
Abstract
Inborn errors of immunity (IEI) are monogenic disorders with a wide spectrum of clinical phenotypes including immune dysregulation, autoimmunity, autoinflammation, and malignancy. IEI may have life-threatening consequences, thus precise and timely genetic diagnosis is crucial for improved access to treatment, genetic counselling and prevention. We aimed to investigate the genotypic findings in a cohort of children with IEI from Romania, in order to understand the diagnostic yield of genetic testing, genetic characterization of IEI and impact of genetic diagnosis. Clinical and genetic investigations were performed in 92 children (50% boys) with IEI phenotype, evaluated between 2018 and 2024, in a tertiary reference genetic center in Timisoara, Romania. Sanger sequencing, next generation sequencing panels with genes associated with IEI (1–474 genes), WES and WGS were used. Disease-causing variants for IEI (pathogenic/likely pathogenic) were identified in 37/92 (40.2%) participants, in 25 genes. 14/92 (15.2%) participants had relevant variants of uncertain significance. Median age at genetic testing was 4.38 (IQR 2–12.8) years. 15/37 (40.5%) of patients with Disease-causing variants had a family history of IEI. NGS gene panels were used in 43/92 (46.7%) patients, WES in 29 (31.5%), while WGS in 20 (21.7%). Twelve patients had more than one genetic test. Most frequent genes with Disease-causing variants identified were: JAGN1(5 patients) and AIRE(3). A great variety of genes were identified as causative for IEI. Considering the highly variable and unspecific phenotype, together with no family history in most cases, prioritizing genetic investigations offers the best option for timely diagnosis and treatment.
Similar content being viewed by others

Introduction
Inborn errors of immunity (IEI) represent a group of disorders affecting normal immune development and function. The previous term “primary immunodeficiencies” has been recently replaced with IEI, in recognition of the diversity of clinical presentations1. IEI are characterized by the presence of susceptibility to infections, autoimmunity, autoinflammatory diseases, allergy, bone marrow failure, and/or malignancy2. Reported collective prevalence of these conditions is likely to range between 1/1000–1/5000 depending on the specific primary genetic defect and geographical region3,4,5. There are different types of primary immune defects: immunodeficiencies affecting cellular and humoral immunity, combined immunodeficiencies, combined immunodeficiencies with syndromic or other features, thymic defects with additional congenital anomalies, antibody defects, diseases of immune dysregulation, congenital defects of phagocyte number or function, defects in intrinsic and innate immunity or complement deficiency1. IEI frequently involve an underlying genetic etiology6. The clinical presentation of IEI can vary widely depending on the specific genetic defect involved. Genetic testing is valuable for risk stratification, treatment decision, genetic counselling and disease prevention.
According to the most recent update by the International Union of Immunological Societies (IUIS) expert committee the last few decades have seen the identification of more than 500 causes of inborn errors of immunity1,7. Next generation sequencing (NGS) has become more accessible worldwide8, and more recently also in Romania9,10,11,12,13,14,15,16. While detailed mapping of the genetic landscape is of great interest for describing founder variants as well as disease prevalence at a country level, population data regarding the genetic etiology of IEI in Romanian people are lacking. In this context, we aimed to investigate the diagnostic yield of genetic testing for IEI among Romanian pediatric patients at a tertiary genetic referral center. Furthermore, we aimed to investigate differences between those with significant genetic findings (disease-causing variants) versus those with negative results, in order to understand if patient selection could be improved, in a setting with limited resources. Also, we aimed to evaluate gene phenotype correlation in Romanian children with IEI, to help improve clinical management.
Methods
Clinical evaluation
Patient’s clinical evaluations were performed between September 2018 and September 2024, in a tertiary hospital in Timisoara, Romania. The study cohort consisted of consecutive pediatric patients with IEI phenotype who received genetic testing. Most patients were unrelated; however, related individuals were not excluded from the study, in order to capture phenotypic variability. IEI was suspected according to the following criteria: pathological susceptibility to infections defined as severe, persistent, unusual, recurrent (acronym SPUR)17, immune dysregulation: granuloma, autoimmunity, recurrent fever, eczema (early onset, atypical, refractory to treatment), lymphoproliferative disorders, inflammatory bowel disease (early onset, atypical, refractory to treatment) (german mnemonic GARFIELD—AWMF guidelines)18, recurrent angioedema and /or a family history of primary immunodeficiency19.
The patient evaluation included: symptom assessment (documented using HPO terms20,21, age at symptoms onset, type of encountered pathogens, complications of vaccination, response to vaccination, non-infectious manifestations (autoinflammations, autoimmunity, cancer, severe allergy), laboratory findings (lymphocyte sub-populations, lymphocyte proliferation, immunoglobulins and specific antibodies), non-immunological differential diagnosis, in order to enable a disease classification among IEI categories. Notably, the mandatory alive vaccines in Romania include Bacillus Calmette–Guérin (BCG) vaccination (live attenuated strain), Poliomyelitis vaccination (inactivated or live attenuated strains), and Measles vaccination (superattenuated live strain), while optional alive vaccines include Varicella-zoster vaccination (live attenuated strain) and Rotavirus Vaccination (live attenuated strains).
Clinical data of the index patients were collected retrospectively from the time of their IEI suspicion. Family history was evaluated in each index patient using a 3 or 4 generations pedigree.
A clinical classification was set for each child with IEI phenotype, based on clinical presentation and laboratory findings (including genetic results if disease causing), using the nine IEI category as classified by the Human inborn errors of immunity: 2024 Update on the classification from the International Union of Immunological Societies Expert Committee, available on the International Union of Immunological Societies website7.
Genetic assessment
In all patients, genomic deoxyribonucleic acid (gDNA) was isolated from whole blood samples using the MagCore® Automated Nucleic Acid Extractor and the MagCore® Genomic DNA Whole Blood Kit (RBC Bioscience, New Taipei City, Taiwan), following the manufacturer’s protocol. gDNA was quantified using both UV–Vis absorbance BioTek Epoch Spectrophotometer (Agilent Technologies Inc., Santa Clara, USA) and Qubit® double-stranded DNA (dsDNA) High Sensitivity (HS) Assay Kit (Invitrogen, Carlsbad, USA).
Genetic investigations used different Illumina NGS methods (Illumina, San Diego, USA), as follows: gene panels with disease-associated IEI genes (ranging between 1–474 genes), whole exome sequencing (WES) and whole genome sequencing (WGS). The patients supported no cost. The choice of method was dependent on the clinical presentation, the suspected type of IEI, and the access to resources. The genetic test was selected after a consensus decision-making discussion between the pediatric immunologist and geneticist. Some patients had more than one test performed, in case of negative results and suggestive clinical phenotype. When multiple tests were performed in a patient, only the most comprehensive one (typically WES or WGS) was reported in this analysis.
Gene panels were selected based on clinical presentation and included various commercially available NGS panels (e.g. Primary Immunodeficiency, Chronic Granulomatous Disease panels) from Invitae (San Francisco, USA), between September 2018–August 2022, and Blueprint Genetics (Espoo, Finland), between September 2022–September 2024.
WES and WGS used Illumina short NGS, performed at Novogene (Cambridge, United Kingdom). DNA fragmentation was performed using sonication. For WES, the library preparation was conducted using the Agilent Technologies SureSelect Human All Exon V6, targeting all protein-coding exons and exon–intron boundaries (± 20 base pairs). Library preparation for WGS utilized NEB Next® Ultra™ DNA Library Prep Kit (Cat No. E7370L) (New England Biolabs Hitchin, United Kingdom). Sequencing was performed on Illumina Novaseq 6000 PE150 high-throughput sequencing platform. Average coverage for WES was 100X, while for WGS it was 30X. Bioinformatics analysis and data interpretation were conducted at the Center of Genomic Medicine, a research laboratory of “Victor Babeș” University of Medicine and Pharmacy, Timișoara, Romania. End-to-end bioinformatics algorithms were implemented, including base alignment, primary filtering of low-quality reads and probable artifacts, and annotation of variants to the GRCh38 reference genome. The DRAGEN v4.0 Illumina software was used for alignment and identification of variants.
Variant interpretation was conducted in the context of population, segregation, computational, functional and phenotypic data available. The Human Phenotype Ontology (HPO)20, was used to integrate patient’s phenotype with sequencing data in order to prioritize causal genes. Databases of human genetic variation used, included the Genome Aggregation Database (gnomAD v4; https://www.gnomad.broadinstitute.org) 22 and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) 23 The Clinical Genome Resource (ClinGen; https://www.clinicalgenome.org/24 was used for the expertly curated reports of clinically relevant genes and their variants. SpliceAI25 and Human Splicing Finder26 were used to investigate the variant effect on alternative splicing. Variants from WGS were analyzed using MOON software (www.diploid.com/moon). All variants with a minor allele frequency (MAF) less than 1% in the gnomAD database and disease-causing variants reported in HGMD®, and ClinVar were considered. The recommendations from American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) were used to classify a genetic variant as benign (B), likely benign (LB), variant of uncertain significance (VUS), likely pathogenic (LP), and pathogenic (P)27. Results were categorized as disease causing/significant (if P or LP), negative (if B or LB), or uncertain (VUS), depending on the classification of the variant identified and the inheritance pattern of the associated condition. Carrier status for autosomal recessive disorders, identified in the present cohort, was not discussed in this article; however, the data is available in supplementary file.
Candidate variants were confirmed by Sanger sequencing in the patient and the parents (where available), using predesigned or self-designed primers and BigDye™ Terminator v3.1 method (Thermo Fisher Scientific, Waltham, USA).
Anonymized clinical information was included in the analysis. This study was conducted in accordance with the declaration of Helsinki and approved by the local ethics committee (11/08.05.2020) form the University of Medicine and Pharmacy 'Victor Babeș' Timișoara). Informed consent for genetic testing and research study was obtained from all the parents of the pediatric participants of the study. The consent form included the option to have secondary findings reported.
Statistical analysis
Clinical and genomic data was collected using a Microsoft Excel file. Continuous variables (non-normally distributed) were expressed as median and interquartile range (IQR) the first and third quartile (Q1-Q3). Categorical variables were presented as numbers (and proportions). Independent samples Mann–Whitney U-tests was used for non-parametric continuous variables. Categorical variables were compared using the chi-squared test. Pearson Chi-Square value was reported for most categorical variables, while Fischer’s Exact Test was reported in case variables in a subgroup were less than 5. Statistical significance was considered if the 2-sided P value was less than 0.05. SPSS v12.0 (IBM SPSS Inc., New York, USA) was used for statistical analysis. The two-mode network (bipartite network) analysis of the relation between gene/disease category and symptoms was conducted using the NetworkX v3.4.2 Python library28 and the Matplotlib v3.10.0 for the graphical presentation29.
Results
A total of 92 consecutive patients (male to female ratio 1:1), age 0–18 years, with a clinical phenotype suggestive for IEI, that had a genetic evaluation, were included in this analysis (Table 1). Median age at IEI at genetic testing was 4.38 (IQR 2.0–12.8) years. More than a quarter of children (27.1%) presented symptoms before the age of 1 year. Median time from symptom onset until genetic testing was 1.67 (IQR 0.3–4.8) years, ranging from neonatal period to 18 years delay. 25/92 (27.1%) patients presented symptoms before age 1 year. One fifth of all patients had a family history of IEI. NGS panels were used in 46.7% patients, WES in 31.5% patients, while WGS in the remaining. Twelve patients had more than one genetic test performed (WGS and WES in nine children, gene panel and WGS in two children, gene panel, WES and WGS in one child).
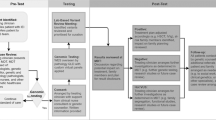
Disease-causing variants for IEI (pathogenic or likely pathogenic) were identified in 40.2% participants. A flowchart showing the results for each type of test and diagnostic yield is presented in Fig. 1.

Flowchart of genetic tests performed and their diagnostic yield. IEI – inborn errors of immunity; VUS—variants of unknown significance; WES – whole exome sequencing; WGS – whole genome sequencing.
Participants having relevant VUS (15.2%) were analyzed together with the ones having negative results. The main clinical cohort characteristics and comparison between those with disease-causing variants versus negative findings (including VUS) in genetic testing are presented in Table 1. Statistically significant differences were observed between the two groups in regards to: earlier age of symptom onset, yet longer time from onset to genetic testing and significant family history for those with disease-causing variants when compared to those with VUS or negative results. Additionally, when considering the clinical diagnosis category, the autoinflammatory disorders were less likely to have a diseases causing variant identified, while the congenital defects of phagocyte number or function had a higher diagnostic yield. A significant difference was observed between groups, pertaining to higher number of deceased children found in the group where disease-causing variants were identified.
We considered that genetic testing created an impact, if it influenced monitoring, treatment or represented an indication for allogeneic hematopoietic stem cell transplant (HSCT). The impact from negative results was more difficult to ascertain, however such results were considered useful by the clinician in the differential diagnosis for primary hemophagocytic lymphohistiocytosis (HLH) in five children and other immune disorders in three children with IEI phenotype.
The patients presented an extremely variable range of symptoms and signs. Frequency of HPO terms is presented in supplementary file. Most frequent terms were recurrent fever (in 25 patients), splenomegaly (in 19), recurrent infections (in 16), neutropenia (in 15), growth delay (10), anemia, autoimmunity, thrombocytopenia (in 9), pancytopenia (in 8). Other symptoms, identified in more than five patients, included: hepatomegaly, hypogammaglobulinemia, intermittent diarrhea, lymphopenia, abdominal pain, vasculitis, arthralgia, decreased circulating IgA level, immunodeficiency, neonatal hypotonia, neurodevelopmental delay, pneumonia, recurrent aphthous stomatitis, sepsis and skin rash. The complexity of sign and symptoms identified is presented in Fig. 2 for inborn errors of immunity category as classified by the IUIS expert committee7 and HPO terms in 92 patients. Node degree shows the number of connections. HPO frequency for the 92 children is presented in detail in supplementary file. Additionally, Fig. 3 presents the genes with disease causing variants in 37 children with IEI, connected in regards to their signs and symptoms (HPO terms).

Network showing the inborn errors of immunity category as classified by International Union of Immunological Societies expert committee7 and symptoms in 92 children with inborn errors of immunity. Red nodes represent inborn error of immunity(IEI) category, while blue nodes represent symptoms. The larger nodes characterize the increased degree of a node in the network (increased number of connections to other nodes). Supplementary file includes the symptom frequency. Some IEI categories were shortened to improve reading of the graph, as follows: Combined immunodeficiencies—Combined immunodeficiencies with associated or syndromic features; Defects of phagocyte—Congenital defects of phagocyte number or function; Immune dysregulation—Diseases of immune dysregulation; Cellular and humoral IEI—Immunodeficiencies affecting cellular and humoral immunity; Phenocopies of IEI- Phenocopies of inborn errors of immunity; Antibody deficiencies—Predominantly antibody deficiencies.

Network showing the genes with disease causing variants in 37 children with inborn errors of immunity, connected in regards to their signs and symptoms. Red nodes represent genes, while blue nodes represent symptoms (HPO terms). The larger nodes characterize the increased degree of a node in the network (increased number of connections to other nodes).
Genetic characterization
Disease-causing variants were identified in people with IEI, in the following 25 genes: AIRE, ATAD3A, ATM, CFTR, CXCR4, CYBB, DNAJC21, FAS, GATA2, ITGB2, JAGN1, LRBA, LYST, MEFV, MSH6, MVK, NCF2, NLRP3, NRAS, PIK3CD, RAB27A, SERPING1, STXBP2, TRNT1, WAS. Most frequent genes with disease-causing variants identified were: JAGN1 (5 patients), AIRE (3), ATAD3A (2), ATM (2), CFTR (2), CYBB (2), FAS (2), and TRNT1 (2), explaining half of genetic causes identified. The pathogenic or likely pathogenic variants are presented in Table 2. Fifteen patients had compound heterozygous, ten patients carried homozygous, nine patients had heterozygous, while three presented hemyzygous disease-causing variants. Nine novel variants (not previously reported in ClinVar) were discovered and reported. Two children received diagnosis after a larger test was performed, showing the DNJAC21 (the gene was not in the initial panel)30 and FAS (deletion was not observed on panel testing) genes with disease causative variants.
One homozygous pathogenic variant c.63G > T (p.Glu21Asp) in JAGN1 was identified in five patients (4 related, one unrelated) of romani ethnicity with congenital neutropenia.
Clinically relevant VUS were identified in 13 genes (in 14 patients), as follows: ATM, CARD11, CARD14, CASP10, IKZF1, JAK1, MSN, NFKB2, NFKBIA, TGFBR2, TLR3, TNFAIP3, TOM1. All variants were identified in heterozygous state, except for two patients with compound heterozygous VUS in ATM and CARD11, presented in the supplementary file.
All patients opted in for secondary findings. One patient with RAS-associated autoimmune leukoproliferative disease (NRAS, heterozygous) had secondary findings in relation to dilated cardiomyopathy risk (TTN, heterozygous). Four patients with negative results for IEI had secondary discoveries: congenital ichthyosis autosomal recessive (TGM1, homozygous), risk for chronic pancreatitis (CFTR, heterozygous), benign familial hematuria (COL4A4, heterozygous), risk for ACTA2 (heterozygous gene deletion) related disorders and cancer predisposing gene (ATM, heterozygous), data presented in supplementary file.
Discussion
Characteristics of Romanian children with IEI phenotype
To the best of our knowledge, this is the largest study that investigated the molecular background of a diverse Romanian IEI pediatric cohort, evaluated in a tertiary genetic center. However, another study from Romania that investigated a single IEI category, (autoinflammatory diseases) in 79 patients, was previously published12. In this Romanian cohort, median age at symptom onset was 2.9 years, with more than a quarter of patients presenting early, before the age of 1 year. Expectantly, positive family history was associated with disease-causing variants in the Romanian children with IEI phenotype. Furthermore, confirming that family history is an important clue for diagnosis in a monogenic disease. Nonetheless, a negative family history should not decrease the suspicion for IEI31.
The diagnostic odyssey was highly variable, with median 4.3 years. The longest time to genetic diagnosis was 18 years in one patient, showing limited resources for genetic testing in Romania over time and disparity in access to genetic testing. Similar disparities in genetic testing for inborn errors of immunity were identified in many countries, showing that even in more developed countries, improved access to genetic testing is needed32,33,34.
The patients presented an extremely variable range of symptoms and signs, consisting of 246 unique specific HPO terms in the Romanian cohort, which were difficult to ascertain with statistical models, yet the brain of a clinical immunologist, is well trained to work with similarly complex connections between symptoms and diseases or groups of disorders. The network graph visually capture the links between IEI category and symptoms reveals the congruence of unspecific symptoms, common to several disease categories (converging nodes in the middle), while diverging nodes show the specific symptoms. Examples of common unspecific terms include recurrent fever, splenomegaly, recurrent infections, neutropenia, growth delay, anemia, autoimmunity and thrombocytopenia. Supplementary file includes the symptom frequency. Furthermore, the network graph showing the genes with disease causing variants in 37 children with IEI, connected in regards to their signs and symptoms reveals outlier genes SERPING1 and MSH6, as particular phenotypes. These results underline the importance of genetic testing to help the clinician in what sometimes may seem as a jumble of disease presentations35.
Among extremely diverse genomic causality in this cohort, a particular finding was one homozygous variant identified in five children with neutropenia. In a recent study with 32 patients with JAGN1 neutropenia, the second most common JAGN1 variant c.63G > T, p.Glu21Asp was identified in eight patients. We found a similar need to receive allogeneic stem cell transplantation due to therapy-refractory neutropenia and severe infections in our patients, compared to the Fekadu-Siebald et al. study (1 in 5, compared to 2 in 8 patients)36.
Diagnostic yield of genetic testing in in IEI
Overall, the diagnostic yield of genetic testing was 40% in this diverse cohort of Romanian children with suspicion of IEI. In a 2019 systematic review, regarding use of NGS in patients with primary immunodeficiency, the diagnostic yield ranged from 15 to 79%37. In a more recent study (1398 patients), the global diagnostic yield using gene panels in the Jeffrey’s Insights Program was 20.3%32. A study from Egypt on 504 patients had a diagnostic yield of 55.9% using gene panels and WES38. Very few reports have investigated the diagnostic yield of IEI using WGS39,40, although current genetic diagnostics in IEI clearly embraces WGS, especially in the context of genomic-based newborn screening41. A study from Genomic Medicine Center Karolinska-Rare Diseases (Sweden) using WGS for 300 patients with IEI, achieved a diagnosis in 29% of patients, similar, with our study42. The higher diagnostic yield in our study, could be explained by more strict selection criteria (such as younger age, more severe phenotype) and possibly more comprehensive genetic testing methods such as WES and WGS. Superior diagnostic yield could be observed in populations with higher prevalence of consanguinity, which in Romania is unusual. As genetic testing is a dynamic process, we highlight the need to reinterpret NGS results and expand the evaluated genes, as this could bring additional diagnostic yield43.
The diagnosis yield in our cohort was the lowest for the monogenic autoinflammatory disorders, similarly to other studies12. Stray-Pedersen et al. found diagnostic yield of 13% for autoinflammatory disorders, while the yield was 100% for patients with severe combined immunodeficiency44. Although more than 30 genes associated with autoinflammatory diseases have been identified, this disease group can also have polygenic or multifactorial origin, with environmental influence modulating the phenotype45.
The study methods did not allow an accurate comparison between diagnostic yields from gene panel, WES and WGS, as cascade testing was applied. One patient clearly benefited from WGS for accurate deletion points involving the FAS gene that was not observed in gene panel testing (no. 15 in Table 2). In addition, the patient with DNAJC21 variants had previous testing in a panel where the gene was not included. Several authors promote moving past the panel, particularly recommending the use of WGS as a first tier test for genetic testing46. Short read WGS provides a higher variant detection accuracy and increased ability to identify copy number and structural variants. Nonetheless, complex structural variations remain challenging for both short read WGS and WES. Additionally, WGS offers the opportunity to evaluate disease causality beyond the established disease genes, possibly also into polygenic risk scores, in the future. Long read sequencing, optical genome mapping and multi-OMICS techniques could potentially increase significantly the diagnostic yield, by offering complete integration of human biomolecular data.
Impact of genetic testing in IEI
Several studies described clinical implications of the genetic findings32,37,39,47. When disease-causing variants are identified, they enable adjustment of the treatment plan (e.g. HSCT or specific therapies), identification of at-risk family members and genetic counselling48. For example, the impact of clinical diagnosis was revised in about half (60/110) and management was directly altered in nearly a quarter (26/110) of families based on molecular findings in a study from Stray-Pedersen et al.44. In the Romanian cohort, the impact was lower, as a few families refused treatment, despite severe disease presentation, due to prolonged denial, religious or cultural rationales.
Surely, uninformative genetic results cannot rule out the diagnosis of IEI, nor a genetic cause for IEI. Nonetheless, coupled with the clinical phenotype, negative findings could be supportive in the differential diagnosis process, as suggested in this study. In addition, clinically relevant VUS (15% in this study) could potentially increase diagnostic yield, by further information from functional studies and registration in collaborative databases49.
Finally, in terms of impact, comprehensive NGS testing incurs the risk for unintended discoveries. Five significant secondary findings were observed in this cohort, which had implications for the patient, but even more for the patient’s family. Secondary findings could be considered a burden or actionable discoveries offering the chance to identify and mitigate disease that may otherwise go unrecognized in a person50,51,52. Opt-in decisions of patients/families referred to WES showed great differences from different countries: 90% of the 41 patients in Romania opted to receive secondary findings, while none of the 98 patients in Luxembourg opted-in in a recent study53. Research is needed to understand the influencing factors53.
In summary, this study shows that NGS (using gene panels, WES and WGS) can identify the genetic etiology for an important percentage of children with IEI. Early precise diagnosis of IEI can improve health outcomes, decrease healthcare costs, and lessen psychological stress for affected families.
Study limitations
At the time of writing, to our knowledge, this the largest reported analysis of genetic variation in patients with diverse IEI from Romania. However, it might not be large enough to be immune to statistical fluctuations that commonly afflict small- to medium-sized cohorts.
Even within well-defined phenotypes, many patients did not have a genetic explanation for their disorder. This study relied on clinical data provided by ordering providers. Clinical data were extremely heterogeneous. Although the reporting is comprehensive, some symptoms may have been overlooked. A limitation is that not all genes were tested in all patients, thus the overall diagnostic yield might be lower due to the limitations of gene panels. Non-coding deep intronic variants were not evaluated in patients with gene panel and WES, although reports have shown the possible role of non-coding variants. The WES analysis did not include copy number analysis. There could be bias in selecting genetic test (gene panel/WES/WGS), as the approach was based on the presenting signs and symptoms and clinical judgment.
Conclusion
Genetic analysis remains fundamental to ameliorate complex IEI diagnosis and medical care. Clinical evaluation helps correlate the patient’s symptoms with possible genetic conditions, ensuring the selected genetic test will lead to diagnosis. By characterizing disease-causing variants in Romanian children with IEI, this study contributes to the understanding of the molecular landscape in IEI. The 40% of this Romanian pediatric IEI study cohort in which disease-causing variants were identified, shows the importance of genetic investigations. A larger number of patients and a different study design is needed to establish which molecular test has the optimal diagnostic yield in the identification of disease-causing variants of IEI, also investigating the cost and benefit ratio for each test type.
Supplementary information
Supplementary file includes phenotypic and genotypic description of all the children analyzed.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Sequence data that support new variants in this study have been deposited in the Clinvar database. Clinvar ID and variant. 3572945 NM_001170535.3(ATAD3A):c.278 T > C (p.Leu93Pro). 3572946 NM_001012339.3(DNAJC21):c.645_646del (p.Arg216fs). 3572947 NM_001364905.1(LRBA):c.7890del (p.Thr2631fs). 3572948 NM_000431.4(MVK):c.82G > T (p.Ala28Ser). 3572949 NM_183235.3(RAB27A):c.240-5_242del. 3572950 NM_000051.4(ATM):c.6666_6668delinsAG (p.Ile2223fs). 3572951 NC_000010.10:g.90533969_90911342del. 3572952 NM_001364905.1(LRBA):c.2746_2747del (p.Leu916fs). 3572953 NM_000433.4:c.(?_-1)_(257 + 1_258-1)del.
References
Tangye, S. G. et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J. Clin. Immunol. 42, 1473–1507 (2022).
Notarangelo, L. D. Primary immunodeficiencies. J. Allergy Clin. Immunol. 125, S182–S194 (2010).
Boyle, J. M. & Buckley, R. H. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J. Clin. Immunol. 27, 497–502 (2007).
Gathmann, B. et al. The German national registry for primary immunodeficiencies (PID). Clin. Exp. Immunol. 173, 372–380 (2013).
Casanova, J.-L. & Abel, L. Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin. Immunol. 36, 1–12 (2018).
Leiding, J. W. & Forbes, L. R. Mechanism-based precision therapy for the treatment of primary immunodeficiency and primary immunodysregulatory diseases. J. Allergy Clin. Immunol. Pract. 7, 761–773 (2019).
Inborn Errors of Immunity Committee (IEI) IUIS [Internet]. IUIS. 2019 [cited 2025 Jan 11]. Available from: https://iuis.org/committees/iei/
Picard, C. & Fischer, A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur. J. Immunol. 44, 2854–2861 (2014).
Tóth, B. et al. Genetic and demographic features of X-linked agammaglobulinemia in Eastern and Central Europe: A cohort study. Mol. Immunol. 46, 2140–2146 (2009).
Gulácsy, V. et al. A novel large deletion and single nucleotide insertion in the Wiskott–Aldrich syndrome protein gene. Eur. J. Haematol. 95, 93–98 (2015).
Sediva, A. et al. Primary immunodeficiencies in Central and Eastern Europe—The power of networking Report on the activity of the Jeffrey Modell Foundation Centers Network in Central and Eastern Europe. Immunol. Res. 67, 358–367 (2019).
Cochino, A. V., Ioan, A. & Farkas, O. M. Autoinflammatory diseases in romanian children: A tertiary center case series study. J. Clin. Rheumatol. 28, 429–432 (2022).
Abolhassani, H. et al. Care of patients with inborn errors of immunity in thirty J Project countries between 2004 and 2021. Front. Immunol. 13, 1032358 (2022).
Tapiz, I. et al. Characterization of novel pathogenic variants leading to caspase-8 cleavage-resistant RIPK1-induced autoinflammatory syndrome. J. Clin. Immunol. 42, 1421–1432 (2022).
Cochino, A. V., Ioan, A., Farkas, O. M., Liu, M. & Lee, P. Y. Novel ADA2 variants in a Romanian case series of DADA2. J. Clin. Immunol. 43, 1788–1791 (2023).
Nicoară, D. et al. A new CECR1 mutation associated with severe hematological involvement in ADA2 deficiency. Immun. Inflamm. Dis. 11, e930 (2023).
Chapel, H. et al. Primary immune deficiencies—Principles of care. Front. Immunol. 5, 627 (2014).
Hausmann, O. & Warnatz, K. Immunodeficiency in adults a practical guide for the allergist. Allergy J. Int. 23, 261–268 (2014).
Mauracher, A. A., Gujer, E., Bachmann, L. M., Güsewell, S. & Pachlopnik, S. J. Patterns of immune dysregulation in primary immunodeficiencies: A systematic review. J. Allergy Clin. Immunol. Pract. 9, 792-802.e10 (2021).
Köhler, S. et al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res. 47, D1018–D1027 (2019).
Haimel, M. et al. Curation and expansion of human phenotype ontology for defined groups of inborn errors of immunity. J. Allergy Clin. Immunol. 149, 369–378 (2022).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Harrison, S. M. et al. Using ClinVar as a resource to support variant interpretation. Curr. Protoc. Hum. Genet. 89, 8.16.1-8.16.23 (2016).
Rehm, H. L. et al. ClinGen—The clinical genome resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535-548.e24 (2019).
Desmet, F.-O. et al. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Hagberg AA, Schult DA, Swart PJ. Exploring Network Structure, Dynamics, and Function using NetworkX. scipy [Internet]. 2008 [cited 2025 Jan 7]; Available from: https://proceedings.scipy.org/articles/TCWV9851
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Chirita-Emandi, A., Petrescu, C-A-M., Zimbru, C. G., Stoica, F., Marian, C., Ciubotaru, A., et al. Case Report: Novel Biallelic variants in DNAJC21 causing an inherited bone marrow failure spectrum phenotype: An odyssey to diagnosis. Front. Genet. https://doi.org/10.3389/fgene.2022.870233
Al Farsi, T. et al. Immune dysregulation in monogenic inborn errors of immunity in oman: Over a decade of experience from a single tertiary center. Front Immunol. 13, 849694 (2022).
Quinn, J. et al. Global expansion of Jeffrey’s insights: Jeffrey modell foundation’s genetic sequencing program for primary immunodeficiency. Front. Immunol. 13, 906540 (2022).
Gilbert, K. M., McLaughlin, H. M., Farmer, J. R. & Ong, M.-S. Disparities in genetic testing for inborn errors of immunity. J. Allergy Clin. Immunol. Pract. S2213–2198(24), 01175–01179 (2024).
Lawrence, M. G., Rider, N. L., Cunningham-Rundles, C. & Poli, M. C. Disparities in diagnosis, access to specialist care and treatment for inborn errors of immunity. J. Allergy Clin. Immunol. Pract. S2213–2198(23), 01196 (2023).
Notarangelo, L. D., Bacchetta, R., Casanova, J. L. & Su, H. C. Human inborn errors of immunity: an expanding universe. Sci. Immunol. 5, eabb1662 (2020).
Fekadu-Siebald, J., Salzmann-Manrique, E., Heusel, J. R., Willasch, A., Hauck, F. & Gonzalez-Granado, L. I., et al. Extended clinical phenotypes and treatment modalities in 32 JAGN1-deficient patients. A multicenter study by EBMT IEWP. Blood Adv. bloodadvances.2024014344 (2025).
Yska, H. A. F. et al. Diagnostic yield of next generation sequencing in genetically undiagnosed patients with primary immunodeficiencies: A systematic review. J. Clin. Immunol. 39, 577–591 (2019).
El Hawary, R. E. et al. Genetic testing in Egyptian patients with inborn errors of immunity: A single-center experience. J. Clin. Immunol. 42, 1051–1070 (2022).
Meyts, I. et al. Exome and genome sequencing for inborn errors of immunity. J. Allergy Clin. Immunol. 138, 957–969 (2016).
Mørup, S. B. et al. Added value of reanalysis of whole exome- and whole genome sequencing data from patients suspected of primary immune deficiency using an extended gene panel and structural variation calling. Front. Immunol. 13, 906328 (2022).
King, J. R., Grill, K. & Hammarström, L. Genomic-based newborn screening for inborn errors of immunity: practical and ethical considerations. Int. J. Neonatal Screen. 9, 22 (2023).
Stranneheim, H. et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 13, 40 (2021).
Vorsteveld, E. E. et al. Clinical exome sequencing data from patients with inborn errors of immunity: Cohort level diagnostic yield and the benefit of systematic reanalysis. Clin. Immunol. 268, 110375 (2024).
Stray-Pedersen, A. et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J. Allergy Clin. Immunol. 139, 232–245 (2017).
Krainer, J., Siebenhandl, S. & Weinhäusel, A. Systemic autoinflammatory diseases. J. Autoimmun. 109, 102421 (2020).
Belkadi, A. et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 112, 5473–5478 (2015).
Arts, P. et al. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med. 11, 38 (2019).
Al-Herz, W. et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front. Immunol. 9, 3146 (2018).
Caballero-Oteyza, A. et al. GenIA, the Genetic Immunology Advisor database for inborn errors of immunity. J. Allergy Clin. Immunol. 153, 831–843 (2024).
Chirita-Emandi, A. et al. Challenges in reporting pathogenic/potentially pathogenic variants in 94 cancer predisposing genes—in pediatric patients screened with NGS panels. Sci. Rep. 10, 223 (2020).
de Wert, G. et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 29, 365–377 (2021).
Katz, A. E. et al. Management of secondary genomic findings. Am. J. Hum. Genet. 107, 3–14 (2020).
Brunfeldt, M., Kaare, M., Saarinen, I., Koskenvuo, J. & Kääriäinen, H. Opt-in for secondary findings as part of diagnostic whole-exome sequencing: Real-life experience from an international diagnostic laboratory. Mol. Genet. Genomic Med. 11, e2180 (2023).
Funding
We acknowledge “Victor Babeș” University of Medicine and Pharmacy Timisoara for their support in covering the costs of publication for this article. The study was supported by a research grant from the University of Medicine and Pharmacy “Victor Babes” Timisoara, Postdoctoral Internal competition, contract no. 5 POSTDOC/2020, Title: Role of Genome sequencing in diagnostics of primary immunodeficiency and rare cancers (02.2020-02.2022). Additionally, funding was received from patient association Romanian Association of Patients with Primary Immunodeficiencies (ARPID).
Author information
Authors and Affiliations
Contributions
ACE and CLP drafted the manuscript, all authors reviewed the manuscript. MB and ACE were involved in patient selection for genetic testing, clinical management and family screening. ACE, CGZ, CVM and BD were involved in genetic testing. BD was involved in data extraction from document files and creation of network graphs. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the form the University of Medicine and Pharmacy 'Victor Babeș' Timișoara, Romania (11/08.05.20200.
Consent to participate
Written informed consent was obtained from the parents.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pantea, CL., Bataneant, M., Zimbru, C.G. et al. Genetic landscape of Romanian children with inborn errors of immunity via gene panels, exome, and genome sequencing. Sci Rep 15, 18830 (2025). https://doi.org/10.1038/s41598-025-03492-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03492-9
