
Abstract
Gastroesophageal reflux disease (GERD) is one of the most frequently diagnosed gastrointestinal disorders, adversely affecting quality of life. Coriandrum sativum has been shown to effectively promote gastrointestinal motility, alleviate gastric discomfort, and positively impact esophageal health, but its mechanism of action remains unclear. This study utilized the TCMSP database to retrieve the components of coriander and the GEO database to identify NERD-related genes. Weighted Gene Co-expression Network Analysis (WGCNA) and machine learning were employed to identify candidate genes, followed by Protein-Protein Interaction (PPI) network analysis and external dataset validation to identify key candidate genes. These genes were further validated through Gene Set Enrichment Analysis (GSEA) and subcellular localization. Finally, molecular docking and molecular dynamics (MD) simulations identified Ammidin, campest-5-en-3beta-ol, Isofucosterol and beta-sitosterol as the key components in Coriandrum sativum for improving NERD. Future research should prioritize experimental validation of these compounds and further investigate potential resistance mechanisms to enhance their therapeutic efficacy and practical applications.
Similar content being viewed by others

Introduction
Gastroesophageal reflux disease (GERD) is a common and debilitating chronic condition that manifests in two primary forms based on esophageal mucosal appearance: reflux esophagitis (RE) and non-erosive reflux disease (NERD).NERD refers to a condition caused by the reflux of gastric or duodenal contents, leading to a series of symptoms and/or complications1.The former is diagnosed based on mucosal damage observed via endoscopic examination, while the latter lacks such damage and accounts for approximately 70% of GERD cases2.The characteristic of NERD is that endoscopic examination does not reveal obvious abnormalities, yet patients frequently experience typical symptoms such as heartburn and reflux3. The pathogenesis of NERD differs from that of erosive esophagitis. A randomized controlled trial (RCT) indicates that approximately 68% of patients experience symptom relapse after discontinuing long-term proton pump inhibitor treatment (Proton pump inhibitor is the most commonly used treatment for GERD)4. This suggests that simply discontinuing Proton pump inhibitor treatment is insufficient for the long-term management of symptoms in patients. From an infection risk perspective, gastric acid serves as a crucial defense against pathogens, a function that is compromised by Proton pump inhibitor use. Data show that NERD patients using proton pump inhibitor have a threefold increased risk of Clostridium difficile infection and a 40% higher likelihood of developing pneumonia compared to those not using the medication3,5. Emerging evidence indicates that non-erosive reflux disease (NERD) may be associated with an increased risk of gastric cancer, potentially mediated by persistent mucosal inflammation, alterations in the gastric microenvironment, and the prolonged use of proton pump inhibitors (PPIs), which can induce biological changes conducive to carcinogenesis6.
Coriandrum sativum, a common culinary seasoning, is often discussed alongside Nigella sativa, as both are frequent ingredients in kitchens worldwide and are integral to the daily diets of various populations7. The fresh leaves commonly known as cilantro or Chinese parsley are extensively employed in eastern cooking and in Indian cuisine as food flavoring or to mask unpleasant odors of certain foods. They are also an important ingredient in Thai and Vietnamese cuisine8,9. As recorded in the Compendium of Materia Medica (Bencao Gangmu), " Coriander has a pungent, warm fragrance that penetrates internally to the heart and spleen and externally reaches the limbs.” Thanks to its unique characteristic of warming and dispersing, coriander effectively promotes gastrointestinal motility, alleviates stomach discomfort, and exerts a positive influence on esophageal health. There is evidence suggesting that coriander may have potential therapeutic effects for NERD10.
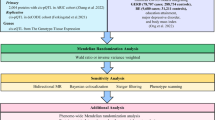
In this study, we obtained microarray datasets of gastric esophageal tissues from patients with NERD through the GEO database. We then employed Construction of Weighted Gene Co-expression Network analysis (WGCNA) and machine learning algorithms to screen samples from both healthy individuals and NERD patients to identify potential biomarkers of NERD. Subsequently, we performed localization analysis of feature genes. Finally, molecular docking and dynamic simulations were used to predict potential therapeutic compounds for NERD in coriander, providing valuable insights for the development of future clinical treatments. The workflow of this study is illustrated in Fig. 1.

The flow chart of this study.
Results
Identification of differentially expressed genes
Fold changes of the expression and P-values were selected as two parameters for screening the transcriptomes of bone marrow tissues of the model and control populations. Finally, 1680 Identification of Differentially Expressed Genes (DEGs) were screened, of which 902 were upregulated genes and 778 were downregulated genes (Fig. 2A). The top 40 upregulated DEGs and the top 40 downregulated DEGs were selected for subsequent visualization of the differential gene heatmap (Fig. 2B).
The gene ontology and Kyoto encyclopedia of genes and genomes analysis
The Gene Ontology (GO) analysis consisted of three categories (Fig. 2C): Biological processes (BP), cellular components (CC), and molecular functions (MF). The BP analysis highlighted terms such as extracellular matrix organization extracellular, structure organization, external encapsulating structure organization, morphogenesis of an epithelium, epidermis development, skin development, epidermal cell differentiation, keratinocyte differentiation, keratinization, intermediate filament organization. The CC analysis revealed enrichment in terms such as collagen-containing extracellular matrix, external side of plasma membrane, basement membrane, collagen trimer, keratin filament, complex of collagen trimer, fibrillar collagen trimer, banded collagen fibril, IgA immunoglobulin complex interstitial, matrix. The MF analysis identified terms including endopeptidase activity, cytokine activity, extracellular matrix structural constituent, cytokine receptor binding, serine-type endopeptidase activity, metalloend opeptidase activity, immune receptor activity, extracellular matrix structural constituent conferring tensile strength, structural constituent of skin epidermis, oxidoreductase activity, acting on the CH-NH2 group of donors.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed the involvement of several key pathways, including cytokine-cytokine receptor interaction, cytoskeleton in muscle cells, viral protein interactions with cytokine and cytokine receptors, protein digestion and absorption, ECM-receptor interaction, amoebiasis, mineral absorption, malaria, inflammatory bowel disease, and type II diabetes mellitus. These pathways are crucial for understanding the underlying mechanisms in the context of the study. (Fig. 2D).

Identification of DEGs in NERD. (A) The volcano plot illustrates the differential expression of DEGs between non-erosive esophagitis and healthy controls. (B) The heatmap displays the top 40 upregulated and downregulated DEGs. (C) The top 10 enriched functions in GO analysis. (D) KEGG of the DEGs.
Construction of weighted gene co-expression network analysis
Construction of Weighted Gene Co-expression Network analysis (WGCNA) was performed on gene expression data from both the model and control groups using a correlation-based weighted value. In this analysis, kME was used to assess the effective connectivity between key genes, serving as a valuable criterion for module membership identification. A clustering analysis was conducted on the 15 samples in the dataset, and the re-clustering significantly differentiated the model group from the control group (Fig. 3A). Genes with kME > 0.8 were selected as module members, as they better represent the overall expression trend of the module (Fig. 3B and C). The data matrix underwent a transformation to generate the adjacency matrix and TOM matrix. Subsequently, gene clustering based on the Topological Overlap Matrix (TOM) matrix, dynamic tree cutting, module clustering, and merging of similar modules were performed(Fig. 3D). Finally, genes were categorized based on their expression patterns, with genes exhibiting similar patterns grouped into the same module. All genes were clustered into 14 modules (Fig. 3E), and the darker-colored modules (closer to blue or red) were identified as key modules distinguishing the model group from the control group, representing the most strongly correlated functional modules. Results indicated that the MEantiquewhite2 module was significantly correlated with the model group (Cor = -0.80, p < 1e-06) (Fig. 3F), the MEmidnightblue module was significantly correlated with the model group (Cor = -0.92, p < 2e-06) (Fig. 3G), and the MEskyblue module was significantly correlated with the model group (Cor = -0.73, p < 0.002) (Fig. 3H). These results highlight the differences between the control and model groups. To identify key genes, Venny diagrams of DEGs were plotted for the MEantiquewhite2, MEmidnightblue, and MEskyblue modules. The results revealed that 195 candidate hub genes were shared among the DEGs of these three modules (Fig. 4A).

WGCNA analysis of GSE182974. (A) Hierarchical clustering dendrogram of samples based on gene expression; (B,C) soft-thresholding power of WGCNA; (D) Gene hierarchical clustering dendrogram and corresponding modules based on TOM; (E) Heatmap of gene expression within modules; (F) Clustering of the antiquewhite2 module; (G) clustering of the midnightblue module; (H) clustering of the skyblue4 module.
Machine learning
We employed two machine learning algorithms: Support Vector Machine-Recursive Feature Elimination (SVM-RFE) and Random Forest. These algorithms were used to address the challenges posed by high-throughput omics data, including small sample sizes, high dimensionality, noise, and nonlinear relationships among variables. The Random Forest algorithm is known for its noise resistance, ability to prevent overfitting, and effectiveness in handling nonlinear relationships. In our study, the Random Forest algorithm identified 40 genes (Fig. 4B). SVM-RFE, a supervised learning algorithm, works by maximizing the “margin” between two classes to establish a decision boundary. Through iterative feature elimination, SVM-RFE identified 21 features that provided optimal cross-validation performance (Fig. 4C), and we present the final 20 significant genes (Fig. 4D). Additionally, during the gene selection process, we set the SVM threshold to 0.911, successfully identifying 85 genes with significant features. Following this, we performed an intersection analysis of the gene sets selected by both SVM-RFE and Random Forest (Fig. 4E). Ultimately, 29 candidate genes were identified, which include SPRR4, DMXL2, PCDHGC3, LIPG, LAMA1, TRPV3, PLEKHG6, SPINT1-AS1, SNTB1, MYH10, MYO10, BX255925.3, LRP12, LCN2, MUC22, ETNK2, KDR, NAV1, FAM3B, FOXN3-AS1, FOXP3, BRCA1, RAPGEF5, COL13A1, ZBED9, PNPLA4, KRT79, VANGL2, and AC044787.1.
Protein–protein interaction network and external dataset validation
In this study, we further investigated the potential interactions among the proteins encoded by 29 candidate genes through PPI network analysis12 and validation with an external dataset, aiming to explore the underlying relationships between these proteins and their roles in disease (Fig. 4F and Table S2). The PPI analysis revealed significant interactions among the proteins encoded by BRCA1, KDR, FOXP3, COL13A1, LAMA1, LCN2, KRT79, MYH10, SPRR4, MYO10, and VANGL2. These interactions may have a substantial impact on cellular signaling, cell cycle regulation, and disease progression. To further validate the biological relevance of these genes, we performed expression level analysis using the GSE 226,303 dataset. The results showed significant differential expression of genes such as KDR, LCN2, LIPG and NAV1 in this dataset (Fig. 4G), which further supports their crucial role in disease processes. By comparing the results from the PPI network analysis and the external dataset validation, we identified KDR and LCN2 as key candidate proteins. These proteins not only interact with other proteins in the PPI network but also exhibit significant expression differences in the GSE226303 dataset. These findings suggest that KDR and LCN2 play central roles in the regulation of biological processes related to esophagitis diseases.

Machine learning for candidate hub genes: (A) A Venny about DEGs and genes across the three modules; (B) Identification of biomarkers based on the Random Forest algorithm; (C,D) Biomarker selection using SVM-RFE; (E) A Venny about Random Forest and SVM-RFE; (F) Interaction with the PPI network; (G) A boxplot for external validation.
Candidate protein localization and subcellular distribution
KDR is prominently expressed in the esophagus, stomach, lungs, and pancreas, with notable expression observed in both the stomach and esophagus (Fig. 5A). LCN2 is predominantly distributed across the esophagus, stomach, lungs, liver, small intestine, bladder, and bone marrow, with significant expression in the stomach and esophagus (Fig. 5B). These findings strongly suggest that KDR and LCN2 play central roles in regulating disease-associated biological processes. Subcellular localization of KDR reveals its presence in the nucleus, endoplasmic reticulum, Golgi apparatus, and vesicles (Fig. 5C). LCN2 is primarily localized in extracellular spaces, exosomes, and specific vesicular compartments (Fig. 5D), providing a basis for future targeted drug delivery strategies. Due to the challenges in detecting and treating NERD, as well as the risk of cancer development if interventions are delayed or inadequately chosen, we applied the Kaplan-Meier method to estimate the survival function. Kaplan-Meier survival curves were constructed, and the log-rank test was used to compare survival rates between the high and low expression groups. The analysis suggests that the KDR and LCN2 genes may influence overall survival (OS) in gastric cancer (Fig. 5E and F).
Gene set enrichment analysis
To identify the overall trends in signaling pathways associated with key genes, single-gene Gene Set Enrichment Analysis (GSEA) enrichment analysis was performed on KDR and LCN2. In the KDR, six signaling pathways were identified, including Dacosta UV response via ERCC3 common DN, which showed an overall upregulation trend; whereas KEGG Medicus reference translation initiation, KEGG ribosome, Reactome eukaryotic translational elongation, Reactome SRP-dependent cotranslational protein targeting to membrane, and Reactome translation displayed an overall downregulation trend (Fig. 5G). In the LCN2, pathways such as KEGG Medicus reference translation initiation, KEGG ribosome, Reactome eukaryotic translation elongation, Reactome response of EIF2AK4 GCN2 to amino acid deficiency, Reactome selenoamino acid metabolism, and WP cytoplasmic ribosomal proteins exhibited an overall upregulation trend (Fig. 5H).

Localization and subcellular distribution of candidate proteins. (A) Distribution of the KDR gene in the human body; (B) Distribution of the LCN2 gene in the human body; (C) KDR subcellular localization; (D) LCN2 subcellular localization; (E) Survival curve analysis of KDR; (F) Survival curve analysis of LCN2; (G) GSEA of KDR; (H) GSEA of LCN2.
Molecular docking
Molecular docking was employed to validate the binding interactions between the compounds in coriander and potential targets, which could provide deeper insights into the potential effects of coriander on diseases. A negative binding energy indicates that the ligand molecule can bind to the receptor target protein. Both lower and higher negative binding energies suggest stronger binding affinity between active compounds and target proteins. The molecular docking results between coriander compounds and key targets are shown in the heatmap (Fig. 6A). Molecular interactions involve van der Waals forces, aromatic stacking, C-H bonds, π-alkyl interactions, conventional hydrogen bonds, and hydrophobic interactions. A binding affinity lower than − 7.0 kcal/mol12,13 indicates strong binding activity. Interestingly, the binding affinity of KDR with Ammidin was − 8.07 kcal/mol, while LCN2 exhibited binding affinities of -11.27 kcal/mol, -10.09 kcal/mol, and − 11.23 kcal/mol with campest-5-en-beta-ol, Isofucosterol, and beta-sitosterol, respectively. The molecular docking results suggest that Ammidin (Fig. 6B), campest-5-en-beta-ol (Fig. 6C), Isofucosterol (Fig. 6D), and beta-sitosterol (Fig. 6E) in coriander may affect metabolic pathways in the body by targeting KDR and LCN2, potentially offering therapeutic effects for NERD.

Molecular docking of KDR and LCN2; (A) Heatmap of molecular docking binding energies; (B) KDR with Ammidin; (C) LCN2 with campest-5-en-beta-ol; (D) LCN2 with Isofucosterol; (E) LCN2 with beta-sitosterol.
Molecular dynamics simulations
To gain a deeper understanding of the stability of protein-ligand complexes, molecular dynamics simulations were conducted in this study. Based on docking results, 100 ns molecular dynamics simulations were performed for KDR-Ammidin and LCN2-campest-5-en-beta-ol complexes to evaluate molecular motion, trajectory, structural features, binding potential, and conformational changes. Root Mean Square Deviation (RMSD) is a reliable indicator for assessing the conformational stability of proteins and ligands, representing the deviation of atomic positions from the initial configuration. A lower deviation indicates better conformational stability. The variation of RMSD values for the complexes was analyzed (Fig. 7A). The RMSD of the KDR-Ammidin complex stabilized after 18 ns, while the RMSD of the LCN2-campest-5-en-beta-ol complex fluctuated early on and stabilized after 23 ns (Fig. 7B). This suggests that the protein conformation did not undergo significant changes, and the complex remained relatively stable. Subsequently, we plotted free energy landscapes using RMSD, radius of gyration (ROC), and binding energy (Fig. 7C and D). Deep blue regions within the broad free energy landscape indicate local minima, which persist as the protein structure transitions to the lowest energy configuration. These local minima are clearly represented by the deep blue regions, elucidating the assembly of chemical moieties in the lowest energy state. A notable feature of this landscape is the consistent presence of a relatively high energy state across these complexes, with energy values ranging from 14 to 16 kJ/mol. This consistency between the complexes suggests that their structural or functional characteristics (e.g., molecular interactions or stability) may exhibit homogeneity. Furthermore, all complexes exhibit stable conformations at energy levels below 2 kJ/mol, as indicated by the deep blue regions on the landscape, confirming their thermodynamic stability. The binding free energies of various complexes were subsequently quantified using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method. The binding energies of the KDR-Ammidin and LCN2-campest-5-en-beta-ol complexes were − 191.513 ± 82.519 kJ/mol and − 193.775 ± 88 KJ/mol (Fig. 7E). Finally, changes in hydrogen bonds during the simulation were analyzed, and hydrogen bonds were formed in all complexes (Fig. 7F). This indicates interactions between the protein and ligand, with the formation of hydrogen bonds contributing to the maintenance of the complexes’ stable structures, thereby confirming the stability of the binding modes.

(A, B) RMSD of KDR and its complex, and RMSD of LCN2 and its complex; (C,D) 3D free energy surface plots for KDR with Ammidin and LCN2 with campest-5-en-β-ol. PC1: RMSD; PC2: ROC; (E) Binding energy plot of the KDR and LCN2 complex; (F) Hydrogen bond diagram of the KDR and LCN2 complex.
Discussion
This study focuses on the potential active ingredients and mechanisms of action of Coriandrum sativum in treating non-erosive reflux disease (NERD). We conducted an in-depth investigation using a combination of WGCNA, machine learning, PPI Network Analysis, external dataset validation, molecular docking, and molecular dynamics simulations. Our results suggest that components such as Ammidin, campest-5-en-beta-ol, Isofucosterol, and beta-sitosterol in coriander have strong affinities for the identified genes KDR and LCN2. These findings provide a crucial theoretical basis for future drug development and clinical applications, though further clinical exploration and validation are needed.
Existing studies indicate that coriander extract can cure acute colitis induced by acetic acid in rats14 and improve appetite in rats15,16. We have also observed positive effects of coriander in treating esophageal diseases17,18. A series of analyses were conducted using the dataset, which led to the identification of the key candidate proteins KDR and LCN2 In subsequent single-gene GSEA analysis, the overall trends of NERD-related pathways presented confirmed the value of KDR and LCN2 as biomarkers for NERD. KDR and LCN2 are widely distributed in multiple organs, including the esophagus and stomach. Subcellular localization studies show that they function in different intracellular regions. Both proteins may influence overall survival in gastric cancer, suggesting their significant roles in regulating NERD-related biological processes and disease progression.
The gene product of KDR is Vascular Endothelial Growth Factor Receptor 2 (VEGFR2), which is activated by VEGF and plays a key role in VEGF-mediated angiogenesis during wound healing. Angiogenesis markers, such as VEGF-A, have been widely shown to play a crucial role in gastrointestinal mucosal repair, such as the healing of gastric ulcers19. In patients, repeated exposure of the esophageal mucosa to reflux substances, such as gastric acid and pepsin, causes local mucosal damage. In response, the body needs to promote angiogenesis to provide additional nutrients and immune cells for the repair of the damaged mucosa20. Abnormal expression of the KDR gene can lead to cancer development21 and may disrupt normal angiogenesis, resulting in impaired esophageal mucosal repair. This causes the mucosa to remain in a damaged state, triggering NERD. KDR and its associated VEGF signaling pathway also play a role in regulating inflammatory responses22,23,24. When the esophageal mucosa is exposed to reflux substances, an inflammatory response is triggered. VEGF, by binding to KDR, regulates the recruitment and activation of inflammatory cells, influencing the release of inflammatory mediators25. If KDR gene function is impaired, it may lead to uncontrolled inflammation, maintaining persistent inflammation in the esophageal mucosa, worsening NERD symptoms, and potentially contributing to gastric cancer development26. The KDR signaling pathway also plays a role in regulating the proliferation and apoptosis of esophageal epithelial cells27. Under normal conditions, the proliferation and apoptosis of esophageal epithelial cells are balanced to maintain the integrity of the esophageal mucosa. In NERD, abnormal expression of the KDR gene may disrupt this balance, leading to abnormal cell proliferation or excessive apoptosis, thereby affecting the normal structure and function of the esophageal mucosa.
LCN2, also known as Neutrophil Gelatinase-Associated Lipocalin (NGAL) or Iron-regulated Transporter Protein, is a peptide produced by macrophages, neutrophils, and other immune and parenchymal cells. It exerts antibacterial functions by binding to catecholate-type iron carriers with high affinity28. Additionally, it is involved in various physiological functions, including the transmembrane transport of hydrophobic ligands, regulation of immune responses, maintenance of iron homeostasis, and promotion of epithelial cell differentiation28,29,30,31. Therefore, LCN2 is a chemically stable protein, making it an intriguing candidate as a biomarker. LCN2 also binds to iron carriers and participates in the regulation of iron metabolism. Reduced expression of LCN2 has been shown to improve asthma in mice32. During inflammatory states, the expression of LCN2 is upregulated33,34,35,36. In NERD, the inflammatory response in the esophageal mucosa alters the local microenvironment. LCN2 modulates iron metabolism, affecting the function of inflammatory cells and the progression of the inflammatory response37,38. For instance, LCN2 can limit bacterial iron uptake, thereby inhibiting bacterial growth and reducing further damage to the esophageal mucosa caused by infection39. However, dysregulation of LCN2 expression may disrupt iron metabolism balance and exacerbate the inflammatory response. LCN2 is also closely related to oxidative stress. Upon reflux substance exposure, the esophageal mucosa produces reactive oxygen species (ROS), triggering oxidative stress40,41. LCN2 alleviates oxidative stress damage to esophageal mucosal cells by regulating antioxidant enzyme activity or directly scavenging ROS40. LCN2 can bind to receptors on the cell surface, activating intracellular signaling pathways42. Abnormal activation or inhibition of related signaling pathways may disrupt the normal physiological functions of esophageal mucosal cells, leading to cellular dysfunction and triggering NERD.
The molecular docking results successfully reveal the strong binding interactions between the components of coriander and the potential targets, KDR and LCN2. The negative binding energies clearly indicate that the ligand molecules are able to bind to the target proteins, and the relatively low negative binding energies further highlight the excellent binding affinity between the active compounds and the target proteins. Specifically, the binding affinity between KDR and Ammidin is -8.07 kcal/mol, while LCN2 binds with campest-5-en-beta-ol, Isofucosterol, and beta-sitosterol with affinities of -11.27 kcal/mol, -10.09 kcal/mol, and − 11.23 kcal/mol, respectively—all values below − 7.0 kcal/mol. The molecular docking results suggest that Ammidin, campest-5-en-beta-ol, Isofucosterol, and beta-sitosterol in coriander may exert therapeutic effects on NERD by targeting KDR and LCN2.
Based on the docking results, we performed 100 ns molecular dynamics simulations on the KDR-Ammidin and LCN2-campest-5-en-β-ol complexes, and analyzed molecular motion, trajectory, structural features, binding potential, and conformational changes in detail. The conformational stability of the protein and ligand was assessed using RMSD. The RMSD of the KDR-Ammidin complex stabilized after 18 ns, while the RMSD of the LCN2-campest-5-en-β-ol complex fluctuated initially but also reached stability after 23 ns. This indicates that the protein conformations of both complexes did not undergo significant changes, maintaining relatively stable structures. Subsequently, we plotted free energy landscapes using RMSF, ROC, and binding energy. Notably, all complexes exhibit relatively stable conformations within a broad free energy landscape, with energy levels ranging from 14 to 16 kJ/mol in the relative maximum energy state. This suggests that they may share similar structural or functional features, such as molecular interactions or stability, Additionally, the binding free energies of the two complexes were quantitatively evaluated using the MM/GBSA method. The binding free energy of the KDR-Ammidin complex was − 191.513 ± 82.519 kJ/mol, while the binding free energy of the LCN2-campest-5-en-β-ol complex was − 193.775 ± 88.889 kJ/mol. These results indicate that both complexes possess strong binding capabilities, further confirming their stability. Finally, we analyzed the changes in hydrogen bonds during the simulation process. Both complexes formed hydrogen bonds, indicating interactions between the protein and ligand. The formation of hydrogen bonds helps maintain the stable structure of the molecules, thus confirming the stability of the binding mode.
Materials and methods
Ingredients of Coriandrum sativum
The keyword “Coriandri Sativi Herba” was used to retrieve the components of coriander from the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) database43 (https://www.tcmsp-e.com/) (table S1).). Components that met the criteria for drug-likeness score (DL) from Molsoft (https://www.molsoft.com) and oral bioavailability (OB) from SwissADME44 (http://www.swissadme.ch/) were selected. Their molecular weights and formulas were obtained from PubChem45 (https://pubchem.ncbi.nlm.nih.gov/), in preparation for molecular docking studies.
Data Preparation
Gene expression data for NERD was collected from the GEO database (http: //www.ncbi.nlm.nih.gov/geo/). The GSE 182,974 dataset19 was downloaded from GPL 24,676, consisting of 15 individual samples, 6 control samples, and 9 NERD samples. Additionally, the GSE 226,303 dataset46 was retrieved from the GPL 18,573 platform, including 12 samples, 8 control samples, and 10 NERD samples.
Extraction of the expression matrix and DEG analysis
The expression matrix file from the GSE 182,974 dataset was extracted, and gene-expression relationships were organized based on the platform file from the dataset. DEGs between the NERD disease group and the control group were selected based on the criteria of p-value < 0.05 and | log2FoldChange| > 1. The “limma” R package was used to identify differentially expressed genes (DEGs) between NERD tissue and normal tissue. A corresponding volcano plot was then generated. The top 40 upregulated and downregulated DEGs were selected based on fold-change values, and a heatmap was generated using the “pheatmap” package in R software.
Gene ontology and Kyoto encyclopedia of genes and genomes
The identified DEGs were subjected to GO47 enrichment and KEGG pathway analysis48 to uncover the biological significance of NERD-associated DEGs. GO analysis49, BP, CC, and MF, was performed using the “GOplot” package in R. KEGG pathway50 analysis was conducted to explore potential pathways, followed by visualization based on key statistical parameters.
Weighted gene co-expression network analysis
A weighted gene co-expression network was constructed for the GSE 182,974 cohort using the scale-free topology criterion-based WGCNA51. The “pickSoftThreshold” function from the WGCNA package was used to determine the soft threshold power and neighborhood52,53. The adjacency matrix was then transformed into a TOM, and the corresponding dissimilarity was calculated for hierarchical clustering analysis. The dynamic tree cut method with a minimum module size of 50 was employed to identify co-expressed gene modules. Gene significance (GS) and module membership (MM) values were used to assess the association between gene modules and NERD, aiming to identify key modules.
Machine learning
Variable selection was performed using SVM-RFE to identify key variables. The SVM-RFE model was fitted using different kernel functions, and model accuracy was compared. The R package “Random Forest” was applied to classify DEGs of hub genes. The random forest model determined the optimal number of variables by calculating the average error rate of candidate hub genes. Based on the determined parameters, a random forest model was constructed. Finally, the feature importance score for each candidate hub gene was determined, and genes with the highest importance scores were selected for subsequent analysis. The intersecting genes from both machine learning algorithms were used as candidate biomarkers for NERD patients.
Protein-protein interaction network analysis
The obtained marker genes were transferred to the online database of the Search Tool for the Retrieval of Interacting Genes (STRING) (http://string-db.org)54 for PPI analysis. The PPI network was constructed using only experimentally validated interactions with a confidence score greater than 0.4. The network was visualized using Cytoscape 3.9.155. Subsequently, an interaction network was constructed for the marker genes. The node color in the network was based on their degree values, with darker colors indicating higher rankings and greater importance of the nodes in the network.
Distribution and subcellular localization of KDR and LCN2
Expression of biomarkers throughout the human body was obtained from the Human Protein Atlas (HPA) database56 (https://www.proteinatlas.org/). Subcellular localization of biomarker proteins was predicted using the COMPARTMENTS database (https://compartments.jensenlab.org/). This provides a foundation for understanding the changes in biomarkers during the disease process.
Survival analysis in gastric cancer
The Kaplan-Meier Plotter57,58 was utilized to perform survival analysis on the final key hub genes. Survival analysis, based on gene expression levels, can also assess the clinical significance of specific genes. Kaplan-Meier survival curves were generated within this database to analyze the correlation between hub genes and the survival rate of gastric cancer patients.
Molecular Docking
The structure was comprehensively characterized using the AlphaFold59, and active sites were identified through the PDB database60,61. The structures of the compounds were downloaded from PubMed and molecular docking was performed using Dockey62. Finally, the top-ranked complex conformations based on docking scores were selected for visualization, which was performed using PyMOL63 and Discovery Studio 4.5 Client64.
Molecular dynamics simulation
Molecular dynamics (MD) simulations were performed for 100 ns to evaluate the stability of the selected complex structures and their interactions with proteins using GROMACS65. We utilized GROMACS 2023.3.Protein parameters were generated using the CHARMM all-atom force field66. The domain topology was constructed using the CHARMM-modified TIP3P water model in GROMACS, while ligand topology was obtained from the CGenFF67. All protein-ligand systems were centered in triclinic boxes and solvated with the simple point charge (SPC) water model. Chloride ions were added to neutralize the protein systems. Finally, MD trajectories were analyzed using GROMACS modules with RMSD, ROG, and the number of intermolecular hydrogen bonds (H-bonds) as evaluation parameters. To further investigate the conformational landscape of the protein-ligand complexes, we constructed the free energy landscape (FEL) based on the MD simulation trajectories. A custom Python script was used to extract and combine RMSD and radius of gyration (Rg) values, generating a two-dimensional data matrix. The Gibbs free energy surface was then calculated, and the final FEL plots were generated in both 2D and 3D formats. Graphical spectra were plotted using Origin 2024 software.
Conclusion
In GERD, NERD accounts for approximately 70%. Current proton pump inhibitor treatments are limited by issues such as relapse upon discontinuation and increased risk of infections, highlighting the need for new therapeutic approaches. This study focuses on the potential active components and mechanisms of coriander in the treatment of NERD. Data from multiple databases were collected, and key candidate proteins KDR and LCN2 were identified through integrated methods, including WGCNA, machine learning, and PPI network analysis. Molecular docking revealed that components such as Ammidin and campest-5-en-beta-ol in coriander have strong binding affinities for KDR and LCN2, and molecular dynamics simulations confirmed the stability of the complexes. Furthermore, the study found that these key proteins are distributed in the esophagus, stomach, and other tissues, participating in various biological processes and signaling pathways, which may influence the overall survival of gastric cancer. Although this study provides a theoretical basis for subsequent drug development and clinical applications, further in vivo and in vitro experiments are needed, and research into resistance mechanisms requires deeper exploration.
Data availability
All data generated or analyzed during this study are included in this manuscript or supplementary information files.
References
Vakil, N. et al. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Official J. Am. Coll. Gastroenterology| ACG. 101, 1900–1920 (2006).
Gyawali, C. P. et al. Nonerosive reflux disease: clinical concepts. Ann. N. Y. Acad. Sci. 1434, 290–303 (2018).
Zhang, J. X. et al. Proton pump inhibitor for non-erosive reflux disease: a meta-analysis. World J. Gastroenterol. 19, 8408–8419. https://doi.org/10.3748/wjg.v19.i45.8408 (2013).
Reimer, C. & Bytzer, P. Discontinuation of long-term proton pump inhibitor therapy in primary care patients: a randomized placebo-controlled trial in patients with symptom relapse. Eur. J. Gastroenterol. Hepatol. 22, 1182–1188. https://doi.org/10.1097/MEG.0b013e32833d56d1 (2010).
Bavishi, C. & Dupont, H. L. Systematic review: the use of proton pump inhibitors and increased susceptibility to enteric infection. Aliment. Pharmacol. Ther. 34, 1269–1281. https://doi.org/10.1111/j.1365-2036.2011.04874.x (2011).
Abrahami, D. et al. Proton pump inhibitors and risk of gastric cancer: population-based cohort study. Gut 71, 16–24 (2022).
Tibebe, D. et al. Evaluation of total phenolic, flavonoid contents, and antioxidant activities of seed extracted from coriander (Coriandrum sativum L.) and black Cumin (Nigella sativa) spices. Food. Anal. Methods. 17, 945–955 (2024).
Aissaoui, A., El-Hilaly, J., Israili, Z. H. & Lyoussi, B. Acute diuretic effect of continuous intravenous infusion of an aqueous extract of Coriandrum sativum L. in anesthetized rats. J. Ethnopharmacol. 115, 89–95. https://doi.org/10.1016/j.jep.2007.09.007 (2008).
Laribi, B., Kouki, K., M’Hamdi, M. & Bettaieb, T. Coriander (Coriandrum sativum L.) and its bioactive constituents. Fitoterapia 103, 9–26 (2015).
Yarnell, E. Herbs for upper digestive overgrowth of flora. Altern. Complement. Ther. 24, 173–179 (2018).
Costa, M. C., Noviello, T. M. R., Ceccarelli, M. & Cerulo, L. Robust Feature Selection strategy detects a panel of microRNAs as putative diagnostic biomarkers in Breast Cancer. bioRxiv, 2007. 2003.547377 (2023). (2023).
Li, B. et al. An investigation of the toxicity and mechanisms of food additives based on network toxicology and GEO databases: A case study of aspartame. Food Anal. Methods, 1–16 (2024).
Leutcha, P. B. et al. Flavonoids and other constituents from Jacaranda mimosifolia: in vitro analysis, molecular docking, and molecular dynamic simulations of antioxidant and anti-inflammatory activities. Biomed. Pharmacother. 182, 117768 (2025).
Heidari, B., Sajjadi, S. E. & Minaiyan, M. Effect of Coriandrum sativum hydroalcoholic extract and its essential oil on acetic acid-induced acute colitis in rats. Avicenna J. Phytomedicine. 6, 205 (2016).
Nematy, M. et al. The effect of hydroalcoholic extract of Coriandrum sativum on rat appetite. Avicenna J. Phytomedicine. 3, 91 (2013).
Anaeigoudari, A. Therapeutic effects of Coriandrum sativum extracts: A review. Asian Pac. J. Trop. Biomed. 14, 181–186 (2024).
Singh, M. B., Singh, P. & Jain, P. in Medicinal Spices and Herbs from India 85–100Apple Academic Press, (2024).
Boudaia, O. et al. Ethnopharmacological investigation and traditional cultural use of anticancer medicinal plants in Morocco’s Casablanca-Settat region. Ethnobotany Res. Appl. 28, 1–49 (2024).
Zhao, Y., Ma, T. & Zou, D. Identification of unique transcriptomic signatures and hub genes through RNA sequencing and integrated WGCNA and PPI network analysis in nonerosive reflux disease. J. Inflamm. Res. 6143–6156 (2021).
Chen, Z. et al. Endothelial β-catenin upregulation and Y142 phosphorylation drive diabetic angiogenesis via upregulating KDR/HDAC9. Cell. Communication Signal. 22, 182 (2024).
He, D. Anti-angiogenic mAb, anti-human VEGFR2 (Cyramza) retards tumor growth in KDR (VEGFR2) humanized mice engrafted with syngeneic vs. xenograft solid tumors. Cancer Res. 83, 4590–4590 (2023).
Misar, P. L. & Otari, K. V. Investigation of the protective effect of cilostazol on acute lung injury-mediated inflammation and in Silico molecular modelling studies of inflammatory signalling pathway: a repurposing study. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1–21 (2025).
Duan, Z. et al. Wumei Wan attenuates angiogenesis and inflammation by modulating RAGE signaling pathway in IBD: network Pharmacology analysis and experimental evidence. Phytomedicine 111, 154658 (2023).
Zhong, Y. & Yu, P. B. Angiogenesis redux: an overall protective role of VEGF/KDR signaling in the microvasculature in pulmonary arterial hypertension. Arterioscler. Thromb. Vasc Biol. 43, 1784–1787 (2023).
Saoud, H., Aflouk, Y., Afia, A. B., Gaha, L. & Jrad, B. B. H. Association of VEGF-A and KDR polymorphisms with the development of schizophrenia. Hum. Immunol. 83, 528–537 (2022).
Liang, R. et al. Dihydroartemisinin inhibits the tumorigenesis and invasion of gastric cancer by regulating STAT1/KDR/MMP9 and P53/BCL2L1/CASP3/7 pathways. Pathology-Research Pract. 218, 153318 (2021).
Zhang, H., Zhang, B., Chen, C. & Chen, J. Circular RNA circlifr regulates the proliferation, migration, invasion and apoptosis of human vascular smooth muscle cells via the miR-1299/KDR axis. Metab. Brain Dis. 37, 253–263 (2022).
Goetz, D. H. et al. The neutrophil Lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell. 10, 1033–1043 (2002).
Kurzrock, R. et al. Interleukin-1 receptor antagonist levels predict favorable outcome after Bermekimab, a first-in-class true human interleukin-1α antibody, in a phase III randomized study of advanced colorectal cancer. Oncoimmunology 8, 1551651 (2019).
Holgersen, K. et al. High-resolution gene expression profiling using RNA sequencing in patients with inflammatory bowel disease and in mouse models of colitis. J. Crohn’s Colitis. 9, 492–506 (2015).
Moniruzzaman, M., Wang, R., Jeet, V., McGuckin, M. A. & Hasnain, S. Z. Interleukin (IL)-22 from IL-20 subfamily of cytokines induces colonic epithelial cell proliferation predominantly through ERK1/2 pathway. Int. J. Mol. Sci. 20, 3468 (2019).
Xu, L., Shi, M. & Lipocalin 2 (LCN2) knockdown regulates Treg/Th17 balance to improve asthma in mice. J. Asthma Allergy, 1323–1332 (2023).
Du, H. et al. Lipocalin-2 alleviates LPS-induced inflammation through alteration of macrophage properties. J. Inflamm. Res. 4189–4203 (2021).
Guardado, S., Ojeda-Juárez, D., Kaul, M. & Nordgren, T. M. Comprehensive review of Lipocalin 2-mediated effects in lung inflammation. Am. J. Physiology-Lung Cell. Mol. Physiol. 321, L726–L733 (2021).
Ma, J. et al. LCN2 mediates skin inflammation in psoriasis through the SREBP2–NLRC4 axis. J. Invest. Dermatology. 142, 2194–2204 (2022).
Al Jaberi, S. et al. Lipocalin-2: structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 142, 112002 (2021).
Lim, D., Jeong, J. & Song, J. Lipocalin 2 regulates iron homeostasis, neuroinflammation, and insulin resistance in the brains of patients with dementia: evidence from the current literature. CNS Neurosci. Ther. 27, 883–894 (2021).
An, H. S. et al. Lipocalin-2 promotes acute lung inflammation and oxidative stress by enhancing macrophage iron accumulation. Int. J. Biol. Sci. 19, 1163 (2023).
Zhao, M. et al. Identification and analysis of a prognostic ferroptosis and iron-metabolism signature for esophageal squamous cell carcinoma. J. Cancer. 13, 1611 (2022).
PishKenari, F. N., Qujeq, D., Bonahi, S. S. M., Kashifard, M. & Hajian-Tilaki, K. Nitric oxide and inducible nitric oxide synthase levels in EE and NERD patients. Gastroenterol. Hepatol. Bed Bench. 15, 79 (2022).
Cheng, X. et al. NeRD: a multichannel neural network to predict cellular response of drugs by integrating multidimensional data. BMC Med. 20, 368 (2022).
Schröder, S. K., Gasterich, N., Weiskirchen, S. & Weiskirchen, R. Lipocalin 2 receptors: facts, fictions, and Myths. Front. Immunol. 14, 1229885 (2023).
Ru, J. et al. TCMSP: a database of systems Pharmacology for drug discovery from herbal medicines. J. Cheminform. 6, 1–6 (2014).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Wang, Y. et al. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37, W623–W633 (2009).
Ustaoglu, A. et al. Identification of novel immune cell signature in gastroesophageal reflux disease: altered mucosal mast cells and dendritic cell profile. Front. Immunol. 14, 1282577 (2023).
Liu, T. et al. Metabolome and transcriptome association study reveals biosynthesis of specialized benzylisoquinoline alkaloids in Phellodendron amurense. Chinese Herb. Medicines (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Wu, T. et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation 2 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Xu, R. et al. Integrated chemical and transcriptomic analyses unveils synthetic characteristics of different medicinal root parts of Angelica sinensis. Chin. Herb. Med. 12, 19–28 (2020).
Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 (2016).
Giulietti, M., Occhipinti, G., Principato, G. & Piva, F. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell. Oncol. 39, 379–388 (2016).
Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Thul, P. J. & Lindskog, C. The human protein atlas: a Spatial map of the human proteome. Protein Sci. 27, 233–244 (2018).
Chen, D. & Hou, X. Aspartame carcinogenic potential revealed through network toxicology and molecular Docking insights. Sci. Rep. 14, 11492 (2024).
Győrffy, B. Transcriptome-level discovery of survival‐associated biomarkers and therapy targets in non‐small‐cell lung cancer. Br. J. Pharmacol. 181, 362–374 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Bold, G. et al. A novel potent oral series of VEGFR2 inhibitors abrogate tumor growth by inhibiting angiogenesis. J. Med. Chem. 59, 132–146 (2016).
Barkovskiy, M., Ilyukhina, E., Dauner, M., Eichinger, A. & Skerra, A. An engineered Lipocalin that tightly complexes the plant poison Colchicine for use as antidote and in bioanalytical applications. Biol. Chem. 400, 351–366 (2019).
Du, L. et al. Dockey: a modern integrated tool for large-scale molecular Docking and virtual screening. Brief. Bioinform. 24, bbad047 (2023).
DeLano, W. L. & Pymol An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92 (2002).
Pawar, S. S. & Rohane, S. H. Review on discovery studio: An important tool for molecular docking. (2021).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291 (2021).
Vanommeslaeghe, K. et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all‐atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Vanommeslaeghe, K. & MacKerell, A. D. Jr Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 52, 3144–3154 (2012).
Funding
Sichuan University Student Innovation Training Program Project and Provincial University Student Entrepreneurship Practice Project of Chengdu University (No. S202411079003S).
Author information
Authors and Affiliations
Contributions
X.S.: contributed towards wrote the original draft and did the experimental work.B.L.: contributed towards analyzed and interpreted the data.Y.C., Y.W., W.Z., M.D.: contributed towards supervision and validationY.X.: contributed towards Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, X., Li, B., Chen, Y. et al. Exploration of the potential therapeutic effects and targets of Coriandrum sativum on non-erosive esophagitis based on bioinformatics and molecular dynamics simulation. Sci Rep 15, 19177 (2025). https://doi.org/10.1038/s41598-025-03945-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03945-1
