
Abstract
Ischemic stroke (IS) is a multifactorial disease caused by the interaction of a variety of environmental and genetic factors, which can lead to severe disability and heavy social burden. This study aimed to find potential biomarkers related to T cell exhaustion (TEX) in IS. Based on the GSE16561 dataset, differentially expressed genes (DEGs) were screened from IS and control groups, and their enriched biological pathways were explored. The TEX enrichment score for each sample was calculated using the GSEA algorithm, and the gene modules with the highest correlation with the TEX score were screened by WGCNA. Then, two machine learning algorithms were used to screen the key genes and test the correlation between the key genes and the level of immune cell infiltration. Potential drugs or molecular compounds that interact with key genes were predicted by searching DGIdb, and the drug-gene interaction network was visualized by Cytoscape software. Using GSE16561 dataset, we performed differential expression analysis and identified 482 DEGs. By weighted gene co-expression network analysis (WGCNA) and machine learning algorithms, we identified five key genes: CD163, LAMP2, PICALM, RGS2 and PIN1. Functional enrichment analysis revealed that these genes were involved in immune response and cellular processes, which were closely related to the level of immune cell infiltration. In addition, potential drug interactions were predicted using the drug-Gene Interaction database, providing avenues for future therapeutic strategies. This study enhances the understanding of TEX-related biomarkers in ischemic stroke and provides insights into the development of novel interventions aimed at improving patient outcomes.
Similar content being viewed by others


Introduction
Ischemic stroke (IS) is a disease caused by the interaction of environmental, genetic and other factors, which can lead to a variety of complications such as hemiplegia, language impairment, and blurred vision, which brings a heavy burden to society and families1,2. At present, the treatment of IS mainly includes drug therapy and surgical treatment, but the treatment effect is not ideal due to postoperative complications and the narrow thrombolysis window of drugs3,4. Therefore, there is an urgent need to explore new biomarkers and therapeutic drugs for IS to improve the prognosis of patients and reduce the burden on families. Some studies have found that the immune microenvironment and inflammatory response are involved in the whole process of the occurrence and development of IS5, and are closely related to the severity and prognosis of IS. Based on this, exploring the immune microenvironment and inflammatory mechanism of IS may provide new ideas and perspectives for the treatment of IS.
Zhang et al. found that T cells play an important role in the inflammatory response of IS patients, and they can infiltrate the brain parenchyma through three pathways6. In addition, the various combinations of chemokines and their receptors on T cell subsets provide spatial and temporal “addresses” for T cell infiltration into the brain parenchyma. T cell exhaustion (TEX) is a newly discovered state of T cell dysfunction characterized by poor effector T cell function, persistent inhibitory receptor expression, and altered transcriptional status that is different from that of functional effector or memory T cells7. It has been found that CD8 T cell subsets at different developmental stages of TEX have different functional and developmental properties and play a crucial role in adaptive immune responses8. These cells can perform cytotoxic functions by recognizing antigens through TCRs and subsequently releasing granzymes and perforins to form pores on target cells and induce apoptosis9. Due to their adaptability, the deleterious effects of CD8 T cells occur mainly in the chronic phase. Zhang et al. found that delaying the depletion of CD8 T cells with CD8 antibody on the 10th day after ischemic stroke significantly improved the recovery of brain function in mice5. Although there have been numerous recent studies exploring the role of TEX in ischemic disease, the acquisition of IS biomarkers from TEX has not been explored in detail.
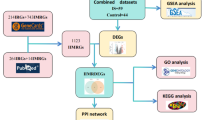
Bioinformatics analysis of microarray results extends previous studies to provide meaningful genetic information. This study aims to use microarray datasets and perform in-depth bioinformatics analysis to explore potential biomarkers associated with TEX in IS. Specifically, differentially expressed genes (DEGs) were screened from IS and controls to explore their enriched biological pathways. Based on the 40 TEX-related genes, the TEX enrichment score of each sample was calculated using the GSEA algorithm, and the gene modules most correlated with the TEX score were screened by WGCNA. Then, two machine learning algorithms were used to screen the key genes and examine the correlation between the key genes and the level of immune cell infiltration. In addition, potential drugs or molecular compounds that interact with key genes were predicted by searching DGIdb, and the drug-gene interaction network was visualized by Cytoscape software. Our study provides new ideas for the diagnosis and treatment of IS.
Materials and methods
Data sources
We downloaded the GSE16561 dataset from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/) using the “GEOquery” R package, which includes 63 samples (IS: 39; Control: 24) The data was obtained from Homo sapiens and the platform GPL6883 [Illumina Human Ref-8 v3.0 expression beadchip]. Forty T cell exhaustion-related genes were obtained from the literature for further analysis9.
Differential expression analysis
The “limma” package10 in R software was utilized to screen DEGs between the IS patients and control samples based on Bayesian calculations of t-values, f-values, and log odds ratios. An adjusted P value of < 0.05 and log2 |Fold Change|> 0.5 were considered statistically significant. A volcano plot of DEGs was generated using the “ggrepel” R package.
Weighted gene co-expression network analysis (WGCNA)
We conducted GSEA analysis on TEX-related gene data using the “GSEA” R package11, calculating enrichment scores for each sample. This scoring data served as input for WGCNA. WGCNA were constructed by correlating gene expression levels with phenotypic characteristics to investigate potential interactions between genes12. Firstly, the outlier samples in the data set were removed to ensure accurate and reliable results. Then, the correlation coefficient between every two genes was calculated to construct a gene expression similarity matrix, which was transformed into an adjacency matrix. Next, the optimal soft threshold was selected to construct the scale-free network, the adjacency value was transformed into the topological overlap matrix (TOM), and the corresponding dissimilarity degree (1-TOM) was calculated. Finally, gene module detection was performed based on average hierarchical clustering and dynamic tree clipping. Correlations between genes and phenotypes in each module were determined by calculating gene significance and module membership (MM).
Functional enrichment analysis
To further analyze the biological functions of DEGs, we utilized the “clusterProfiler” R package11 for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, visualizing results with the “ggplot2” R package13. The P-value of less than 0.05 was identified as a significant term.
Machine learning for key gene selection
We employed Random Forest (RF) and Least Absolute Shrinkage and Selection Operator (LASSO) machine learning algorithms to further filter candidate genes. Random Forest is an ensemble predictive method adept at handling numerous input variables while assessing variable importance. LASSO is a regression method that excels in evaluating high-dimensional data14. We used the “randomForest” and “glmnet” R packages for RF analysis and LASSO regression15. The intersection of genes identified by both algorithms yielded key genes, and ROC curves were plotted to demonstrate the sensitivity and specificity of these key genes in relation to the disease.
Immune infiltration analysis
Single-sample gene set enrichment analysis (ssGSEA) was applied to score 28 immune cell types16, resulting in a scoring file representing the immune cell composition of each individual sample. The immune cell scores of IS and control groups were statistically analyzed using t-tests, with results visually represented in box plots. Correlation tests were conducted to assess relationships between key genes and immune cells, visualized using the “ggplot” package17.
Potential drug identification
The drug-gene interaction database (DGIdb, https://www.dgidb.org/) is a resource for drug targeting and sensitive genomic interactions. We utilized DGIdb to predict potential drugs or molecular compounds interacting with key genes, visualizing drug-gene interaction networks using Cytoscape 3.9.118software.
Statistical analysis
All analyses were performed in R version 4.3.3. For significance testing among various values (expression levels, infiltration ratios, and other features), we employed Wilcoxon rank-sum tests or t-tests for comparisons between two groups, setting statistical significance at P < 0.05.
Results
Identification of DEGs and IS-related genes
A total of 482 DGEs were identified using the “limma” R package, of which 277 genes were up-regulated and 205 genes were down-regulated (|log2FC|> 0.5; P < 0.05; Fig. 1A). TEX enrichment scores were calculated for each sample by GSEA analysis based on 40 TEX-related genes. After preprocessing, a gene expression matrix was obtained, and the top 5000 genes with the highest absolute median differences were selected for further analysis. After filtering out abnormal samples and genes, we constructed a sample clustering dendrogram (Fig. 1B). With a soft threshold of 8, we established a scale-free network (Fig. 1C). Following the construction of an adjacency matrix and topology overlap matrix, we identified 14 modules through average hierarchical clustering and dynamic tree cutting (Fig. 1D). The blue module, highly correlated with IS and TEX scores, was defined as the IS-related module (Fig. 1E,F). 1188 genes in this module were identified as IS-related genes.

Identification of DEGs and IS-related genes (A) Volcano plot of DEGs. The cut-off criteria were |log2FC|> 0.5 and P < 0.05. (B) Sample clustering was conducted to detect outliers. (C) Scale-free fit index analysis and average connectivity analysis of each soft threshold power. (D) Clustering dendrogram of genes, various colors represent different modules. (E) Heatmap of the association between modules and different cluster. P values and correlation coefficients are represented by numbers inside and outside parentheses, respectively. (F) Correlation chart between MM and GS of the clustered genes in the module.
Biological pathway enrichment of intersection genes
The DEGs and IS-related genes were intersected, and 225 intersection genes were obtained for subsequent analysis (Fig. 2A). We explored the biological functions and pathways enriched by these intersection genes. GO analysis indicated that these genes predominantly focused on biological processes such as lymphocyte differentiation, immune response-regulating cell surface receptor signaling pathways, and T cell differentiation. Cellular component alterations were mainly associated with the external side of the plasma membrane, ribosomes, and ribosomal subunits, while molecular function changes centered on structural constituents of ribosomes, MHC protein complex binding, and NADH dehydrogenase activity (Fig. 2B). KEGG analysis revealed that these genes were primarily enriched in Th17 cell differentiation, T cell receptor signaling pathways, and oxidative phosphorylation (Fig. 2C)19,20. The enrichment of intersection genes in immune-related pathways may suggest that the differentiation of immune cells and the mechanism involved are closely related to the occurrence of IS.

Biological pathway enrichment of intersection genes. (A) Venn diagram showing the numbers of intersection genes. (B) GO functional analysis showing enrichment of intersection genes. (C) KEGG pathway enrichment analysis of intersection genes.
Identification of key genes
To further identify key genes with high diagnostic value, we applied two machine learning algorithms to screen key genes. 14 feature genes were identified by selecting the minimum criteria of LASSO classifier (Fig. 3A,B). Random forest algorithm was applied to select the top 20 genes in terms of importance defined as feature genes (Fig. 3C,D). The intersection of the genes screened by the two algorithms was taken to obtain 5 key genes (CD163, LAMP2, PICALM, RGS2, PIN1) (Fig. 3E).

Identification of key genes. (A) Cross-validation to select the optimal tuning parameter log (λ) in LASSO regression analysis. (B) LASSO coefficient profiles of key genes. (C) RF prediction error curves based on tenfold cross-validation curve. (D) The scoring plot of each gene. (E) Venn diagram showing the numbers of key genes.
Immune infiltration analysis
In order to investigate the differences in immune cell composition between IS patients and normal controls, we employed the ssGSEA algorithm to calculate immune cell scores for each sample, encompassing 28 immune cell types. Box plots of these scores were generated using the “ggplot2” package, with t-tests performed between disease and control groups, indicating significant differences in 11 immune cell types, including Neutrophil, Plasmacytoid dendritic cell, Activated CD8 T cell, Effector memeory CD8 T cell, CD56 dim natural killer cell, Activated dendritic cell, Regulatory T cell, Activated B cell, T follicular helper cell, Natural killer cell, Eosinophil (Fig. 4A). Strong interactions also exist between different immune cells (Fig. 4B). Additionally, we assessed correlations between key genes and immune cells, revealing strong correlations of CD163, LAMP2, PICALM, and RGS2 with neutrophils, activated dendritic cells, and natural killer cells, while PIN1 correlated strongly with activated CD8 T cells and effector memory CD8 T cells (Fig. 4C).

Immune infiltration analysis. (A) Expression of 28 immune cells in IS and control groups. (B) Correlation between different immune cells. (C) Heat map of correlation between key genes and immune cells, *P < 0.05, **P < 0.01, ***P < 0.001, ns indicates that the difference is not statistically significant.
Expression and diagnostic value of key genes
We analyzed the expression levels of key genes in the GSE16561 dataset, observing up-regulation of CD163, LAMP2, PICALM, and RGS2 in the IS group, whereas PIN1 was down-regulated (Fig. 5A). ROC curve analysis of key genes demonstrated high sensitivity and specificity, with AUC values exceeding 0.9 for CD163, LAMP2, PICALM, RGS2, and PIN1 (Fig. 5B–F).

Expression and diagnostic value of key genes. (A) Box plots of differential expression analysis of key genes in IS and control samples. (B) The AUC of CD163. (C) The AUC of LAMP2. (D) The AUC of PICALM. (E) The AUC of PIN1. (F) The AUC of RGS2, ***P < 0.001.
Identification of potential drug
Utilizing DGIdb, we identified 22 drugs capable of interacting with PIN1, 2 drugs with CD163, and 1 drug each for RGS2 and PICALM. Interaction networks were visualized using Cytoscape software 3.9.1 (Fig. 6).

Identification of potential drug.
Discussion
IS is a craniocerebral disease caused by temporary or permanent blockage or thrombosis of cerebral arteries, resulting in a further reduction of blood flow to specific brain regions, which triggers pathophysiological processes such as inflammation and oxidative stress, and ultimately leads to neuronal cell death and brain damage in patients21,22,23,24. Current treatments are limited and the prognosis of patients is poor, and there is an urgent need to explore new treatments to improve the prognosis and prolong the survival of patients.
Neal et al. found that regulatory T cells have immunomodulatory and neuroprotective effects on stroke25, because there is a persistent inflammatory state in stroke patients, which leads to a decline in T cell function and changes in metabolic pathways, thus accelerating T cell exhaustion, and the depleted T cells in turn lead to dysregulation of the inflammatory response, and the enhancement of the function of regulatory T cells can inhibit the occurrence of the TEX response, thus playing an immunomodulatory role. The enhancement of regulatory T cell function can inhibit the occurrence of TEX response and thus play an immunomodulatory role, but whether it has the same role in IS is not yet known. Based on this, this study screened 225 IS-related TEX genes by WGCNA analysis and differential analysis results. KEGG analysis showed that these genes were mainly enriched in signaling pathways such as T cell receptor signaling pathway, oxidative phosphorylation, and Th17 cell differentiation. Based on the above results, these genes may contribute to IS by affecting the function of T cells and the signaling pathways they participate in.
PIN1, a peptidyl proline isomerase that regulates p53 transactivation in response to stress, is involved in the pathogenesis of IS through the Notch signaling pathway26. PIN1 has also been found to play a key role in neuronal cell death and neurological deficits induced by cerebral ischemia27. Further exploration of IS in this study revealed that five genes (CD163, LAMP2, PICALM, RGS2, and PIN1) were identified as key genes for IS, and the AUCs of all five genes were greater than 0.9, indicating that they have high sensitivity and specificity for the disease and are expected to be diagnostic biomarkers for IS. In addition, the study further identified potential therapeutic drugs or molecular compounds for IS by screening five genes, and found that 22 drugs bound to PIN1, two drugs bound to CD163, and one binding drug was detected for RGS2 and PICALM.
This study predicts targets and potential biomarkers for improving the inflammatory status of IS patients but our study has several limitations. First, the small sample size may limit generalizability, and the clinical applicability of the identified biomarkers requires prospective validation. Second, the complexity of T cell exhaustion involves many other factors that are not explored here. Finally, functional validation of key genes is needed to confirm their biological relevance. Addressing these limitations is critical to improving our understanding and treatment of IS.
Conclusion
In this study, five biomarkers closely related to IS, including CD163, LAMP2, PICALM, RGS2 and PIN1, were identified by bioinformatics analysis for future IS diagnosis and screening.
Data availability
The GEO datasets used in this study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the following data accession identifers: GSE16561.
References
Falcone, G. J., Malik, R., Dichgans, M. & Rosand, J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. 13, 405–418. https://doi.org/10.1016/s1474-4422(14)70029-8 (2014).
Yamada, Y. et al. Identification of six polymorphisms as novel susceptibility loci for ischemic or hemorrhagic stroke by exome-wide association studies. Int. J. Mol. Med. 39, 1477–1491. https://doi.org/10.3892/ijmm.2017.2972 (2017).
Powers, W. J. et al. 2018 guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American heart association/American stroke association. Stroke 49, e46–e110. https://doi.org/10.1161/str.0000000000000158 (2018).
Zheng, S. et al. GRP per capita and hospital characteristics associated with intravenous tissue plasminogen activator adherence rate: Evidence from the Chinese Stroke Center Alliance. Stroke Vasc. Neurol. 6, 337–343. https://doi.org/10.1136/svn-2020-000633 (2021).
Zera, K. A. & Buckwalter, M. S. The local and peripheral immune responses to stroke: Implications for therapeutic development. Neurother. J. Am. Soc. Exp. NeuroTher. 17, 414–435. https://doi.org/10.1007/s13311-020-00844-3 (2020).
Schuhmann, M. K. et al. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J. Neuroinflamm. 14, 18. https://doi.org/10.1186/s12974-017-0792-y (2017).
Wherry, E. J. T cell exhaustion. Nat. Immunol. 12, 492–499. https://doi.org/10.1038/ni.2035 (2011).
Barsch, M. et al. T-cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J. Hepatol. 77, 397–409. https://doi.org/10.1016/j.jhep.2022.02.032 (2022).
Zhang, Z. et al. Pan-cancer landscape of T-cell exhaustion heterogeneity within the tumor microenvironment revealed a progressive roadmap of hierarchical dysfunction associated with prognosis and therapeutic efficacy. EBioMedicine 83, 104207. https://doi.org/10.1016/j.ebiom.2022.104207 (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics (Oxford, England) 38, 3844–3846. https://doi.org/10.1093/bioinformatics/btac409 (2022).
Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 16, 385–395. https://doi.org/10.1002/(sici)1097-0258(19970228)16:4%3c385::aid-sim380%3e3.0.co;2-3 (1997).
Liu, C. et al. Identification of novel potential homologous repair deficiency-associated genes in pancreatic adenocarcinoma via WGCNA coexpression network analysis and machine learning. Cell Cycle 22, 2392–2408. https://doi.org/10.1080/15384101.2023.2293594 (2023).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Jo, L. & Kajala, K. ggPlantmap: An open-source R package for the creation of informative and quantitative ggplot maps derived from plant images. J. Exp. Bot. 75, 5366–5376. https://doi.org/10.1093/jxb/erae043 (2024).
Zhu, M. et al. Exploring the mechanism of aloe-emodin in the treatment of liver cancer through network pharmacology and cell experiments. Front. Pharmacol. 14, 1238841. https://doi.org/10.3389/fphar.2023.1238841 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672-d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Barthels, D. & Das, H. Current advances in ischemic stroke research and therapies. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165260. https://doi.org/10.1016/j.bbadis.2018.09.012 (2020).
Cheng, H. Y. et al. miR-195 has a potential to treat ischemic and hemorrhagic stroke through neurovascular protection and neurogenesis. Mol. Ther. Methods Clin. Dev. 13, 121–132. https://doi.org/10.1016/j.omtm.2018.11.011 (2019).
Kuriakose, D. & Xiao, Z. Pathophysiology and treatment of stroke: Present status and future perspectives. Int. J. Mol. Sci. 21, 7609. https://doi.org/10.3390/ijms21207609 (2020).
He, J. et al. Oxidative stress, inflammation, and autophagy: potential targets of mesenchymal stem cells-based therapies in ischemic stroke. Front. Neurosci. 15, 641157. https://doi.org/10.3389/fnins.2021.641157 (2021).
Neal, E. G., Acosta, S. A., Kaneko, Y., Ji, X. & Borlongan, C. V. Regulatory T-cells within bone marrow-derived stem cells actively confer immunomodulatory and neuroprotective effects against stroke. J. Cereb. Blood Flow. Metab. 39, 1750–1758. https://doi.org/10.1177/0271678x18766172 (2019).
Arumugam, T. V. et al. Notch signaling and neuronal death in stroke. Prog. Neurobiol. 165–167, 103–116. https://doi.org/10.1016/j.pneurobio.2018.03.002 (2018).
Baik, S. H. et al. Pin1 promotes neuronal death in stroke by stabilizing Notch intracellular domain. Ann. Neurol. 77, 504–516. https://doi.org/10.1002/ana.24347 (2015).
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Yajun Gao: data analysis, writing original draft. Ruyu Bai: data analysis, methodology. Bo Gao: software. Li Ma: conceptualization, funding acquisition, resources, writing review and editing, supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The data in this study were obtained from the GEO public database, ethical approval was not required, and all methods were performed in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, Y., Bai, R., Gao, B. et al. Bioinformatics prediction of function of T-cell exhaustion related genes in ischemic stroke. Sci Rep 15, 18863 (2025). https://doi.org/10.1038/s41598-025-03724-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03724-y
