
Abstract
Neoplastic transformation is accompanied by critical changes in cell mechanical properties, including reduced cell elasticity. By leveraging such mechanical flaw, exposure to low intensity therapeutic ultrasounds (LITUS) has been proposed as a tool for selective killing of cancer cells. Here, we have developed dynamic models to address the morpho-mechanical differences between prostate cancer and non-tumoral counterparts and studied the effects of LITUS on cell viability. We show that LITUS exposure (1 MHz) leads to cancer-selective cytoskeletal disruption associated to loss of nuclear envelope integrity, DNA damage marked by γH2AX and 53BP1 foci, and release of DNA into the cytosol with activation of the cGAS–STING signaling cascade. Mechanistically, the LINC complex, which connects the cytoskeleton to nucleoskeleton and chromosomes, is critical to mediate nuclear rupture triggered by LITUS. Accordingly, genetic ablation of the LINC component SUN2 tuned down DNA damage and cGAS–STING signaling while the inactivation of the endosomal sorting complex (ESCRT), required for the transport machinery that preserves the nuclear envelope integrity, enhanced cell killing by LITUS. In conclusion, LITUS induce cancer cell DNA damage and an innate immune response, this suggesting LITUS treatment as a mechanobiology-driven anti-neoplastic strategy.
Similar content being viewed by others

Introduction
Malignant transformation involves critical changes in the cell mechanical properties, most cancer cells showing an elastic stiffness decrease up to 70% with respect to the healthy counterpart1,2,3,4,5,6,7,8,9. Reduced cell stiffness can be positively selected in malignant transformation, since it fosters cancer cell motility, adhesion, invasion and metastatization2,6,10,11,12,13,14,15,16,17. Indeed, mechanical stiffness inversely correlates with migration and invasion of cancer cells10. Accordingly, the ability to deform may facilitate cell intravasation and extravasation during metastasis formation6. Tumor cells, as well as tumor cell nuclei, soften during migration and soft cells have been shown to be more tumorigenic and metastatic than stiff ones14,15,17.
Cancer cell softening is mediated by the alteration of the cytoskeleton, cellular/nuclear morphology, DNA content, and nuclear/cell and nucleolar/nuclear volume ratios13. Hence, cancer cells feature an impairment in the expression of nuclear structural proteins, lamin A/C depletion and downregulation of LINC complex12,18,19. The resulting re-distribution of intracellular mechanical stresses kindles a cascade of events, including nuclear softening furthering cell migration, aberration of nuclear morphology, chromatin reorganization, as well as alteration of nuclear/cell and nucleolar/nuclear volume ratios. In turn, this affects nucleus-cytoskeleton coupling and mechano-transduction pathways12,13,19,20.
Measurement of cellular mechanical properties may serve to discriminate between normal and cancer cells4. Atomic Force Microscopy (AFM) represents one of the most adopted biophysical techniques to probe cell mechanical phenotype and measure the elastic differences between cancer and normal cells1,2,21.
The different mechanical properties of cancer cells compared to normal ones has suggested the possibility of applying mechanical-based strategies to selectively target cancer cells2,13,22,23,24,25,26,27. Previous studies have demonstrated that cancer cells are more sensitive than normal ones to ultrasound (US) exposure28,29. Based on well-known thermal, cavitation and sonoporation effects of US on tissues, both High Intensity Focused Ultrasounds (HIFU) and Low Intensity Therapeutic Ultrasounds (LITUS, typically characterized by intensities below 5 W/cm2 and frequencies < 3 MHz) have been considered for possible anti-neoplastic applications28,30,31,32,33,34,35,36.
Cell mechanical models have been developed to explore the effects that LITUS produces on cell structures. These models suggest that stiffness differences entail distinct in-frequency response, both at single-cell level and in cell aggregates, ultimately resulting in cancer cell toxicity22,23,24,37,38. Selective ablation of cancer cells, named as ”oncotripsy”23,25, has been experimentally confirmed by using pulsed LITUS (0.5–0.67 MHz, pulse duration > 20 ms) to induce selective disruption of suspended breast, colon, and leukemia cancer cells. Mechanistically, the formation of acoustic standing waves and cell-seeded cavitation leads to cytoskeletal disruption and cancer cell death13. Other studies have demonstrated the reduced viability of various cancer cell lines in suspension upon US excitation21,26,39,40. However, despite the studies highlighting the selective effects of US on cancer cells, the molecular mechanisms underlying such effects remained elusive.
Prostate cancer is the most common male malignancy, and it is potentially amenable to selective ablative therapies41. AFM measurements have shown that prostate cancer cells are more easily deformable and feature a reduced elastic modulus with respect to normal prostate cells3,42. Here, we have studied the molecular mechanisms underlying the effects of LITUS on prostate cancer cells. Initially, we assessed the morphology and stiffness of adhering prostate cancer cells via AFM. This allowed us to generate ad hoc dynamic models to tailor LITUS in terms of power, frequency and time duration to maximize the vibration of cancer cells, while minimizing the effects on non-tumoral cells. Our data indicate that LITUS led to a cancer cell-selective cytoskeletal disruption, which was translated by the LINC protein complex into nuclear envelope breakage, accumulation of DNA damage and release of DNA fragments into the cytosol, thereby triggering activation of the cGAS–STING innate immunity signaling.
Materials and methods
Cell culture
Human DU145 cells were from American Type Culture Collection (ATCC, Manassas, VA, USA) and were grown in EMEM supplemented with 10% fetal bovine serum (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA); LNCaP cells were from ATCC and were grown in RPMI-1640 supplemented with 10% fetal bovine serum (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA); BPH1 cells were from European Collection of Authenticated Cell Cultures (ECACC, Porton Down, Salisbury, UK) and were cultured in RPMI-1640 supplemented with 20% FBS; VCap cells were from ATCC and were cultured in DMEM supplemented with 10% FBS; PNT1A were from ATCC and were cultured using Prostate Epithelial Cell Growth Kit (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA). For transfection, DU145 cells were transfected using LIPOFECTAMINE 2000, accordingly to manufacturer instructions (#11668-019, INVITROGEN, Carlsbad, CA, USA). IFN-beta-GFP reporter plasmid was kindly donated by Dr. Roger Greenberg43. Lamin A/C mCherry plasmid was purchased from Addgene (Plasmid #55068). DU145 and BPH1 were transfected with siRNA targeting SUN2 and BAF (see below) using RNAimax lipofectamine accordingly to manufacturer instructions (#13778100, Thermo Fisher Scientific).
AFM-based measurements of cell stiffness and morphology
The stiffness of the PNT1A, BPH1, DU145 and VCap cell lines was measured by exploiting an ad hoc assembled experimental station for performing AFM on samples submerged in liquid media. An AFM instrument (NT-MDT NTEGRA Prima) was integrated with an inverted optical microscope (Olympus IX71) equipped with a 10x objective (Olympus PLN 10x), to position the stand-alone AFM head directly on the top of the optical microscope to observe cells during testing procedures. Suitable soft colloidal and silicone probes with nominal cantilever spring constant ranging between 0.01 and 0.77 N/m (TipsNano CPC_SiO2-A and B and CSG10) were used for measurements; a top view camera (5MP CMOS sensor) was used for visualizing in real-time both the cantilever and the adherent cells in the culture medium within a Petri dish. This allowed the correct placement for the topology and stiffness scanning, which was performed in contact mode. For each dish, different regions, comprising one or more cells, were probed and height and stiffness maps were thus obtained (see Fig. 1A and B).

(A–D) AFM-based measurements and static schemes. (A) AFM scanning of cell cultures and data collection; snapshots during AFM acquisition and geometrical 3D-profiles of cells are shown. The histogram reports the comparison between the stiffness data of DU145 cancer cells (purple) and PNT1A non-tumoral cells (cyan). (B) Morpho-mechanical post-processing analyses of single cells performed to reconstruct cell geometries and associated stiffness maps (orthogonal axes indicate the directions along which profiles of cell geometry and stiffness are extracted). (C) Cartoon of the equivalent static scheme used for estimating nuclear and cytoskeletal stiffness constants informed by AFM measurements (see also Supplementary Information). (D) (Left) Geometry-based reconstruction of regularized cell profiles and evaluation of shape and position of the internal nuclei. (Right) Combination of morphological and mechanical schemes was used to estimate the spatially inhomogeneous elastic response of different cell districts upon AFM measurements, i.e. of cell nucleus (NUC), nuclear envelope (NE) and cytoskeletal regions (CYT). (E, F) Dynamic modelling and in-frequency response of cancer and non-tumoral cells. (E) Illustration of a vibrating adherent single cell submerged in liquid medium, and the associated structural scheme of the single-cell dynamic model highlighting the cytoskeleton and nucleus viscoelastic elements vibrating under the weight of a liquid column. (F) Charts with theoretical predictions of the harmonic response of DU145 and PNT1A cells illustrated in (B). Two resonance-like peaks were identified for each cell type, with frequency of 1 MHz suggesting 1 MHz-LITUS as the optimal treatment for selectively attacking cancer cells minimizing effects on non-tumoral cells.
Cell treatment with ultrasounds
At day 0, 20,000 cells were seeded on plates. The day after seeding, cells were sonicated by using ultrasounds (US) at 1 MHz and 0.1 W/cm2 for 20 min. Sonication was implemented according to the scheme and the actual experimental setup described in Supplementary information (Supplementary Information, Fig. S1). Specifically, an arbitrary waveform generator (Tektronix AFG31022 25 MHz 2 channels) was exploited for generating a continuous sinusoidal signal at a frequency of 1 MHz and amplitude A = 75 mV. This was then amplified through a 50x voltage amplifier (Falco System WMA-300 High voltage amplifier) working in the needed bandwidth (i.e. DC − 5 MHz) and transmitted to a piezoelectric contact transducer (Olympus V106-RM 2.25 MHz central frequency) producing longitudinal US waves. In this way, an average density power P = A2/(2 π Z R2) = 0.1 W/cm2 was obtained, Z = 50 Ohm being the nominal input impedance of the transducer and R = 0.65 cm its nominal effective radius. The Petri dishes containing cells were placed directly on the top of the transducer by using a US gel as coupling medium for optimizing wave transmission. The proper US transmission was verified by employing a PVDF hydrophone (RP acoustics PVDF hydrophone, Type s), connected to an amplifier (Femto 40/60 dB hydrophone signal amplifier HVA-10 M-60-F) and, finally, to a digital oscilloscope (Tektronix TBS2074 70 MHz 4 channels). This allowed us to estimate an amplitude of the pressure wave stimulating seeded cells in the order of units of kPa, a value comparable with the order of magnitude of cells’ Young moduli. Such a measurement was preliminarily performed before each experimental campaign by using an uncovered Petri dish filled only with culture medium -in the same amount used for cell cultures- to preserve sterility conditions of the cells during the tests and avoid compromising their viability. Preliminary analyses with a thermocouple were conducted for ensuring that any significant temperature variation occurred for the considered sonication parameters.
Cell proliferation assay
DU145, LNCaP, VCap, PNT1A and BPH1 (20,000/well) were seeded in p30 plates and sonicated (1 MHz, 0.1 W/cm2 for 20 min). After treatment, cells were counted daily for 6 consecutive days. The medium was changed every 2 days.
Clonogenic assay
Untreated or sonicated cells were counted, and 1,000 cells were plated in 6-well plate and grown for 12 days. Colonies were then stained with a solution of Crystal Violet (#C6158, Sigma-Aldrich, Merck KGaA, Darmstadt, Germania) in 20% Methanol, 10% Acid Acetic for 20 min. The colonies were counted after extensive washing with 20% Methanol and 10% Acid Acetic.
Immunofluorescence
For indirect immunofluorescence, cells plated on glass coverslips were sonicated and fixed in Methanol 100% for 10 min at − 20 °C (for γH2AX) or in Paraformaldehyde 4% for 20 min at room temperature (depending on the protein to visualize). Then, cells were washed in Phosphate Buffered Saline (PBS 1X), permeabilized with 0.2% Triton X-100 (5 min) and incubated with the appropriate antibodies for 1 h at room temperature. Slides were then washed with PBS 1X and incubated with secondary antibodies for 30 min in the dark (Alexa Fluor anti-mouse #A32723 and anti-rabbit #A32740, Thermo Fisher Scientific, Waltham, MA, USA). Cells were finally washed with PBS 1X, counterstained with DAPI (#P36962, Thermo Fisher Scientific, Waltham, MA, USA). and analyzed with a confocal fluorescence microscope Leica (LSM700) using the ZenBlue software. Images were acquired with laser scanning confocal microscope (LSM 700; Carl Zeiss MicroImaging, Inc.) equipped with a planapo 63x oil-immersion (NA 1.4). In all experiments, pinhole diameter size was fixed at one Airy unit, and images were collected using the same setting (laser power, detector gain, and threshold of fluorescence intensity). Three-dimensional reconstructions were performed with Zeiss ZenBlue software collecting the Z-slices from the top to the bottom of the cells. Morphometric analyses were performed using ImageJ software, exploiting nuclear descriptor tool. Specifically, area, perimeter, and roundness (that is how closely nucleus shapes a perfect circle) were measured by drawing ROI (Region of Interest) around nucleus labeled by Lamin A/C the perimeter around the entire nucleus as revealed by Lamin A/C labeling.
The percentage of cells showing abnormal Tubulin signal or aberrant Lamin A/C signal or cGAS accrual signal was measured by analyzing fifteen different fields, each containing at least 5 cells. For the analysis of γH2AX or 53bp1 foci, at least 50 different cells were analyzed.
Western blotting
Protein lysates were prepared according to standard procedures. Briefly, cells were lysed in a buffer containing 50 mM N-2- hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES; pH 7.5), 1% (vol/vol) Triton X-100, 150 mM NaCl, 5 mM EGTA, 50 mM NaF, 20 mM sodium pyrophosphate, 1 mM sodium vanadate, 2 mM phenylmethylsulphonyl fluoride (PMSF) and 1 ug/ml aprotinin and clarified by centrifugation at 10,000 xg for 15 min. Protein concentration was estimated with a modified Bradford assay (Bio-Rad, Munich, Germany) and lysates were subjected to Western blot analysis. Immune complexes were detected with the enhanced chemiluminescence kit (Thermo Fisher Scientific, Waltham, MA, USA).
RNA extraction and RT-PCR
Total RNA was isolated from BPH1 and DU145 cells by using TRIzol reagent (Invitrogen, #15596026). Two µg of RNA was reverse-transcribed with M-MLV Reverse Transcriptase (ThermoFisher Scientific, # 28025021). qRT-PCR reactions were done in triplicate using SYBR green (Biorad, #1725271), and fold changes were calculated with the following formula: 2^− (sample 1 ΔCt – sample 2 ΔCt), where ΔCt is the difference between the amplification fluorescence threshold of the mRNA of interest and the mRNA of the GAPDH used as an internal reference. The sequence of the primers used for qRT-PCR are:
-
BAF Forward TGGCTGGGATTGGTGAAGTCCT.
-
BAF Reverse CGCCACAAGTGTCTTTCAGCCA.
-
GAPDH Forward GCCATCAATGACCCTTCATTT.
-
GAPDH Reverse TTGACGGTGCCATGGAATTTT.
Flow cytometry
Cells were fixed and permeabilized using FOXP3 staining buffers (#130-093-142, Milteny Biotec, Bergisch Gladbach, Germany), subsequently incubated with primary antibodies and then with secondary antibodies at 4 °C, for 1 h each. Cells were then analyzed by flow cytometry using an Accuri C6 apparatus (Becton Dickinson Bioscience, Franklin Lakes, NJ, USA). Collected data were analyzed using the Accuri C6 software.
List of primary antibodies
-
Mouse Tubulin: #T5168 (Sigma-Aldrich, Merck KGaA, Darmstadt, Germania).
-
Rabbit Lamin A/C: #108,595 (Abcam, Cambridge, UK).
-
Rabbit Lamin B: #32,535 (Abcam, Cambridge, UK).
-
Rabbit γH2AX: #53-9865-82 (R&D systems, Minneapolis, MN, USA).
-
Rabbit 53BP: #4937 (Cell signaling technology Danvers, MA, USA).
-
Rabbit cGAS #E5V3W (Cell signaling technology Danvers, MA, USA).
-
Rabbit pIRF3 (Ser386): #37,829 (Cell signaling technology, Danvers, MA, USA).
-
Rabbit pTBK1 (Ser172): #5483 (Cell signaling technology, Danvers, MA, USA).
-
Rabbit Anti-SUN2: #EPR6557 (Abcam, Cambridge, UK).
List of SiRNA
-
SUN2 (1) CCGCATCGGGCTGGCAGACTA.
-
SUN2 (2) CACCCGATGTTCTGAGACCTA.
-
BAF (1) TACGACAATAGCAATCTTT.
-
BAF(2) TTCATCTTTCTTTAGCACC.
Statistics
Two-tailed unpaired Student’s t test was used as detailed in the Figure legend, as indicated. Differences were considered significant when p was less than 0.05. All statistical analyses were carried out using GraphPad Instat software (version 3.06.3).
Results
Engineering design of US treatment to selectively attack cancer cells
Alterations in cell morphology and elastic response are recognized as physical hallmarks of neoplastic cells, this suggesting the possibility of exploiting mechanical vibrations induced by ultrasounds to selectively damage cancer cells1,2,3,4,5,6,7,8,9,13,22,24,37.
In order to demonstrate the feasibility of this principle and investigate the associated molecular mechanisms, we followed a series of experimental and theoretical steps aimed at obtaining engineering predictions of US frequencies and powers to make the treatment as effective and selective as possible. We selected three human prostate cancer cell lines, DU145 (refractory to anti-androgen drugs) and LNCaP and VCap (both sensitive to anti-androgen drugs). As controls, we used BPH1, from a human benign prostate hyperplasia and PNT1A from a human normal gland42. To dose LITUS in terms of power, frequency and time duration, and optimize the sonication protocol for maximizing vibration of cancer cells while minimizing the effects on non-tumoral cells, we first obtained morphology and stiffness maps of cells seeded in culture dish via AFM (Fig. 1A). Direct tumor-to-normal comparison confirmed that non-tumoral prostate cells were stiffer than the malignant ones, with PNT1A and BPH1 normal cells showing stiffness values of 9.1 ± 4.6 mN/m and 4.1 ± 1.1 mN/m, respectively, and VCap and DU145 cancer cells showing stiffness values of 1.3 ± 1.0 mN/m and 2.3 ± 0.8 mN/m, respectively (Supplementary information, Fig. S2).
Then, through a reverse engineering method that considered the inherent heterogeneity of single cells, we used point-wise height and stiffness AFM data to obtain quantitative estimations of the mechanical properties of relevant cell districts, i.e. elastic stiffness of cell nucleus, nuclear envelope and cytoskeleton (Fig. 1B and Supplementary information, Fig. S3). A static equivalent scheme (Fig. 1C) and regularized cell 3D reconstructions (Fig. 1D) were used to obtain virtual replicas of the cells including the key morpho-mechanical features of the intracellular components derived from AFM measurements. Such a procedure allowed us to gain estimations of stiffness of nuclear (15.06 ± 1.89 mN/m vs. 7.44 ± 4.10 mN/m), cytoskeletal (13.5 ± 1.87 mN/m vs. 2.87 ± 0.17 mN/m) and peri-nuclear (18.25 ± 3.39 mN/m vs. 7.65 ± 5 mN/m) material in PNT1A and DU145 cells, respectively (Fig. 1D).
Subsequently, to predict the in-frequency response and the maximum expected oscillations of cancer and non-tumoral single cells under US treatment, we built up a cell-specific and experimentally informed dynamic model by integrating morphological and viscoelastic features (Fig. 1E and F and Supplementary information). With respect to DU145 and PNT1A, Fig. 1F shows that frequency peaks move as result of the interplay of cell size, adhesion and micro-mechanical cell properties. At low frequencies (< 100 kHz), oscillation peaks of cancer and normal cells featured a gap of about 30 kHz while, at higher frequencies, cancer cells exhibited maximum oscillations at about 1 MHz compared to 500 kHz in the case of non-tumoral cells. Therefore, we selected 1 MHz as the operational LITUS frequency to amplify vibrations of cancer cells with the lowest impact on non-tumoral ones. In agreement with actual pressures (units of kPa) registered by hydrophone in the culture medium, an input power of 0.1 W/cm2 was set to avoid undesired thermal effects. Finally, we estimated 20 min in continuous wave (CW) as suitable time length of US radiation for inducing dismounting of cytoskeleton and disruption of cellular and sub-cellular structures, due to the cooperation of mechanical resonance at 1 MHz and associated fatigue-like phenomena. Calculations indeed allowed us to anticipate that vibration-induced stress cycles of the order of 109 could be adequate to damage cancer cell microstructures44.
LITUS exposure reduces human prostate cancer cell viability
Preliminarily, sub confluent cells seeded in culture dish were treated with LITUS (Supplementary information, Fig. S3) (1 MHz, CW 20 min, 0.1 W/cm2) and counted. Number of cancer cells was reduced by about 20% soon after sonication, while virtually no effect was measured in non-tumoral cells (Fig. 2A). To better measure LITUS effect, cells were treated and then counted at regular time intervals up to 6 days. The curves reported in Supplementary information (Fig. S4A and B) show that sonication selectively reduced the growth of cancer cells. Finally, a clonogenic assay was performed; upon LITUS treatment, cells were counted and then plated (1,000/well). Colonies were counted after 12 days. Again, while the LITUS effect was negligible in benign BPH1 and PNT1A cells, colony forming ability was clearly reduced (by about 20%) in the case of cancer cells (DU145 and LNCap) (Fig. 2B). To verify whether multiple treatments could increase the effect of LITUS on cells we repeated LITUS every 12 h for 4 times, in both DU145 and BPH1. We observed that each single treatment was inducing a similar decrease in cell count (around 15–20% of cell compared to control) in DU145 thereby featuring an additive but not synergistic effect in cancer cells. On the other hand, multiple treatments were not inducing any reduction in cell count in BPH1 cells (Supplementary information, Figure S4C).

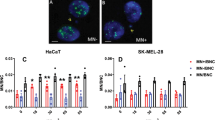
(A) Counts (percentage) of the indicated cells after treatment with LITUS (1 MHz, CW 20 min, 0.1 W/cm2) compared to untreated (NT) cells. Each point represents one individual experiment. (B) Left: representative images of colony-formation assays of the indicated cell lines treated or not with LITUS (1 MHz, CW 20 min, 0.1 W/cm2) and then grown for 12 days. Colonies were counted after crystal violet staining. Right: number of colonies counted in each well. *p < 0.05; **p < 0.01; each dot represents a different experiment. (C) Left: Representative confocal immunofluorescent images of the indicated cell lines treated or not with LITUS (1 MHz, CW 20 min, 0.1 W/cm2). Soon after sonication cells were fixed and stained with anti-Tubulin antibody (green signal) and DAPI (blue signal). Arrows indicate cells presented at higher magnification in Supplementary information (Fig. S5A). Right: Cells showing abnormal Tubulin signal were counted and represented in the bar graph as the percentage of the total number of cells in each field. Each dot represents a different field. 3 independent experiments were performed. Scale bar 10 μm. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
All together, these findings indicate that low intensity ultrasound sonication causes a selective damage of human prostate cancer cells.
LITUS causes cytoskeleton and nuclear envelope rupture with accumulation of DNA damage
Disassembly of microtubules has been observed in HeLa cells upon treatment with LITUS45. Thus, we applied indirect immunofluorescence to check cytoskeleton damage induced by LITUS on prostate cancer cells. Cells were plated on glass coverslips, sonicated and stained with anti-tubulin antibodies. As shown in Fig. 2C and Supplementary information (Fig. S5A), the treatment induced a partial disassembly of microtubules, with thinning and depolymerization of microtubules, and loss of their canonical radial distribution from the centrosome toward the cell periphery in about 20–30% of cancer cells. Again, this effect was virtually undetectable in non-tumoral controls (Supplementary information, Fig. S5B).
The nucleus is the largest organelle and a major player in cell mechanics6,11. Nucleus morphology and size are altered in neoplastic transformation, typically with an increased nucleus/cytoplasm ratio12. The nuclear envelope (NE) is molecularly connected to the cytoskeleton, thus allowing extracellular mechanical forces to be transmitted to the nucleus46. The primary structural scaffold of the NE is represented by the lamina, composed of two types of lamin proteins (LmnA/C and LmnB)47,48. Thus, we asked whether LITUS exposure caused any change to NE. Prostate cancer cells and controls were sonicated and stained by immunofluorescence with LmnA/C antibodies. A fraction (between 10 and 30%, depending on the cell line) of the cancer cells featured nuclear membrane protrusions (blebs) as well as formation of micronuclei (Fig. 3A and Supplementary information, Fig. S6A). This effect was negligible in BPH1 and PNT1A non tumoral controls (Supplementary information, Fig. S6B).

(A) Left: Representative images of Lamin A/C immunofluorescence in untreated (left) or treated (1 MHz, CW 20 min, 0.1 W/cm2) cancer cells. Arrows indicate cells presented at higher magnification in Supplementary information (Fig. S6A) After sonication, cells were fixed and subjected to confocal immunofluorescence with anti-Lamin A/C antibody (red signal) and DAPI (blue signal). Right: Cells showing aberrant Lamin A/C signal were counted and represented as the percentage of the total number of cells observed in each field. Each dot represents a different field. 3 independent experiments were performed. (B) Left: Representative images of confocal immunofluorescent analysis of DU145, LNCap and VCap cell lines treated or not with LITUS (1 MHz, CW 20 min, 0.1 W/cm2). After 6 h from sonication, cells were fixed and subjected to immunofluorescence with anti- γH2AX antibody (green signal) and DAPI (blue signal). Right: The number of γH2AX signal (foci) per nucleus were counted in at least fifty different cells and represented in the bar graph. 3 independent experiments were performed. Scale bar 10 μm. *p < 0.05; **p < 0.01; ****p < 0.0001.
Moreover, to better define the alterations of nuclear structure we performed a deep morphometric analysis using ImageJ software (see Methods), measuring area, perimeter and roundness and we found significant variations in DU145, but not in BPH1 treated cells with respect to perimeter and roundness (Supplementary information, Fig. S6C and D). No change in terms of nuclear area was found (data not shown). Similar effects were noted in DU145, but not BHP1 cells, stained with LmnB antibodies (Supplementary information, Fig. S7A and B). Chromatin herniation was detectable within NE blebs by DAPI staining. Thus, upon transient expression of Histone 2B-GFP in DU145 cancer cells, a fluorescent signal was observed within such blebs (Supplementary information, Fig. S7C).
We also tested whether ultrasound-induced mechanical stress to the nucleus and/or loss of NE integrity may be accompanied by DNA damage. Cells were exposed to ultrasounds and then, after 6 h, stained for γ-H2AX, a marker of DNA double-strand breaks49. Sonication induced intense DNA damage (on average, 9–19 γ-H2AX foci/nucleus compared to 3–5 foci/nucleus in untreated cells) in prostate cancer but not in non-tumoral cells (Fig. 3B and Supplementary information, Fig. S8A). The staining of DU145 cancer and BPH1 normal cells with 53BP1, another marker of DNA damage, confirmed such results (Supplementary information, Fig. S8B). DNA damage foci (γ-H2AX or 53BP1) directly correlated with the presence of NE ruptures, being more abundant in cells displaying blebs and/or micronuclei (revealed either with IF for Lamin A/C or by expressing with mCherry-tagged Lamin A/C) than in cells with an intact NE, either immediately after the treatment or after 6 h (Supplementary information, Fig. S8C, S9A and B).
All together, these findings indicate that low intensity ultrasound sonication causes a disassembly of the microtubule cytoskeleton with NE rupture, associated to formation of blebs and micronuclei, and accumulation of DNA damage.
LITUS-associated DNA leakage in the cytoplasm mediates activation of cGAS–STING signaling
Disruption of the NE may cause the release of genomic DNA into the cytoplasm50 and cytosolic DNA triggers an innate immune response, being recognized as a damage-associated molecular pattern (DAMP) by the pattern recognition receptor (PRR) cGAS51,52,53. Mechanistically, cytosolic DNA drives the formation of cytoplasmic foci containing cGAS and these condensates promote cGAS activity54. Active cGAS functions as a cyclic GMP-AMP synthase, thereby producing cGAMP, a second messenger that, in turn, triggers the activation of the adapter STING at the endoplasmic reticulum. Active STING translocates to the Golgi, where it recruits TANK binding kinase 1 (TBK1) and the transcription factor interferon regulatory factor 3 (IRF3). Finally, IRF3 enters the nucleus and promotes the expression of pro-apoptotic type 1 interferon response and of other immune-stimulated genes51,52,53. This system works as a critical anti-cancer immune surveillance55 (Fig. 4A).

(A) Schematic representation of cGAS-STING pathway. Upon binding cytosolic DNA, the cyclic GMP–AMP synthase (cGAS) produces 2′3′ cyclic GMP–AMP (cGAMP); cGAMP interacts with the stimulator of interferon genes (STING) which, by recruiting TANK-binding kinase 1 (TBK1), promotes TBK1 autophosphorylation, STING phosphorylation and the recruitment of interferon regulatory factor 3 (IRF3). The phosphorylation of IRF3 by TBK1 enables IRF3 dimerization and translocation to the nucleus to induce gene expression of type I interferons, interferon-stimulated genes (ISGs), and several other inflammatory mediators, pro-apoptotic genes and chemokines (see text). (B) Left: Representative images of confocal immunofluorescent analysis of BPH1 and DU145 cells treated or not with LITUS (1 MHz, CW 20 min, 0.1 W/cm2). After 12 h from sonication, cells were fixed and stained with anti-cGAS antibody (green signal) and DAPI (blue signal). Right: Cells showing cGAS accrual signal were counted and represented as the percentage of the total number of cells observed in each field. Each dot represents a different field. 3 independent experiments were performed. Scale bar 10 μm. (C) Left: anti phospho-TBK1 staining of DU145 cells treated as described in (B) and subjected to cytofluorimetric analysis 12 h after treatment. Right: bar graph representing the mean fluorescence intensity (MFI) of phospho-TBK1 signal compared to untreated (NT cells). Each point represents one individual experiment. (D) Left: anti phospho-IRF3 staining of DU145 cells treated as described in (B) and subjected to cytofluorimetric analysis. Right: bar graph representing the MFI of phosho-IRF3 signal compared to untreated (NT cells). Each point represents one individual experiment 12 h after treatment. (E) DU145 cells were transiently transfected with IFN beta-GFP (IFN GFP) reporter and after 48 h treated or not with LITUS (1 MHz, CW 20 min, 0.1 W/cm2). Cells were fixed and subjected to cytofluorimetric analysis 12 h after treatment. GFP-positive cells were counted and reported in the bar graphs as percentage of total number of cells. *p < 0.05; **p < 0.01; ****p < 0.0001; NS: not significant.
Thus, we checked cGAS activation and signaling upon sonication. A few hours after LITUS exposure, DU145 and LNCaP cancer cells, but not BPH1 controls, featured an increased formation of cGAS foci in 10–20% cells (Fig. 4B and Supplementary information, Fig. S10A). In addition, by expressing mCherry-tagged Lamin A/C we could demonstrate that cGAS staining was predominant in cells displaying aberrant lamina (Supplementary information, Fig S10B). Consistently, cytofluorimetric analysis of phospho-TBK1 and -IRF3 in DU145 cells revealed a similar increase of positive cells upon sonication (Fig. 4C and D).
To further confirm the activation of the cGAS-STING signaling pathway, we used a reporter plasmid in which GFP expression was controlled by the IFN-beta promoter, a well-known target of IRF3 transcription factor. As shown in Fig. 4E, sonication of DU145 cells was able to induce GFP expression, indicating the activation of the IRF3 transcriptional activity.
All together, these findings indicate that low intensity ultrasound exposure elicits an innate immunity response with activation of the cGAS-STING signaling.
LINC complex is critical for the transmission of LITUS effects to the nuclear envelope and DNA damage accumulation
Mechanical forces can be transmitted from microtubules to the lamina, thereby causing nucleus deformation. The NE hosts the LINC (Linker of Cytoskeleton and Nucleoskeleton) complex, that mechanically connects the cytoskeleton to the nucleus48,56. LINC is comprised of an outer nuclear membrane protein containing a KASH domain and an inner nuclear membrane protein containing a SUN domain. Nesprins, belonging to the KASH domain protein family, bind to microtubules, intermediate filaments, and actin in the cytoplasm and to the SUN proteins (SUN1 and SUN2) in the perinuclear space57. In turn, SUN proteins bind to the nuclear lamina and chromatin58.
Thus, we asked whether the LINC complex functions as the molecular linker between ultrasound-induced cytoskeleton damage and the nuclear lamina and DNA damage that promotes activation of cGAS-STING signaling. To this aim, we set-up a RNAi based approach to silence SUN2 protein expression and tested effects of SUN2 silencing on ultrasound-induced damage of NE and DNA, and cGAS signaling. SUN2 downregulation in DU145 cells (Supplementary information, Fig. S11A) blunted NE disruption upon sonication but had no effect on the ultrasound-mediated damage of microtubules, thus functionally placing LINC downstream microtubules and upstream NE (Fig. 5A). Importantly, SUN2 silencing also reduced formation of γ-H2AX and cGAS foci compared to control cells upon treatment with LITUS (Fig. 5B and C, and Supplementary information Fig. S11B and C).
All together, these findings point to LINC complex as a molecular linker between ultrasound-induced cytoskeleton damage and nuclear rupture.

(A) Left: Representative images of Lamin A/C and Tubulin confocal immunofluorescence in siSUN2 DU145 cells compared to control (siCTR). DU145 were transfected with siCTR or siSUN2; after 48 h, cells were treated with LITUS (1 MHz, CW 20 min, 0.1 W/cm2), immediately fixed and subjected to immunofluorescence. Right: cells showing abnormal Tubulin and Lamin A/C signal were counted and plotted in the bar graph as the percentage of cells compared to total number of cells in each field. Each dot represents a different field. 3 independent experiments were performed. Scale bar 10 μm. (B) Number of γH2AX foci in siCTR and siSUN2 DU145 cells. Cells were treated as described in (A) and subjected to anti-γH2AX immunofluorescence 6 h after treatment. (C) Percentage of cells positive for cGAS foci; each dot represents the percentage of cells showing cGAS accrual per field; siCTR and siSUN2 DU145 cells were treated as described in (A) and subjected to immunofluorescence with the anti-cGAS antibody 12 h after treatment. (D) DU145 were transfected with siRNA against BAF (siBAF) or control (siCTR). After 48 h, cells were subjected to LITUS (1 MHz, CW 20 min, 0.1 W/cm2), immediately fixed and subjected to immunofluorescence and counted. Bar graphs representing the percentage of cells showing aberrant Lamin A/C signal. Each dot represents a different field. 3 independent experiments were performed. (E) Colony-formation assays of siCTR and siBAF cells treated or not with 1 MHz and then grown for 12 days. Colonies were counted after crystal violet staining; bar graph representing the number of colonies in each well. 3 independent experiments were performed. *p < 0.05; **p < 0.01; ***p < 0.001.
Blockade of NE repair enhances LITUS effects
NE integrity is preserved by the activity of the endosomal sorting complex required for transport machinery (ESCRT). This is a highly conserved membrane remodeling complex constituted by charged multivesicular body proteins called CHMPs59. CHMP7 protein and its inner nuclear membrane binding partner, LEMD2, are specifically involved in NE reformation and repair60. Upon NE ruptures the autointegration factor (BAF) rapidly binds to the exposed chromatin to recruit the inner nuclear membrane protein LEMD2 and CHMP7 thus promoting the repair of the damaged membranes48. We hypothesized that the inhibition of such a membrane resealing process may render cancer cells more sensitive to LITUS-mediated mechanical stress forces. Thus, we silenced BAF in DU145 and BPH1 cells (Supplementary information, Fig. S12A) and monitored NE alterations (by Lamin A/C immunofluorescence) and cell viability after low intensity ultrasound exposure. After LITUS treatment, BAF-silenced (siBAF) DU145 cells showed a significant increase of aberrant Lamin A/C signal compared to control cells (Fig. 5D and Supplementary information, Fig. S12B). Consistently, siBAF cells showed a strong reduction in the number of colonies after 10 days of treatment compared to siCTR cells (Fig. 5E and Supplementary information, Fig. S12C). On the contrary, BAF silencing did not significantly sensitize BPH1 cells to LITUS treatment (Supplementary information, Fig. S12D and E).
These data indicate that the NE repair system is necessary to minimize the harmful consequences of LITUS-mediated NE ruptures and protect cells from mechanical stress forces.
Discussion
Reduced cell stiffness is a general hallmark of malignant transformation. This feature may render cancer cells exquisitely sensitive to mechanical damage elicited by specific low intensity ultrasounds (LITUS)22,23. Accordingly, previous reports have shown that malignant cells are susceptible to LITUS-mediated damage2,13,26,27,28,29,30. Here, we have studied the effects of LITUS on prostate cancer cells with respect to non-tumoral ones. Our data show that malignant prostate cells are sensitive to cytotoxic and cytostatic effects of LITUS. This was associated to microtubule cytoskeleton disruption, which was transduced to the nuclear lamina through the LINC protein complex. In turn, nuclear deformation, alongside the formation of blebs and micronuclei, was associated to DNA damage and release in the cytosol with activation of the cGAS-STING innate immunity response.
It is worth to highlight that as US pressure acts interfering with the intrinsic proper frequency of the tumor cells, this amplifying the mechanical signal and thus inducing – over time – a progressively growing vibration amplitude in all structures inside the cell (i.e. cytoskeleton and nucleus). As a consequence of the time duration of the treatment (20 min for the case at hand), the increasing oscillations lead to dismounting of cytoskeletal architecture, followed by an increase of mechanical stresses inside both microtubules and NE due to concomitant factors, which can be mainly ascribed to the transition from small to large structural deformations and to fatigue-induced phenomena associated to the high number of load cycles (of the order of 109). As a matter of fact, this strong dynamic excitation of microtubules and nucleus membrane creates additional mutual forces exchanged across nucleus and cytoskeleton elements, further increasing the internal forces in microtubules and NE. Therefore, the triggering factor provoking microtubules disruption and NE damaging must be ascribed to the cooperative action of resonance and cyclic fatigue loading that cannot be obtained by disrupting directly microtubules, for example using nocodazole.
Reduced cell viability, cytoskeleton disruption and cGAS-STING activation were observed in only a fraction (no more than 30%) of cultured prostate cancer cells. It has been proposed that interaction with ECM, genomic variation, or flow during different phases of cell cycle may contribute to variabilities in mechanical properties of cancer cells23. Clarifying the role of these variables may allow depicting strategies to enhance LITUS mediated cancer cell death. In this context, inactivation of the NE repair machinery by interfering with BAF expression enhanced the killing effect of ultrasound increasing the number of damaged cells.
The role of the cGAS-STING pathway in cancer is complex. On one side, genomic instability-mediated cGAS-STING signaling induces a pro-tumorigenic inflammatory response. On the other, the same pathway promotes the secretion of several immune-stimulatory factors, collectively termed senescence-associated secretory phenotype (SASP), which can attenuate, in a cell-autonomous manner, cancer cell growth or promote antitumor immune surveillance51,52,61. For instance, cGAS-STING induced type I IFNs are known to act as antiproliferative signals51. Accordingly, several evidence indicates that cGAS-STING signaling is critical for the response to major cancer therapies, including radio-, chemo-, and immune-therapy51. Importantly, the capability of cGAS-STING of recruiting immune cells has been suggested to be critical not only for the local tumor response to radiotherapy but also to mediate tumor regression outside of the irradiated field (the so-called “abscopal” effect)43,62. Several strategies may enhance cGAS-STING-mediated anti-tumor effects, such as use of chemotherapeutics to induce chromosome instability, Aurora B kinase inhibitors to increase micronuclei formation, chemical STING agonists to potentiate the pathway, and immune checkpoint blockers to foster anti-tumor immunity43,51,52. It should be explored whether these strategies are able to potentiate anti-tumor effects of low intensity ultrasounds in vivo.
Prostate cancer is characterized by a variable natural history and clinical behavior, with many patients bearing a low-risk disease and deserving only a low intensity treatment41. Several minimally invasive therapies have been proposed to treat localized prostate cancer while mitigating extensive tissue damage. These include cryotherapy, high intensity focused ultrasound, photodynamic therapy, interstitial laser thermotherapy, electroporation, and stereotactic radiotherapy41. Our study suggests LITUS as a potential novel tool to be explored to selectively control prostate localized malignant lesions while sparing normal surrounding tissues.
Dealing with tissues or macroscopic masses, the dynamic response to US is expected to become consequently more complex, all the constituent structures being solicited at different scales and thus vibrating involving a wider spectrum of frequencies37,38,63,64. However, regardless of the complexity of the elastic or viscoelastic structures and the number and micro-organization of their constituents, there always exists a set of frequencies at which one can selectively make resonant one of those components37,38, provided that a sufficient accuracy of both the micro-mechanical model of the tissue and the experimental measurements of masses and stiffness involved in the dynamics can be ensured. This would lead to create a bioengineering platform (integrating theoretical and computational models, biological analyses, imaging and mechanical testing) aimed at designing patient/tissue-specific LITUS protocols, opening the way for envisioning mechanics-based therapeutic strategies potentially less invasive or even more effective than current ones, either used alone or synergistically with drugs.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Change history
26 June 2025
The original online version of this Article was revised: The Acknowledgements section in the original version of this Article was incomplete. It now reads:
“All Authors gratefully thank Gaetano Manfredi for the fruitful discussion. This work was supported by the PROSCAN (PON: National Operative Project) grant (ARS01_ 01384) and by the Project CIRO (Campania Imaging Infrastructure for Research in Oncology (B61G17000190007). Authors also acknowledge the support by Italian Ministry of University and Research (MUR), under the complementary actions to the NRRP “Fit4MedRob - Fit for Medical Robotics” Grant (# PNC0000007). A.C. and ARC also acknowledge the financial support under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender No. 1409 published on 14.9.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU, under the Projects n P2022MXCJ2 CUP E53D23018000001 - Grant Assignment Decree No. 1379, and P2022M3KKC MECHAVERSE – CUP E53D23017310001 - Grant Assignment Decree No. 1385, adopted on 1.9.2023 by the Italian Ministry of University and Research (MUR).”
References
Tao, N. J., Lindsay, S. M. & Lees, S. Measuring the microelastic properties of Biological-Material. Biophys. J. 63, 1165–1169 (1992).
Cross, S. E., Jin, Y. S., Rao, J. & Gimzewski, J. K. Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol. 2, 780–783 (2007).
Faria, E. C. et al. Measurement of elastic properties of prostate cancer cells using AFM. Analyst 133, 1498–1500 (2008).
Lekka, M. et al. Cancer cell recognition - Mechanical phenotype. Micron 43, 1259–1266 (2012).
Prabhune, M., Belge, G., Dotzauer, A., Bullerdiek, J. & Radmacher, M. Comparison of mechanical properties of normal and malignant thyroid cells. Micron 43, 1267–1272 (2012).
Alibert, C., Goud, B. & Manneville, J. B. Are cancer cells really softer than normal cells? Biol. Cell. 109, 167–189 (2017).
Korayem, M. H., Sooha, Y. H. & Rastegar, Z. MCF-7 cancer cell apparent properties and viscoelastic characteristics measurement using AFM. J. Braz. Soc. Mech. Sci. 40 (2018).
Korayem, M. H., Shahali, S. & Rastegar, Z. Experimental determination of folding factor of benign breast cancer cell (MCF10A) and its effect on contact models and 3D manipulation of biological particles. Biomech. Model. Mechanobiol. 17, 745–761 (2018).
Korayem, M. H., Heidary, K. & Rastegar, Z. The head and neck cancer (HN-5) cell line properties extraction by AFM. J. Biol. Eng. 14 (2020).
Swaminathan, V. et al. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 71, 5075–5080 (2011).
Zwerger, M., Ho, C. Y. & Lammerding, J. Nuclear mechanics in disease. Annu. Rev. Biomed. Eng. 13, 397–428 (2011).
Denais, C. & Lammerding, J. Nuclear mechanics in cancer. Adv. Exp. Med. Biol. 773, 435–470 (2014).
Mittelstein, D. R. et al. Selective ablation of cancer cells with low intensity pulsed ultrasound. Appl. Phys. Lett. 116 (2020).
Rianna, C., Radmacher, M. & Kumar, S. Direct evidence that tumor cells soften when navigating confined spaces. Mol. Biol. Cell. 31, 1726–1734 (2020).
Lv, J. et al. Cell softness regulates tumorigenicity and stemness of cancer cells. EMBO J. 40, e106123 (2021).
Donis-Keller, H. et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 2, 851–856 (1993).
Roberts, A. B. et al. Tumor cell nuclei soften during transendothelial migration. J. Biomech. 121 (2021).
Fischer, E. G. Nuclear morphology and the biology of Cancer cells. Acta Cytol. 64, 511–519 (2020).
Singh, I. & Lele, T. P. Nuclear morphological abnormalities in cancer: A search for unifying mechanisms. Results Probl. Cell. Differ. 70, 443–467 (2022).
Zink, D., Fischer, A. H. & Nickerson, J. A. Nuclear structure in cancer cells. Nat. Rev. Cancer. 4, 677–687 (2004).
Sun, S. et al. Ultrasound-mediated immune regulation in tumor immunotherapy. Mater. Today Adv. 14 (2022).
Fraldi, M., Cugno, A., Deseri, L., Dayal, K. & Pugno, N. M. A frequency-based hypothesis for mechanically targeting and selectively attacking cancer cells. J. R Soc. Interface. 12, 20150656 (2015).
Heyden, S. & Ortiz, M. Oncotripsy: targeting cancer cells selectively via resonant harmonic excitation. J. Mech. Phys. Solids. 92, 164–175 (2016).
Fraldi, M. et al. Small-on-Large fractional Derivative-Based Single-Cell model incorporating cytoskeleton prestretch. J. Eng. Mech. 143 (2017).
Heyden, S. & Ortiz, M. Investigation of the influence of viscoelasticity on Oncotripsy. Comput. Method Appl. M. 314, 314–322 (2017).
Schibber, E. F. et al. A dynamical model of Oncotripsy by mechanical cell fatigue: selective cancer cell ablation by low-intensity pulsed ultrasound. Proc. R. Soc. A 476 (2020).
Landgraf, L. et al. Focused ultrasound treatment of a spheroid in vitro tumour model. Cells 11 (2022).
Lejbkowicz, F. & Salzberg, S. Distinct sensitivity of normal and malignant cells to ultrasound in vitro. Environ. Health Perspect. 105 (Suppl 6), 1575–1578 (1997).
Mizrahi, N. et al. Low intensity ultrasound perturbs cytoskeleton dynamics. Soft Matter. 8, 2438–2443 (2012).
Lejbkowicz, F., Zwiran, M. & Salzberg, S. The response of normal and malignant cells to ultrasound in vitro. Ultrasound Med. Biol. 19, 75–82 (1993).
Tomizawa, M., Ebara, M., Saisho, H., Sakiyama, S. & Tagawa, M. Irradiation with ultrasound of low output intensity increased chemosensitivity of subcutaneous solid tumors to an anti-cancer agent. Cancer Lett. 173, 31–35 (2001).
Wood, A. K. et al. The antivascular action of physiotherapy ultrasound on murine tumors. Ultrasound Med. Biol. 31, 1403–1410 (2005).
Chumakova, O. V., Liopo, A. V., Evers, B. M. & Esenaliev, R. O. Effect of 5-fluorouracil, optison and ultrasound on MCF-7 cell viability. Ultrasound Med. Biol. 32, 751–758 (2006).
Schuster, A. et al. Cell specific ultrasound effects are dose and frequency dependent. Ann. Anat. 195, 57–67 (2013).
Li, F. et al. The dual effect of ultrasound-targeted microbubble destruction in mediating Recombinant adeno-associated virus delivery in renal cell carcinoma: transfection enhancement and tumor Inhibition. J. Gene Med. 16, 28–39 (2014).
Wood, A. K. W. & Sehgal, C. M. A review of Low-Intensity ultrasound for Cancer therapy. Ultrasound Med. Biol. 41, 905–928 (2015).
Fraldi, M., Cutolo, A., Carotenuto, A. R., Palumbo, S. & Pugno, N. A lesson from earthquake engineering for selectively damaging cancer cell structures. J. Mech. Behav. Biomed. 119 (2021).
Cutolo, A. et al. Ultrasound waves in tumors via needle irradiation for precise medicine. Sci. Rep. 12, 6513 (2022).
Geltmeier, A. et al. Characterization of dynamic behaviour of MCF7 and MCF10A cells in ultrasonic field using modal and harmonic analyses. PLoS ONE 10 (2015).
Tamboia, G. et al. A comparative analysis of low intensity ultrasound effects on living cells: from simulation to experiments. Biomed. Microdevices. 24, 35 (2022).
Sandhu, S. et al. Prostate cancer. Lancet 398, 1075–1090 (2021).
Bastatas, L. et al. AFM nano-mechanics and calcium dynamics of prostate cancer cells with distinct metastatic potential. Biochim. Biophys. Acta. 1820, 1111–1120 (2012).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017).
Zitnay, J. L. et al. Accumulation of collagen molecular unfolding is the mechanism of Cyclic fatigue damage and failure in collagenous tissues. Sci. Adv. 6 (2020).
Hrazdira, I., Skorpikova, J. & Dolnikova, M. Ultrasonically induced alterations of cultured tumour cells. Eur. J. Ultrasound. 8, 43–49 (1998).
Maniotis, A. J., Chen, C. S. & Ingber, D. E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. U S A. 94, 849–854 (1997).
Gruenbaum, Y., Margalit, A., Goldman, R. D., Shumaker, D. K. & Wilson, K. L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell. Biol. 6, 21–31 (2005).
Halfmann, C. T. et al. Repair of nuclear ruptures requires barrier-to-autointegration factor. J. Cell. Biol. 218, 2136–2149 (2019).
Nakamura, A. J., Rao, V. A., Pommier, Y. & Bonner, W. M. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell. Cycle. 9, 389–397 (2010).
Nader, G. P. D. et al. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 184, 5230– (2021).
Li, T. & Chen, Z. J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-Sensing cGAS-STING pathway in Cancer. Cancer Discov. 10, 26–39 (2020).
Ghosh, M., Saha, S., Li, J. & Montrose, D. C. Martinez, p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell. 83, 266–280e266 (2023).
Du, M. & Chen, Z. J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017).
Hatch, E. M. & Hetzer, M. W. Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell. Biol. 215, 27–36 (2016).
Khilan, A. A., Al-Maslamani, N. A. & Horn, H. F. Cell stretchers and the LINC complex in mechanotransduction. Arch. Biochem. Biophys. 702, 108829 (2021).
Lityagina, O. & Dobreva, G. The LINC between mechanical forces and chromatin. Front. Physiol. 12, 710809 (2021).
Y. Olmos, The ESCRT Machinery: Remodeling, repairing, and sealing membranes. Membranes 12 (2022).
Olmos, Y., Perdrix-Rosell, A. & Carlton, J. G. Membrane binding by CHMP7 coordinates ESCRT-III-Dependent nuclear envelope reformation. Curr. Biol. 26, 2635–2641 (2016).
Hong, C. et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 607, 366–373 (2022).
Barker, H. E., Paget, J. T., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer. 15, 409–425 (2015).
Fraldi, M. & Angelo, R. Carotenuto. Cells competition in tumor growth poroelasticity. J. Mech. Phys. Solids. 112, 345–367 (2018).
Blanco, B. et al. Modeling low-intensity ultrasound mechanotherapy impact on growing cancer stem cells. Math. Comput. Simul. 228, 87–102 (2025).
Acknowledgements
All Authors gratefully thank Gaetano Manfredi for the fruitful discussion. This work was supported by the PROSCAN (PON: National Operative Project) grant (ARS01_ 01384) and by the Project CIRO (Campania Imaging Infrastructure for Research in Oncology (B61G17000190007). Authors also acknowledge the support by Italian Ministry of University and Research (MUR), under the complementary actions to the NRRP “Fit4MedRob - Fit for Medical Robotics” Grant (# PNC0000007). A.C. and ARC also acknowledge the financial support under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender No. 1409 published on 14.9.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU, under the Projects n P2022MXCJ2 CUP E53D23018000001 - Grant Assignment Decree No. 1379, and P2022M3KKC MECHAVERSE – CUP E53D23017310001 - Grant Assignment Decree No. 1385, adopted on 1.9.2023 by the Italian Ministry of University and Research (MUR).
Author information
Authors and Affiliations
Contributions
G.F., A.R.C., A.C., St.P., M.M., and Si.P. performed research; M.S. and T.R. analyzed data; M.F. and F.C supervised the research and wrote the paper.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Federico, G., Carotenuto, A.R., Cutolo, A. et al. Ultrasound-induced mechanical damage of cancer cell cytoskeleton causes disruption of nuclear envelope and activation of cGAS-STING. Sci Rep 15, 18037 (2025). https://doi.org/10.1038/s41598-025-03317-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03317-9
