing only one specific type of splicing event. IntronR events were the most prevalent, followed by A3SS and A5SS events (Fig. 6B and C). Figure 6D shows the standard deviation ratio between upregulated and downregulated differential ASEs across all samples, revealing that the ratio for upregulated A3SS&ES events was significantly greater than that for their downregulated counterparts, whereas the ratios for other event types remained comparable. Furthermore, when known versus novel predicted splicing events across ten categories were compared, the proportion of novel predicted variants was significantly greater than that of known variants (Fig. 6E). The pie chart shows that protein-coding genes constitute the majority of AS genes, followed by long noncoding RNAs (lncRNAs) (Fig. 6F). GO pathway enrichment analysis revealed that genes whose expression significantly changed in ASEs after STAU1 knockdown were enriched predominantly in pathways related to the regulation of cell division and control of the cell cycle (Fig. 6G). Furthermore, by ranking the alternative splicing events based on P-values and considering factors such as supporting read counts, ratio values, and the magnitude of change, we identified that STAU1 regulates the alternative splicing of several cancer-related genes, including SNHG17, UPP1, RNH1, UCHL5, BIN1, LINC01234, and AHSA2P (Fig. 6H and Supplementary Fig. 4). Notably, BIN1 and SNHG17 are particularly associated with lung cancer and exhibit distinct alternative splicing variants. To validate these findings, we performed PCR analysis of exon skipping (ES) events for BIN1 and SNHG17, and the results were consistent with those obtained from RNA-Seq (Fig. 6H).
Discussion
The dysregulation of STAU1 expression typically disrupts the delicate balance among oncogenes, tumor suppressor genes, proapoptotic genes, and antiapoptotic genes by modulating mRNA translation, splicing, and decay, thereby influencing the progression of cancer14,17,18,22. Previous studies have demonstrated that STAU1 is highly expressed in lung cancer tissues16,19; however, the underlying molecular mechanisms remain poorly understood. In this study, we found that the knockdown of STAU1 inhibited the proliferation, invasion, and migration of A549 cells, potentially through the regulation of downstream target RNA expression and alternative splicing.
In this study, the DEGs regulated by the downregulation of STAU1 were associated primarily with cell adhesion and cell cycle regulation. Previous reports indicate that CFHR123, KLF224,25, RHOB26,27,28, and BTG229,30 are expressed at reduced levels in lung cancer tissues, potentially inhibiting the proliferation and migration of lung cancer cells. MESD has been shown to inhibit the Wnt/β-catenin signaling pathway and suppress cancer cell proliferation in human breast cancer HS5-78 T cells as well as in prostate cancer PC-3 cells30,31. The downregulation of HABP4 promotes the proliferation, migration, and invasion of renal cancer cells and is considered a candidate tumor suppressor gene in colorectal cancer32,33. SFRP4 inhibits proliferation and migration while activating apoptotic pathways in mesothelioma34. BBC3 is recognized as a novel tumor suppressor gene across various human cancers35. SNAP25 is significantly downregulated in prostate cancer and may influence the activation, differentiation, and migration of immune cells36. In our study, we observed that following STAU1 knockdown in A549 cells, genes with inhibitory effects on cell proliferation or invasion were upregulated. Conversely, overexpression of MASTL has been shown to promote chromosomal instability and metastasis in breast cancer cells37, whereas STC2 facilitates metastasis and progression in both lung and pancreatic cancers38,39. Notably, genes associated with promoting invasion and migration were downregulated in A549 cells. Consistent with these findings, the database results indicated that STAU1 was positively correlated with MASTL and STC2 but negatively correlated with KLF2 and BTG2. These findings support the positive role of STAU1 in promoting the proliferation and invasion of A549 cells through its regulated target genes, suggesting that STAU1 may possess oncogenic properties. In addition to this study, a previous literature analysis of 602 cases of lung squamous cell carcinoma (LUSC, a subtype of non-small cell lung cancer) identified STAU1, ADGRF1, ATF7IP2, MALL, and KRT23 as poor prognostic factors [16]. However, another study on large cell lung cancer found that STAU1 was a protective factor for lung cancer [19]. Currently, there are no real-world studies reporting the relationship between STAU1 expression and the prognosis of lung cancer. We believe that the reason for such contradictory results may be attributed to different pathological types of lung cancer on the one hand; on the other hand, the clinical samples in the real world are highly complex and heterogeneous, which may lead to deviations between the predicted results and the actual clinical situation. Therefore, further rigorous clinical validation is still needed to clarify the exact relationship between STAU1 expression and the overall survival rate of lung cancer patients.
Furthermore, GSEA revealed that STAU1 knockdown significantly affected the epithelial‒mesenchymal transition (EMT) pathway. EMT is characterized by a process in which epithelial cells detach from neighboring cells and acquire mesenchymal traits, which are associated with enhanced migratory capacity. The activation of EMT is a critical mechanism in cancer cell metastasis, during which epithelial cells adopt mesenchymal features, thereby increasing their mobility and migratory ability40. Similarly, researchers have reported that STAU1 is involved in lncTCL6-miR-155-Src/Akt-mediated EMT, as well as in the network-regulated progression and metastasis of renal cancer41. These findings suggest that STAU1 may also exert a pro-oncogenic effect by regulating EMT in lung cancer.
In addition to its role in regulating gene expression levels, alternative splicing (AS) is crucial for ensuring the diversity of gene products. Increasing evidence suggests that AS significantly influences various oncogenic processes, including cancer initiation, progression, angiogenesis, and immune evasion42. However, current research on AS in lung adenocarcinoma (LUAD) remains limited. In this study, we discovered that STAU1 knockdown regulated a substantial number of differentially spliced events. Genes identified through Gene Ontology pathway enrichment analysis were associated primarily with pathways related to cell division regulation and the cell cycle. We organized other types of variable splicing events (NIR variable splicing events) by P value in descending order and selected six genes exhibiting altered splicing following STAU1 knockdown on the basis of supporting read counts, ratio values, change amplitudes, and relevant literature.
It has been reported that the selective splicing of BIN1, SNHG17, and RNH1 in human cancers is induced specifically and closely correlates with patient prognosis43,44,45. BIN1 expression is reduced in non-small cell lung cancer (NSCLC)46, where its absence increases susceptibility to cancer during aging—particularly lung cancer47. SNHG17 regulates proliferation and migration in human NSCLC cells48. RNH1 is expressed at lower levels in LUAD tissues and inhibits LUAD cell proliferation while promoting apoptosis49. The upregulation of LINC01234 can enhance metastasis in non-small cell lung cancer cells30. UCHL5 is upregulated in LUAD and is correlated with poor clinical prognosis; silencing UCHL5 significantly inhibits LUAD cell proliferation while reducing the expression of key cell cycle proteins50. UPP1 promotes the progression of lung adenocarcinoma through epigenetic regulation of glycolysis or by inducing an immunosuppressive microenvironment that facilitates tumor advancement51,52. The findings of the aforementioned study indicate that STAU1 regulates alternative splicing events in A549 cells and that the genes affected by STAU1 regulation that undergo these splicing events are associated with lung cancer progression, particularly those that play crucial roles in the proliferation and migration of cancer cells. In summary, we hypothesize that STAU1 modulates alternative splicing of these genes to produce different protein isoforms, thereby influencing the onset and development of lung cancer. However, the effects of these alternative splicing variants on lung cancer remain unknown; thus, further investigation is needed to elucidate the mechanisms by which STAU1 regulates alternative splicing and its impact on lung cancer.
Our findings suggest that STAU1 may regulate both the expression and alternative splicing of genes associated with cell adhesion and cell cycle processes, thereby contributing to the proliferation, invasion, and migration of lung adenocarcinoma cells. This study enhances our understanding of the role of STAU1 in carcinogenesis and offers new insights into potential diagnostic and therapeutic strategies for lung adenocarcinoma.
Data availability
Sequence data that support the findings of this study have been deposited in the GEO database under the primary accession code GSE279930 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE279930), which is currently maintained in private status: azwzuqguzjcldal.
References
Thandra, K. C., Barsouk, A., Saginala, K., Aluru, J. S. & Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. (Pozn). 25, 45–52. https://doi.org/10.5114/wo.2021.103829 (2021).
Wadowska, K., Bil-Lula, I. & Trembecki, Ł. Śliwińska-Mossoń, M. Genetic markers in lung Cancer diagnosis: A review. Int. J. Mol. Sci. 21 https://doi.org/10.3390/ijms21134569 (2020).
Nooreldeen, R. & Bach, H. Current and future development in lung Cancer diagnosis. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22168661 (2021).
Hao, D. et al. The Single-Cell Immunogenomic landscape of B and plasma cells in Early-Stage lung adenocarcinoma. Cancer Discov. 12, 2626–2645. https://doi.org/10.1158/2159-8290.Cd-21-1658 (2022).
Kratzer, T. B. et al. Lung cancer statistics, Cancer 130, 1330–1348, (2023). https://doi.org/10.1002/cncr.35128 (2024).
Jiang, S. et al. STAU1 promotes adipogenesis by regulating the alternative splicing of Pparγ2 mRNA. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 1868, 159293. https://doi.org/10.1016/j.bbalip.2023.159293 (2023).
Abouward, R. & Schiavo, G. Walking the line: mechanisms underlying directional mRNA transport and localization in neurons and beyond. Cell. Mol. Life Sci. 78, 2665–2681. https://doi.org/10.1007/s00018-020-03724-3 (2021).
Yamaguchi, Y., Naiki, T. & Irie, K. Stau1 regulates Dvl2 expression during myoblast differentiation. Biochem. Biophys. Res. Commun. 417, 427–432. https://doi.org/10.1016/j.bbrc.2011.11.133 (2012).
Alluri, R. K., Li, Z. & McCrae, K. R. Stress Granule-Mediated oxidized RNA decay in P-Body: hypothetical role of ADAR1, Tudor-SN, and STAU1. Front. Mol. Biosci. 8, 672988. https://doi.org/10.3389/fmolb.2021.672988 (2021).
Zhong, Y. et al. STAU1 selectively regulates the expression of inflammatory and immune response genes and alternative splicing of the nerve growth factor receptor signaling pathway. Oncol. Rep. 44, 1863–1874. https://doi.org/10.3892/or.2020.7769 (2020).
Ruan, X. et al. LncRNA LINC00665 stabilized by TAF15 impeded the malignant biological behaviors of glioma cells via STAU1-Mediated mRNA degradation. Mol. Ther. Nucleic Acids. 20, 823–840. https://doi.org/10.1016/j.omtn.2020.05.003 (2020).
Almasi, S. et al. Differential regulation of autophagy by STAU1 in alveolar rhabdomyosarcoma and nontransformed skeletal muscle cells. Cell. Oncol. (Dordr). 44, 851–870. https://doi.org/10.1007/s13402-021-00607-y (2021).
Bonnet-Magnaval, F. & DesGroseillers, L. The Staufen1-dependent cell cycle Regulon or how a misregulated RNA-binding protein leads to cancer. Biol. Rev. Camb. Philos. Soc. 96, 2192–2208. https://doi.org/10.1111/brv.12749 (2021).
Marcellus, K. A., Parks, C., Almasi, T. E., Jasmin, B. J. & S. & Distinct roles for the RNA-binding protein Staufen1 in prostate cancer. BMC Cancer. 21, 120. https://doi.org/10.1186/s12885-021-07844-2 (2021).
Jing, F. et al. The PABPC5/HCG15/ZNF331 feedback loop regulates vasculogenic mimicry of glioma via STAU1-Mediated mRNA decay. Mol. Ther. Oncolytics. 17, 216–231. https://doi.org/10.1016/j.omto.2020.03.017 (2020).
Wang, K., Li, Y., Wang, J., Chen, R. & Li, J. A novel 12-gene signature as independent prognostic model in stage IA and IB lung squamous cell carcinoma patients. Clin. Transl Oncol. 23, 2368–2381. https://doi.org/10.1007/s12094-021-02638-1 (2021).
Almasi, S. & Jasmin, B. J. The multifunctional RNA-binding protein Staufen1: an emerging regulator of oncogenesis through its various roles in key cellular events. Cell. Mol. Life Sci. 78, 7145–7160. https://doi.org/10.1007/s00018-021-03965-w (2021).
Chen, C. et al. LncCCLM inhibits lymphatic metastasis of cervical cancer by promoting STAU1-mediated IGF-1 mRNA degradation. Cancer Lett. 518, 169–179. https://doi.org/10.1016/j.canlet.2021.07.005 (2021).
Bonnet-Magnaval, F. et al. High level of Staufen1 expression confers longer recurrence free survival to Non-Small cell lung Cancer patients by promoting THBS1 mRNA degradation. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms23010215 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360. https://doi.org/10.1038/nmeth.3317 (2015).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. https://doi.org/10.1038/nbt.1621 (2010).
Almasi, S. et al. Staufen1 controls mitochondrial metabolism via HIF2α in embryonal rhabdomyosarcoma and promotes tumorigenesis. Cell. Mol. Life Sci. 80, 328. https://doi.org/10.1007/s00018-023-04969-4 (2023).
Wu, G. et al. CFHR1 is a potentially downregulated gene in lung adenocarcinoma. Mol. Med. Rep. 20, 3642–3648. https://doi.org/10.3892/mmr.2019.10644 (2019).
Jiang, W. et al. Methylation of kruppel-like factor 2 (KLF2) associates with its expression and non-small cell lung cancer progression. Am. J. Transl Res. 9, 2024–2037 (2017).
Yin, L. et al. Downregulation of Kruppel-like factor 2 is associated with poor prognosis for non-small cell lung cancer. Tumor Biol. 36, 3075–3084. https://doi.org/10.1007/s13277-014-2943-4 (2015).
Podmirseg, S. R., Vosper, J. & Hengst, L. p27(Kip1) - p(RhoB)lematic in lung cancer. J. Pathol. 248, 3–5. https://doi.org/10.1002/path.5218 (2019).
Wang, J. et al. CERS4 predicts positive anti-PD-1 response and promotes Immunomodulation through Rhob-mediated suppression of CD8(+)Tim3(+) exhausted T cells in non-small cell lung cancer. Pharmacol. Res. 194, 106850. https://doi.org/10.1016/j.phrs.2023.106850 (2023).
Calvayrac, O., Pradines, A. & Favre, G. RHOB expression controls the activity of serine/threonine protein phosphatase PP2A to modulate mesenchymal phenotype and invasion in non-small cell lung cancers. Small GTPases. 9, 339–344. https://doi.org/10.1080/21541248.2016.1234429 (2018).
Zhang, X. Z., Chen, M. J., Fan, P. M., Jiang, W. & Liang, S. X. BTG2 serves as a potential prognostic marker and correlates with immune infiltration in lung adenocarcinoma. Int. J. Gen. Med. 15, 2727–2745. https://doi.org/10.2147/ijgm.S340565 (2022).
Chen, Z. et al. Upregulated LINC01234 promotes non-small cell lung cancer cell metastasis by activating VAV3 and repressing BTG2 expression. J. Hematol. Oncol. 13 https://doi.org/10.1186/s13045-019-0842-2 (2020).
Lin, C. et al. Mesd is a general inhibitor of different Wnt ligands in Wnt/LRP signaling and inhibits PC-3 tumor growth in vivo. FEBS Lett. 585, 3120–3125. https://doi.org/10.1016/j.febslet.2011.08.046 (2011).
Liu, Y., Hu, X., Hu, L., Xu, C. & Liang, X. Let-7i-5p enhances cell proliferation, migration and invasion of CcRCC by targeting HABP4. BMC Urol. 21, 49. https://doi.org/10.1186/s12894-021-00820-9 (2021).
Melo-Hanchuk, T. D. et al. Intracellular hyaluronic acid-binding protein 4 (HABP4): a candidate tumor suppressor in colorectal cancer. Oncotarget 11, 4325–4337. https://doi.org/10.18632/oncotarget.27804 (2020).
Perumal, V., Dharmarajan, A. M. & Fox, S. A. The Wnt regulator SFRP4 inhibits mesothelioma cell proliferation, migration, and antagonizes Wnt3a via its netrin-like domain. Int. J. Oncol. 51, 362–368. https://doi.org/10.3892/ijo.2017.4011 (2017).
Chen, W. & Li, X. MiR-222-3p promotes cell proliferation and inhibits apoptosis by targeting PUMA (BBC3) in Non-Small cell lung Cancer. Technol. Cancer Res. Treat. 19, 1533033820922558. https://doi.org/10.1177/1533033820922558 (2020).
Di, L. et al. SNAP25 is a potential prognostic biomarker for prostate cancer. Cancer Cell. Int. 22, 144. https://doi.org/10.1186/s12935-022-02558-2 (2022).
Rogers, S. et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 37, 4518–4533. https://doi.org/10.1038/s41388-018-0295-z (2018).
Liu, Y. N. et al. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int. J. Cancer. 145, 1609–1624. https://doi.org/10.1002/ijc.32487 (2019).
Lin, C., Sun, L., Huang, S., Weng, X. & Wu, Z. STC2 is a potential prognostic biomarker for pancreatic Cancer and promotes migration and invasion by inducing Epithelial–Mesenchymal transition. Biomed. Res. Int. 2019 (8042489). https://doi.org/10.1155/2019/8042489 (2019).
Pastushenko, I. et al. Fat1 deletion promotes hybrid EMT State, tumor stemness and metastasis. Nature 589, 448–455. https://doi.org/10.1038/s41586-020-03046-1 (2021).
Kulkarni, P. et al. A LncRNA TCL6-miR-155 interaction regulates the Src-Akt-EMT network to mediate kidney Cancer progression and metastasis. Cancer Res. 81, 1500–1512. https://doi.org/10.1158/0008-5472.Can-20-0832 (2021).
Marzese, D. M., Manughian-Peter, A. O., Orozco, J. I. J. & Hoon, D. S. B. Alternative splicing and cancer metastasis: prognostic and therapeutic applications. Clin. Exp. Metastasis. 35, 393–402. https://doi.org/10.1007/s10585-018-9905-y (2018).
Wang, J. et al. SRSF1-dependent alternative splicing attenuates BIN1 expression in non-small cell lung cancer. J. Cell. Biochem. 121, 946–953. https://doi.org/10.1002/jcb.29366 (2020).
Mhatre, A. et al. Multiomics analysis of the Indian ovarian cancer cohort revealed histotype-specific mutation and gene expression patterns. Front. Genet. 14, 1102114. https://doi.org/10.3389/fgene.2023.1102114 (2023).
Engel, M. L., Hines, J. C. & Ray, D. S. The crithidia fasciculata RNH1 gene encodes both nuclear and mitochondrial isoforms of RNase H. Nucleic Acids Res. 29, 725–731. https://doi.org/10.1093/nar/29.3.725 (2001).
Wang, J. et al. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene 36, 6235–6243. https://doi.org/10.1038/onc.2017.217 (2017).
Chang, M. Y. et al. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 67, 7605–7612. https://doi.org/10.1158/0008-5472.Can-07-1100 (2007).
Xu, T. et al. Gene Amplification-Driven long noncoding RNA SNHG17 regulates cell proliferation and migration in human Non-Small cell lung Cancer. Mol. Ther. Nucleic Acids. 17, 405–413. https://doi.org/10.1016/j.omtn.2019.06.008 (2019).
Zhao, W., Liu, Y., Yang, Y. & Wang, L. New link between RNH1 and E2F1: regulates the development of lung adenocarcinoma. BMC Cancer. 24, 635. https://doi.org/10.1186/s12885-024-12392-6 (2024).
Zhang, J. et al. Deubiquitinase UCHL5 is elevated and associated with a poor clinical outcome in lung adenocarcinoma (LUAD). J. Cancer. 11, 6675–6685. https://doi.org/10.7150/jca.46146 (2020).
Wang, X. et al. UPP1 promotes lung adenocarcinoma progression through epigenetic regulation of Glycolysis. Aging Dis. 13, 1488–1503. https://doi.org/10.14336/ad.2022.0218 (2022).
Li, Y. et al. UPP1 promotes lung adenocarcinoma progression through the induction of an immunosuppressive microenvironment. Nat. Commun. 15, 1200. https://doi.org/10.1038/s41467-024-45340-w (2024).
Acknowledgements
We thank all the members of our laboratory for their support and helpful discussions. This work was supported by the Youth Talent Development Program in Medical Science and Technology for Healthcare in the Xinjiang Uygur Autonomous Region (NO. WJWY-202415) and the Science and Technology Support Project of the Xinjiang Uygur Autonomous Region (NO. 2022E02127) .
Author information
Authors and Affiliations
Contributions
Conceptualization: Ling Zhang, Zhen PengData curation: Haixia Wu, Wei Ding, Rong GuoFunding acquisition: Ling Zhang, Xuemei WeiMethodology: Ling Niu, Dewei LiProject administration: Ling Niu, Xuemei WeiSupervision: Ling Niu, Xuemei WeiWriting: Ling Zhang, Zhen Peng, Dewei LiAll authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Peng, Z., Ding, W. et al. STAU1 exhibits oncogenic characteristics and modulates alternative splicing and gene expression in lung adenocarcinoma cells. Sci Rep 15, 17031 (2025). https://doi.org/10.1038/s41598-025-01883-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01883-6
