
Abstract
Uremic cardiomyopathy (UCM) is a leading cause of death in patients with chronic kidney disease. This study aimed to explore the role and underlying mechanisms of neutrophil extracellular traps (NETs) in UCM. A UCM mouse model was constructed using 5/6 nephrectomy and treated with the NET inhibitor DNaseI. Cardiac fibroblasts were incubated with 200 ng/mL of NETs to establish an in vitro model. Physiological parameters of mice were measured, and commercial kits were used to detect MPO-DNA, IL-1β, and IL-6 levels. Hematoxylin-eosin and TUNEL staining were performed to evaluate myocardial injury and apoptosis. Immunofluorescence was used to detect MPO, CitH3, and caspase-1 expression. Sytox Green staining and CCK-8 assay were applied to assess NET formation and determine the optimal NET concentration. Western blot was conducted to detect collagen1, α-SMA, NLRP3, GSDMD-N, and caspase-1 expression. Transmission electron microscopy was performed to observe pyroptosis. UCM mice exhibited impaired kidney and cardiac function, indicating the successful establishment of the UCM model. MPO-DNA levels were elevated in UCM mice, suggesting NET formation. DNaseI treatment significantly reduced MPO and CitH3 expression, inhibiting NET formation in UCM mice. NET inhibition improved renal function and alleviated cardiac injury. Furthermore, NET inhibition reduced inflammation, apoptosis, and pyroptosis in UCM mice. Neutrophils isolated from UCM mice, as well as PMA, induced NET formation. NETs significantly enhanced fibrosis and pyroptosis in myocardial fibroblasts. NETs may promote UCM progression by inducing pyroptosis in myocardial fibroblasts.
Similar content being viewed by others


Introduction
Patients with chronic kidney disease (CKD) experience a gradual decline in kidney function, leading to the accumulation of uremic toxins and ultimately resulting in uremia1. Uremic toxin-related factors trigger signal transduction and metabolic changes that contribute to capillary dysfunction and cardiomyocyte hypertrophy in CKD2. Consequently, uremic cardiomyopathy (UCM) is a common complication of CKD, characterized by left ventricular hypertrophy and diastolic dysfunction, with a normal ejection fraction (EF)3. The incidence of UCM is rising due to the increasing prevalence of CKD, and sudden death and heart failure caused by UCM contribute to the high mortality in patients with end-stage renal disease4,5. However, current clinical treatments for UCM remain limited.
Pyroptosis is an inflammasome sensor-induced programmed cell death, which disrupts the integrity of the plasma membrane6. During this process, caspase-1/4/5/11 cleave gasdermin D (GSDMD) to produce GSDMD-N, which forms pores in the plasma membrane, while membrane protein NINJ1 further ruptures the membrane7. Inflammation in cardiovascular diseases (CVD) is closely linked to pyroptosis, and various cells in the cardiovascular system, such as cardiac fibroblasts, cardiomyocytes, neutrophils, and macrophages, participate in CVD-related pyroptosis8. Recent studies have shown that certain drugs can improve diabetic cardiomyopathy and septic cardiomyopathy by inhibiting myocardial pyroptosis9,10.
Neutrophil extracellular traps (NETs) are extracellular antimicrobial structures produced following neutrophil activation, playing a unique role in innate immunity and various pathological processes11. Specifically, NETs are neutrophil DNA webs coated with histones and antimicrobial proteins, and their antimicrobial properties are associated with a range of histones, granule proteases, myeloperoxidase (MPO), and lactotransferrin12. Evidence suggests that citrullination of histone H3 (CitH3), which is catalyzed by peptidyl arginine deiminase (PAD), triggers NET formation13. NETs are considered important factors in the pathological processes of CVD. They can promote thrombosis by inducing fibrin deposition, and the crosstalk between inflammasome and NETs has been reported in several diseases, as pro-inflammatory factors generated by inflammasome are key mediators of CVD14. NET formation is closely related to myocardial infarction and affects the prognosis of patients with this condition15. Studies have shown that NETs in myocardial tissue may contribute to cardiac dysfunction in patients with heart failure with dilated cardiomyopathy by modulating mitochondrial dysfunction16. Colchicine significantly improves myocardial injury during cardiac surgery by inhibiting NET formation17. USP47 promotes pyroptosis through the NLRP3 pathway, impairing cardiac function after myocardial infarction and aggravating inflammatory response, with upregulated NETs involved in this process18. Furthermore, NET formation is dependent on the pyroptosis-executing protein GSDMD, which is activated by neutrophil protease during NET production, affecting protease activation and nuclear amplification in a feed-forward loop19. NETs can promote pyroptosis in various diseases, including interstitial lung disease and diabetic nephropathy20,21. Moreover, ficolin-A/2 enhances NET formation through GSDMD-mediated pyroptosis22.
However, the roles of NETs and pyroptosis in UCM remain unclear. Herein, UCM mouse models were established to determine whether NETs are formed in UCM, and DNaseI was used to assess the impact of NET inhibition on pathological injury and pyroptosis in these mice. Additionally, the effect of NETs on fibrosis and pyroptosis in cardiac fibroblasts was examined.
Methods
Animals
Twenty-five C57BL/6 J mice (male, 10 weeks) were purchased from the Experimental Animal Center of Yangzhou University. The mice were housed in a specific pathogen-free room under controlled environmental conditions, with a 12 h/12 h light-dark cycle. Animals had free access to food and water. The study included experiments 1 and 2. In experiment 1, ten mice were randomly divided into two groups (n = 5): Sham and Model groups. In experiment 2, fifteen mice were randomly assigned to three groups (n = 5): Sham, Model, and Model + DNaseI groups. 10% pentobarbital sodium solution was injected intraperitoneally into the mice and then the anesthetized mice were placed in a supine position. In Model group, an incision was made on the left side of the mouse abdomen to expose the left kidney. The kidney was separated from the perirenal tissue, ligated with a 4/0 suture (approximately 1/3 of the kidney), and both poles of the kidney were cut off. The incision was sutured following disinfection. After one week, the right kidney was exposed in the same way, a clamp was placed at the renal hilum, and the ureter was ligated. The incision was sutured after resection of the right kidney23. The Sham group underwent the same operation as the Model group but without kidney resection. In Model + DNaseI group, treatment was given immediately after modeling, 10 mg/kg DNaseI (Yuanye, S33238) was injected intravenously into mice every 12 h, for a total of 72 h24. All animal experiments complied with the ARRIVE guidelines and were approved by the Experimental Animal Welfare Ethics Committee of Yangzhou University (approval number: 202407036). All experimental procedures were performed in accordance with relevant guidelines.
Measurement of physiological parameters
The weight of the mice was measured every 6 days during the experiment. Blood and urine samples were collected from mice on days 0 and 48. Blood was centrifuged at 1000 g for 10 min to obtain serum. Serum creatinine (Scr) and blood urea nitrogen (BUN) levels were measured by a fully automated biochemical analyzer to assess the degree of kidney injury. On day 48, echocardiograms were captured to assess cardiac function. Additionally, systolic blood pressure (SBP) and diastolic blood pressure (DBP) were measured using the BP-2000 blood pressure analysis system (Visitech, BP-2000), and the data were analyzed with BP-2000 analysis software (Visitech Systems, Inc.). After measurements, 2.5 Vol% Isoflurane was used for anesthesia, and the mice were then euthanized using a rat guillotine device. Subsequently, the myocardial tissue was obtained for further experiments.
Immunofluorescence
Mouse myocardial tissues were fixed in 4% paraformaldehyde (Beyotime, P0099) for 24 h. The samples were dehydrated in graded alcohol, embedded in paraffin, and then sectioned to a thickness of 4–7 μm25. Sections were placed at 92–96 °C for 10–15 min for antigen retrieval. After blocking with 5% BSA at 37℃ for 60 min, the sections were incubated with primary antibodies against CitH3 (1: 200, Abcam, ab1791), MPO (1: 100, Abcam, ab45977), and caspase-1 (1: 100, ABclonal, A0964) overnight at 4℃. After washing with PBS, sections were incubated with secondary antibodies (1: 200, Beyotime, China) at room temperature for 60 min. The sections were then stained with DAPI working solution (Beyotime, C1005) for 10 min. Images were captured using a laser confocal microscope (Leica, TCS SP8).
ELISA
Commercial kits were used to detect the level of MPO-DNA (Roche, 11774425001) in serum samples of mice, according to the manufacturer’s instructions. Additionally, the levels of IL-6 (Beyotime, PI326) and IL-1β (Beyotime, PI301) in myocardial tissue were assessed. The absorbance was measured using a microplate reader (Wuxi Hiwell Diatek, DR-3518G).
Hematoxylin-Eosin (HE) staining
Following dewaxing and hydration, the sections were stained using an HE staining kit (Beyotime, C0105S). Pathological changes in the sections were observed under a microscope (Leica, DMi3000 B).
TUNEL staining
Proteinase K working solution (50 µL) was added to sections of mouse myocardial tissue at 37 °C for 30 min. After washing with PBS, 5 µL TdT enzyme, 45 µL fluorescent labeling solution, and 50 µL TUNEL detection solution (Beyotime, C1086) were mixed thoroughly and added to each sample at 37 °C for 60 min. A fluorescence microscope (Leica, TCS SP8) was used to observe the stained sections.
Isolation of murine neutrophils
Fresh anticoagulant blood from mice was mixed with normal saline for injection and erythrocyte sedimentation solution in a 1:1:1 ratio, and the supernatant was obtained after standing at room temperature for 30–40 min. The cell suspension was spread on the surface of separation solution (Solarbio, P9201), and centrifuged at 600–1000 g for 25–30 min at room temperature. Neutrophils were collected from the surface of separation solution and washed for further experiments.
Cell culture and treatment
Murine neutrophils were cultured in RPMI 1640 (Gibco, 11875119) containing 3% fetal bovine serum (Gibco, 16140071), while primary murine cardiac fibroblasts (Pro-cell, CP-M074) were cultured in DMEM (Gibco, 6123034) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Hyclone, SV30010) at 37℃ with 5% CO2.
Verification of NET formation
Phorbol myristate acetate (PMA), an ester derivative of croton oil, can activate and recruit neutrophils, thereby inducing NET formation26,27. Murine neutrophils isolated from sham and model mice were seeded into plates (1 × 106) and divided into Control, Model, and PMA groups. In PMA group, PMA (100 nmol/L, Solarbio, P6741) was added to neutrophils from sham mice for 4 h, while the other groups were cultured normally. The culture medium was removed, and NETs were treated with restriction enzymes for 1 h. These restriction enzymes included BseRI (New England Biolabs, R0581 V), PacI (New England Biolabs, R0547 V), NdeI (New England Biolabs, R0111 V), and BslI (New England Biolabs, R0555 V), which cleaved DNA and effectively isolated the NETs. The supernatant containing NET fragments was collected and centrifuged to remove the remaining cell debris. NET formation was verified by staining with Sytox Green (Keygentec, KGA261).
Cell counting kit-8 (CCK-8) assay
Cardiac fibroblasts were seeded into 96-well plates. Different concentrations of NETs (0, 100, 200, 300, 400, and 500 ng/mL) obtained from model mice were incubated with cardiac fibroblasts for 24 h. CCK-8 solution (10 µL, Beyotime, C0037) was added to each well and incubated for 2 h. The absorbance at 450 nm was measured using a microplate reader (Wuxi Hiwell Diatek, DR-3518G).
Western blot
NETs (200 ng/mL) obtained from model mice were incubated with cardiac fibroblasts for 24 h, and the cells were then collected for Western blot. Myocardial tissue and cells were lysed using RIPA lysis buffer, and the protein concentrations of samples were detected by a bicinchoninic acid assay kit (Solarbio, PC0020). Proteins were transferred to polyvinylidene fluoride membranes (Beyotime, FFP24) after electrophoresis. Membranes were blocked with skim milk (Beyotime, P0216) for 1 h and then incubated with primary antibodies against collagen1 (1: 1000, Abcam, ab138492), α-SMA (1: 1000, CST, 19245), NLRP3 (1: 1000, CST, 19245), caspase-1 (1: 1000, Abcam, ab207802), GSDMD-N (1: 1000, Abcam, ab207802), and GAPDH (1: 10000, Abcam, ab181602) at 4℃ overnight. The following day, membranes were incubated with a secondary antibody (1: 10000, Beyotime, A0208) for 60 min. Protein bands were detected using an enhanced chemiluminescence kit (Pierce, 34579).
Transmission electron microscope
Cardiac fibroblasts were fixed in a 2.5% glutaraldehyde solution. After dehydration with graded alcohol and acetone, and embedding, the cell mass was made into sections using an ultra-thin microtome (Leica, RM2245). The sections were stained with 2% sodium acetate-lead citrate and observed under a transmission electron microscope (Hitachi, HT7800) at 80 kV.
Statistical analysis
Data are presented as mean ± SD. The t test and one-way analysis of variance with Tukey’s post hoc test were used to compare the differences between groups. All statistical analyses were performed on GraphPad 8.0 software, and P < 0.05 was considered statistically significant.
Results
NET formation occurred in UCM mice
Physiological parameters of mice were measured. The results showed that no significant difference was observed in Scr and BUN levels between UCM and sham mice on day 0, but these levels were markedly elevated in UCM mice on day 48 compared to controls (Fig. 1A). Moreover, UCM mice showed significantly lower cardiac E/A ratios and higher myocardial performance index (MPI) compared to Sham group, while ejection fraction (EF) and left ventricular diastolic anterior wall thickness (LVAWD) showed no significant difference between the two groups (Fig. 1B). As shown in Fig. 1C, the SBP of UCM mice was significantly higher than that of controls, with DBP having no significant difference. The above results indicated the successful establishment of UCM mouse model. Furthermore, the level of MPO-DNA, a DNA and neutrophil-derived protein complex, was significantly increased in Model group compared to Sham group (Fig. 1D), suggesting NET formation in UCM mice.

NETs were formed in UCM mice (A) Scr and BUN levels were detected by a fully automated biochemical analyzer. (B) Representative echocardiography and parameters including EF, E/A, MPI, and LVAWD. (C) Systolic and diastolic BP. (D) MPO-DNA levels in serum were detected by commercial kits. NET, neutrophil extracellular traps; UCM, uremic cardiomyopathy; Scr, serum creatinine; BUN, blood urea nitrogen; EF, ejection fraction; MPI, myocardial performance index; LVAWD, left ventricular diastolic anterior wall thickness; BP, blood pressure; MPO, myeloperoxidase, **P < 0.01.
DNaseI treatment inhibited NET formation in UCM mice
Immunofluorescence was used to detect NET component (MPO and CitH3) levels. The results demonstrated that the levels of MPO and CitH3 were significantly increased in model mice compared to controls, while they were decreased after DNaseI treatment (Fig. 2), indicating that DNaseI reduced NET levels in UCM mice.

DNaseI reduced NET formation in UCM mice. The levels of MPO and CitH3 were detected by immunofluorescence. Scale bar: 20 µm. DNaseI, deoxyribonuclease I; DAPI, 4’, 6-diamidino-2-phenylindole; CitH3, citrullinated histone H3, **P < 0.01.
The Inhibition of NET formation improved kidney injury and myocardial damage in UCM mice
As shown in Fig. 3A, compared to Sham group, UCM mice showed slower weight gain, while they gained more weight after DNaseI treatment. UCM mice had significantly greater heart weight than Sham mice, which was inhibited after DNaseI treatment (Fig. 3B). The levels of BUN and Scr, both indicators of renal function, had no significant difference on day 0 between groups (Fig. 3C). However, they were markedly increased on day 48 in UCM mice compared to Sham group, and DNaseI treatment reversed these changes (Fig. 3C). The myocardial tissue in Sham group was intact, with normal cell morphology and no injury, whereas the myocardial tissue of UCM mice exhibited severe injury, cell atrophy, and partial cell death, and DNaseI treatment alleviated myocardial injury in UCM mice (Fig. 3D). Furthermore, the levels of IL-1β and IL-6 were significantly elevated in UCM mice compared to controls, while they were decreased after DNaseI treatment (Fig. 3E).

The inhibition of NETs improved kidney injury and myocardial damage in UCM mice (A) Body weight of mice. (B) Heart weight of mice. (C) Scr and BUN levels were detected by a fully automated biochemical analyzer. (D) Representative pathological images from hematoxylin-eosin staining. Scale bar: 20 μm. (E) IL-1β and IL-6 levels in myocardial tissue were detected by commercial kits. IL, interleukin, *P < 0.05, **P < 0.01.
The Inhibition of NET formation reduced pyroptosis in myocardial tissue of UCM mice
TUNEL staining showed that, compared to Sham group, the level of apoptosis in myocardial tissue of UCM mice was significantly increased, but it was decreased after DNaseI treatment (Fig. 4A). Immunofluorescence results showed that the expression of caspase-1, a key pyroptosis-related protein, was significantly increased in Model group compared to controls, and DNaseI treatment altered caspase-1 expression (Fig. 4B). Moreover, the protein expression of NLRP3, GSDMD-N, and caspase-1 was significantly increased in UCM mice compared to Sham mice, while DNaseI treatment reduced these protein levels in UCM mice (Fig. 4C, Supplementary Fig. 1). These findings suggest that inhibiting NET formation reduces pyroptosis in UCM mice.

The inhibition of NETs reduced pyroptosis in myocardial tissue of UCM mice (A) Apoptosis rate of myocardial tissue was measured by TUNEL staining. Scale bar: 20 μm. (B) Caspase-1 expression in myocardial tissue was detected by immunofluorescence. Scale bar: 20 μm. (C) The expression of NLRP3, GSDMD-N, and caspase-1 in myocardial tissue was detected by Western blot, *P < 0.05, **P < 0.01.
Neutrophils isolated from UCM mice induced NET formation
Neutrophils were isolated from mice in Sham and Model groups, and PMA was added to neutrophils from Sham mice. The results showed that NETs were formed in both Model and PMA groups, with no significant difference between them, while no NETs were observed in Control group (Fig. 5A). Cardiac fibroblasts were treated with different concentrations of NETs induced by neutrophils from UCM mice. The results demonstrated that cell viability elevated with the increase in NET concentration, with 300 ng/mL of NETs reaching the maximum cell viability (Fig. 5B). The IC50 value for NETs was 193.9 ng/mL.

Neutrophils isolated from UCM mice induced NET formation in cardiac fibroblasts (A) NET formation was observed by Sytox Green staining. Scale bar: 10 μm. (B) The optimal NET concentration was determined by cell counting kit-8. PMA, phorbol 12-myristate 13-acetate; IC50, half-maximal inhibitory concentration, **P < 0.01.
NET treatment induced pyroptosis in cardiac fibroblasts
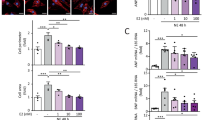
Cardiac fibroblasts were treated with 200 ng/mL of NETs for 24 h. As shown in Fig. 6A (Supplementary Fig. 2), the expression of fibrosis-related proteins (collagen1 and α-SMA) was significantly increased in NET-treated cardiac fibroblasts compared to controls. TEM results showed intact and normal cell membrane morphology in Control group, while NETs group exhibited swollen and ruptured cell membranes, with damaged nuclear structures and organelles (Fig. 6B), indicating the tendency of pyroptosis. The expression of NLRP3, GSDMD-N, and caspase-1 was markedly increased in NETs group compared to controls (Fig. 6C, Supplementary Fig. 3), suggesting that NET induced pyroptosis in cardiac fibroblasts.

NET treatment induced fibrosis and pyroptosis in cardiac fibroblasts (A) The expression of collagen1 and α-SMA was detected by Western blot. (B) Pyroptosis was observed under a transmission electron microscope. Red arrowheads indicate the vesicles released from the plasma membrane. Scale bar: 2 μm. (C) The expression of NLRP3, GSDMD-N, and caspase-1 was detected by Western blot. α-SMA, α-smooth muscle actin; NLRP3, NOD-like receptor protein 3; GSDMD, gasdermin D; GAPDH, glyceraldehyde-3-phosphate dehydrogenase, **P < 0.01.
Discussion
In this study, UCM mouse models were successfully established, and NET formation was observed in these mice. The inhibition of NETs by DNaseI treatment alleviated renal function, myocardial injury, and pyroptosis in UCM mice. Additionally, in vitro experiments confirmed that NET treatment promoted fibrosis and pyroptosis in cardiac fibroblasts, suggesting that NET-mediated pyroptosis may be a therapeutic target for UCM.
CKD increases the risk of UCM and is associated with myocardial changes28. The accumulation of uremic retention solutes is a sign of impaired renal excretion, and these uremic toxins can cause pathological cardiac remodeling and dysfunction due to their direct cardiotoxicity29. The 5/6 nephrectomy is a commonly used surgical model to induce CKD for exploring the pathological mechanisms of UCM30. Decreased serum urea and creatinine concentrations, increased left ventricular wall thickness, and diastolic dysfunction were observed in a 5/6 nephrectomy-induced CKD rat model31. Similarly, we observed increased Scr and BUN levels, decreased E/A ratios, elevated MPI, and increased SBP in UCM mice compared to controls, suggesting impaired kidney and cardiac function. Additionally, fibrosis-associated protein expression was increased in NET-induced myocardial fibroblasts, confirming the successful establishment of UCM models.
Activated neutrophils release NETs in response to stimulation, with NET formation being driven by type 4 protein-arginine deiminase. This process releases intracellular granular components that trap and destroy microorganisms, playing a crucial role in various diseases32. We found increased expression of MPO-DNA in the myocardial tissue of UCM mice, indicating NET formation in these mice. Additionally, neutrophils isolated from UCM mice, as well as PMA, induced NET formation. Consistent with our findings, MPO expression was increased, and massive NETs were formed in myocardial injury models induced by coxsackievirus B333. CaSR-mediated stimulation in polymorphonuclear neutrophils leads to an increase in NET formation, thus inducing cytotoxicity in cardiomyocytes34. Although these studies highlighted the role of NETs in myocardial injury and provided new insights into the pathological mechanisms of heart-related disease, the disease models they studied were not UCM, and our study revealed NET formation as an important factor influencing UCM progression.
NET clearance and effective removal of extracellular DNA, enzyme proteins such as MPO, and histones are necessary for maintaining tissue homeostasis. NETs are cleaved by the synergistic action of extracellular and secreted DNaseI and are degraded intracellularly by macrophages35. Consistent with this, our results showed that DNaseI treatment effectively inhibits NET formation in UCM mice. However, while DNaseI has been shown to reduce NET formation, its therapeutic effect on UCM remains unexplored. We further found that UCM mice demonstrated slower weight gain, decreased renal function, and severe myocardial tissue injury accompanied by inflammation, and DNaseI treatment improved these pathological changes, suggesting that inhibiting NET formation may be an effective treatment for UCM. Similarly, PSTPIP2 has been shown to improve renal dysfunction in aristolochic acid nephropathy through IL-19-mediated NET formation, and elevated NET levels have been observed in heart failure patients and mice, while NET inhibitor treatment improved heart function36,37. These findings highlight the potential of targeting NET formation to halt disease progression, but they primarily focus on changes in a single organ, whereas our study assessed changes both in kidney function and heart damage to better understand renal-related complications.
The proper structure and function of the cardiovascular system require a balance of cell proliferation and cell death, and excessive pyroptosis usually leads to tissue and organ dysfunction38. NLRP3 inflammasome-mediated pyroptosis of cardiomyocytes has been confirmed to be closely related to myocardial dysfunction and dilated cardiomyopathy39. However, this study only explored the classical pyroptosis pathway and did not further investigate its upstream mechanisms. The regulatory relationship between NETs and pyroptosis has been uncovered. NETs promote erectile dysfunction in diabetic rats through NLRP3-mediated pyroptosis, and microvesicles derived from pyroptotic macrophages can induce NET formation via mitochondrial transfer40,41. Zhang et al. found that NETs promote liver inflammation/fibrosis by inducing AIM2 inflammasome-dependent pyroptosis in macrophages42. Consistent with these findings, we revealed that pyroptosis in myocardial tissue was significantly increased in UCM mice, while DNaseI treatment inhibited pyroptosis in these mice. Moreover, NETs induced pyroptosis in myocardial fibroblasts. The above studies identified the regulatory role of NETs in pyroptosis and further clarified how NETs contribute to disease development. However, the upstream and downstream mechanisms involved in NET-mediated pyroptosis in myocardial fibroblasts remain unclear. A previous study has shown that PAI-1 deficiency promotes NET-mediated pyroptosis in acute lung injury by activating the PI3K/MAPK/AKT axis43. Moreover, alpha-linolenic acid pretreatment alleviates NET-induced alveolar macrophage pyroptosis by inhibiting pyrin inflammasome activation in ALI/ARDS mouse models44. As pro-inflammatory cell death, pyroptosis can activate cardiac fibroblasts and release pro-inflammatory factors such as IL-1β and IL-18, inducing myocardial interstitial fibrosis and promoting cardiac dysfunction45. We hypothesize that these mechanisms may play a role in NET-mediated pyroptosis to regulate UCM progression.
This study has some limitations. First, the effects of inhibiting NET formation on myocardial fibroblasts were not explored in vitro. Additionally, the findings of this study require further clinical validation.
Conclusion
In this study, we demonstrated that NETs were formed in UCM mice, and the NET inhibitor DNaseI inhibited their formation. The results revealed that NETs promoted UCM progression by inducing pyroptosis in myocardial fibroblasts. Therefore, targeting NET formation is a promising therapy for treating UCM, and our study highlights the need for further exploration to fully elucidate the underlying mechanisms by which NETs affect UCM.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Lim, Y. J., Sidor, N. A., Tonial, N. C., Che, A. & Urquhart, B. L. Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets. Toxins 13(2), (2021).
Kaesler, N. et al. Mapping cardiac remodeling in chronic kidney disease. Sci. Adv. 9 (47), eadj4846 (2023).
Arcari, L., Camastra, G., Ciolina, F., Danti, M. & Cacciotti, L. T(1) and T(2) mapping in uremic cardiomyopathy: an update. Cardiac Fail. Rev. 8, e02 (2022).
Garikapati, K., Goh, D., Khanna, S. & Echampati, K. Uraemic cardiomyopathy: A review of current literature. Clin. Med. Insights Cardiol. 15, 1179546821998347 (2021).
Law, J. P. et al. Clinical potential of targeting fibroblast growth Factor-23 and ΑKlotho in the treatment of uremic cardiomyopathy. J. Am. Heart Association. 9 (7), e016041 (2020).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol. Immunol. 18 (5), 1106–1121 (2021).
Ai, Y., Meng, Y., Yan, B., Zhou, Q. & Wang, X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol. Cell. 84 (1), 170–179 (2024).
Yarovinsky, T. O. et al. Pyroptosis in cardiovascular diseases: pumping gasdermin on the fire. Semin. Immunol. 69, 101809 (2023).
Wang, F. et al. Ghrelin inhibits myocardial pyroptosis in diabetic cardiomyopathy by regulating ERS and NLRP3 inflammasome crosstalk through the PI3K/AKT pathway. J. Drug Target. 32 (2), 148–158 (2024).
Duan, F. et al. Cortistatin protects against septic cardiomyopathy by inhibiting cardiomyocyte pyroptosis through the SSTR2-AMPK-NLRP3 pathway. Int. Immunopharmacol. 134, 112186 (2024).
Ravindran, M., Khan, M. A. & Palaniyar, N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules 9(8), (2019).
Burgener, S. S. & Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 12(7), (2020).
Neeli, I., Khan, S. N. & Radic, M. Histone deimination as a response to inflammatory stimuli in neutrophils. Journal of immunology (Baltimore, Md: 2008;180(3):1895 – 902. (1950).
Bonaventura, A., Vecchié, A., Abbate, A. & Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 9(1), (2020).
Li, X. et al. Analysis and validation of hub genes in neutrophil extracellular traps for the long-term prognosis of myocardial infarction. Gene 914, 148369 (2024).
Ichimura, S. et al. Neutrophil extracellular traps in myocardial tissue drive cardiac dysfunction and adverse outcomes in patients with heart failure with dilated cardiomyopathy. Circulation Heart Fail. 17 (6), e011057 (2024).
Pan, H. D. et al. Colchicine prevents perioperative myocardial injury in cardiac surgery by inhibiting the formation of neutrophil extracellular traps: evidence from rat models. Eur. J. cardio-thoracic surgery: official J. Eur. Association Cardio-thoracic Surg. 66(4), (2024).
Wu, Z., Li, W., Wang, S. & Zheng, Z. Role of deubiquitinase USP47 in cardiac function alleviation and anti-inflammatory immunity after myocardial infarction by regulating NLRP3 inflammasome-mediated pyroptotic signal pathways. Int. Immunopharmacol. 136, 112346 (2024).
Sollberger, G. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3(26), (2018).
Zhao, P. et al. Neutrophil extracellular traps induce pyroptosis of pulmonary microvascular endothelial cells by activating the NLRP3 inflammasome. Clin. Exp. Immunol. 217 (1), 89–98 (2024).
Zheng, F. et al. Neutrophil extracellular traps induce glomerular endothelial cell dysfunction and pyroptosis in diabetic kidney disease. Diabetes 71 (12), 2739–2750 (2022).
Huang, L. et al. Ficolin-A/2 aggravates severe lung injury through neutrophil extracellular traps mediated by gasdermin D-Induced pyroptosis. Am. J. Pathol. 194 (6), 989–1006 (2024).
Xu, Y. et al. Mechanism of lncRNA-ANRIL/miR-181b in autophagy of cardiomyocytes in mice with uremia by targeting ATG5. PloS One. 16 (9), e0256734 (2021).
Heuer, A. et al. Therapeutic targeting of neutrophil extracellular traps improves primary and secondary intention wound healing in mice. Front. Immunol. 12, 614347 (2021).
Zhou, P. et al. Uncoupling protein 2 alleviates myocardial ischemia/reperfusion injury by inhibiting cardiomyocyte ferroptosis. J. Vasc. Res. 61 (3), 109–121 (2024).
Lin, C. C., Hsieh, N. K., Liou, H. L. & Chen, H. I. Niacinamide mitigated the acute lung injury induced by phorbol myristate acetate in isolated Rat’s lungs. J. Biomed. Sci. 19 (1), 27 (2012).
Branitzki-Heinemann, K. et al. Formation of neutrophil extracellular traps under low oxygen level. Front. Immunol. 7, 518 (2016).
Wollenhaupt, J. et al. Pro-oxidative priming but maintained cardiac function in a broad spectrum of murine models of chronic kidney disease. Redox Biol. 56, 102459 (2022).
Lekawanvijit, S. Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal Syndrome. Toxins 10(9), (2018).
Soppert, J. et al. A systematic review and meta-analysis of murine models of uremic cardiomyopathy. Kidney Int. 101 (2), 256–273 (2022).
Dinh, H. et al. Role of the kisspeptin-KISS1R axis in the pathogenesis of chronic kidney disease and uremic cardiomyopathy. GeroScience 46 (2), 2463–2488 (2024).
Mutua, V. & Gershwin, L. J. A review of neutrophil extracellular traps (NETs) in disease: potential Anti-NETs therapeutics. Clin. Rev. Allergy Immunol. 61 (2), 194–211 (2021).
Li, B. et al. Interleukin-37 alleviates myocardial injury induced by coxsackievirus B3 via inhibiting neutrophil extracellular traps formation. Int. Immunopharmacol. 113 (Pt A), 109343 (2022).
Zeng, J. et al. Calcium-sensing receptor and NF-κB pathways in TN breast cancer contribute to cancer-induced cardiomyocyte damage via activating neutrophil extracellular traps formation. Cell. Mol. Life Sci. 81 (1), 19 (2024).
Demkow, U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int. J. Mol. Sci. 24(5), (2023).
Du, C. et al. PSTPIP2 ameliorates aristolochic acid nephropathy by suppressing interleukin-19-mediated neutrophil extracellular trap formation. eLife 13, (2024).
Mang, G. et al. Von Willebrand factor exacerbates heart failure through formation of neutrophil extracellular traps. Eur. Heart J. 45 (37), 3853–3867 (2024).
Zhaolin, Z., Guohua, L., Shiyuan, W. & Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 52 (2), e12563 (2019).
Zeng, C. et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 34, 101523 (2020).
Xu, Y., Ren, Y., Zou, W., Ji, S. & Shen, W. Neutrophil extracellular traps promote erectile dysfunction in rats with diabetes mellitus by enhancing NLRP3-mediated pyroptosis. Sci. Rep. 14 (1), 16457 (2024).
Kuang, L. et al. Pyroptotic Macrophage-Derived microvesicles accelerate formation of neutrophil extracellular traps via GSDMD-N-expressing mitochondrial transfer during Sepsis. Int. J. Biol. Sci. 20 (2), 733–750 (2024).
Zhang, Y. et al. Neutrophil extracellular traps facilitate liver inflammation/fibrosis progression by entering macrophages and triggering AIM2 inflammasome-dependent pyroptosis. Cell. Communication Signaling: CCS. 22 (1), 556 (2024).
Aji, N. et al. PAI-1 Deficiency Promotes NET-mediated Pyroptosis and Ferroptosis during Pseudomonas Aeruginosa-induced Acute Lung Injury by Regulating the PI3K/MAPK/AKT Axis. Inflammation. (2024).
Liu, C. et al. Alpha-linolenic acid pretreatment alleviates NETs-induced alveolar macrophage pyroptosis by inhibiting Pyrin inflammasome activation in a mouse model of sepsis-induced ALI/ARDS. Front. Immunol. 14, 1146612 (2023).
Geng, X. F. et al. The mechanism and promising therapeutic strategy of diabetic cardiomyopathy dysfunctions: focus on pyroptosis. J. Diabetes Complicat. 38 (10), 108848 (2024).
Funding
This research was funded by the Project of Scientific Research Foundation of Chinese Medicine (2022ZB035) and the General Project of the Medical and Health of Zhejiang Province (2022 KY550, 2024 KY629, 2023 KY043).
Author information
Authors and Affiliations
Contributions
Conception and design of the research: Wei Shen; Acquisition of data: Wenli Zou; Analysis and interpretation of data: Xianyun Ye and Minmin Wang; Statistical analysis: Yan Ren; Obtaining funding: Wei Shen and Ying Xu; Drafting the manuscript: Ying Xu; Revision of manuscript for important intellectual content: Wei Shen.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All animal experiments complied with the ARRIVE guidelines and were approved by the Experimental Animal Welfare Ethics Committee of Yangzhou University (approval number: 202407036). All experimental procedures were performed in accordance with relevant guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Y., Ren, Y., Zou, W. et al. Neutrophil extracellular traps aggravate uremic cardiomyopathy by inducing myocardial fibroblast pyroptosis. Sci Rep 15, 17509 (2025). https://doi.org/10.1038/s41598-025-02383-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02383-3
Keywords
This article is cited by
-
Chronic kidney disease-associated cardiomyopathy: clinical features, pathophysiology and treatment
Nature Reviews Cardiology (2026)
