
Abstract
Chagas disease, also known as American Trypanosomiasis, is a zoonosis with global distribution caused by the parasite Trypanosoma cruzi, primarily transmitted through the feces of infected triatomines. The emergence of new cases highlights the importance of early pathogen detection in vectors and reservoirs to generate effective control strategies and establish preventive policies. The objective of this study was to design and validate a detection system of T. cruzi based on specific DNA cleavage, activation of Cas12a and trans-cleavage, targeting the genes Cytochrome B (Cytb), 18 S ribosomal subunit (SR18 s), and histone (H2 A). This system was validated for their uses in both vectors and reservoirs of the parasite. The initial step involved performing a bioinformatic analysis of the target genes, followed by the design of RNA guides specific to each cleavage site, along with primers for amplifying the target region through PCR and RPA. Subsequently, we sequenced the amplified DNA target and validated the detection system using T. cruzi DNA extracted from naturally infected Rhodnius pallescens in the metropolitan area of Bucaramanga, Colombia. After standardizing the method, we tested the CRISPR/Cas system with Silvio X10 laboratory strain of T. cruzi and scaled up to blood samples of naturally infected Didelphis marsupialis. As a result, we observed DNA cleavage using the CRISPR/Cas system with the Cytb guide, achieving a detection sensitivity of 118 parasite equivalents/mL in PCR and 116 parasite equivalents/mL with RPA amplification. Sequencing of the Cytb gene showed no mutations in the cleavage site. However, point mutations and indels were found in SR18S and H2 A, avoiding the formation of the CRISPR/LbCas12 complex. Furthermore, we introduce the design of a fluorescent detection prototype with CRISPR/LbCas12a called “Tropical Diseases Detector” (TropD-Detector). This device operates with an excitation wavelength of 480 nm emitted by an LED and a high-pass light filter with a cutoff wavelength of 500 nm. We detected positive samples using any photographic camera system. The TropD-Detector provides a visual, viable, and sensitive method for detecting T. cruzi in both vectors and reservoirs from endemic areas.
Similar content being viewed by others


Introduction
Chagas disease (CD) is historically considered a rural and semi-rural health issue in Latin America1. However, the reports show the spread of the disease to other continents where it was previously non-existent, positioning it as an emerging and expanding public health concern2,3. Trypanosoma cruzi is the etiological agent of CD, which affects millions of people, particularly in developing countries from America4. Vectorial transmission of the parasite occurs through contact with the feces of infected insects from the Triatominae subfamily5. In addition to insect transmission, other ways of infection include contaminated food ingestion, infected blood transfusion, laboratory accidents, and organ transplants have been reported. CD’s current control and prevention efforts focus on vectorial control, early disease detection, and access to treatment1,6. While a wide range of sensitive and specific diagnostic techniques exist in humans, in vectors and reservoirs of T. cruzi, the available methods for diagnosis are limited and costly, reducing the likelihood of conducting epidemiological studies in these hosts.
In vector diseases, the One Health concept comes with an integrated perspective of control that considers the interconnectedness of human, animal, and ecological health. This holistic view has gained relevance for its technical application in CD due to the significant rise in acute cases over recent decades, with more than 70% of records from oral transmission in which infected reservoirs and vectors have been involved7. This finding suggests that current preventive strategies must expand beyond vectorial control based on documented outbreaks, even without vectorial insects8. In this context, the reservoirs of Didelphis genus, take relevance due to their role in the oral transmission of T. cruzi through contaminated food from infected secretions of their anal glands9. Considering that increasing deforestation in endemic regions from Latin America has facilitated the migration of reservoirs and vectors of T. cruzi to urban and peri-urban areas, it is crucial to establish new systems of diagnosis and surveillance in these hosts to help at health entities to reduce the incidence of CD cases in urban centers with prevention programs.
In light of the above, to implement prevention programs, it is crucial to establish screening and diagnostic systems targeting species involved in the parasite’s life cycle across all geographical regions affected by CD10,11. In this way, early detection of the parasite in vectors or reservoirs can identify active transmission cycles and pinpoint high-risk areas for communities, thus enabling the implementation of localized preventive measures11,12. Although there is significant progress in the detection systems for T. cruzi, it is essential to highlight that most current methods primarily focus on humans. Moreover, depending on each technique, concerns regarding sensitivity, reliability, and high costs exist. For instance, in Latin America, the most simple and rapid detection test is the peripheral blood smear, which can be a viable initial screening tool with an effectiveness of 68% as long as it is applied in patients with the acute phase of the disease1. More complex techniques include serological tests, which have higher detection capacity and are common in hospitals. Nonetheless, false positives with CD have been reported with other parasites responsible for leishmaniasis, especially in geographic regions endemic to both diseases13,14. In addition to these techniques, it is necessary to employ confirmatory diagnostic essays such as indirect immunofluorescence (IFI) or molecular tests.
Among molecular techniques, endpoint PCR is considered the “Gold Standard” test for molecular identification of T. cruzi. While this method is highly reliable, it is prone to false positives due to cross-contamination between samples. Its effectiveness depends on the appropriate primer design and the operator’s technical expertise15. Despite the diagnostic system’s advantages in CD, false negatives could also appear in host low parasitic loads16,17. More sensitive techniques, such as real-time PCR (qPCR) or digital PCR (dPCR), have been conducted in various diseases, including CD. Given its high sensitivity, qPCR is an alternative to PCR in chronic cases, which tends to produce false negatives in such instances18,19. However, the downside of these methods lies in the need for robust, expensive equipment with highly trained personnel and having limited field applicability. Additionally, it is necessary to bring the samples to a third-level laboratory, which results in extended detection times to get a diagnosis.
Given the context mentioned above, it is essential to develop new diagnostic techniques that are accurate, confirmatory, rapid, cost-effective, and adaptable to settings with limited infrastructure, such as rural areas in Latin America where the CD is endemic20. Addressing these diagnostic necessities before explained, CRISPR/LbCas12a systems adapted as detection sensors are a new, sensible, low-cost, programmable, and specific alternative to detect target regions within DNA. Although CRISPR systems were originally a genome editing tool, their application in disease diagnosis has increased in recent years, mainly due to their role in rapid COVID-19 tests during the pandemic21. The CRISPR-based detection methodology involves a few steps. Initially, amplification of the target site is necessary, followed by a Cas12a scanning step to recognize the PAM site, which enables hybridization of the gRNA spacer region to the target sequence within the amplicon22. Conventional techniques, such as PCR, achieve pre-amplification; however, new isothermal amplification methods like RPA offer substantial advantages in terms of time efficiency, low instrumentation requirements, high sensitivity, and suitability for field deployment23. Subsequently, Cas protein and designed guide-RNA are complexed to locate the target site. Once identified, the Cas protein produces the first (Cis) cleavage at a specific site on the amplified DNA. Consecutively, a second (Trans) random cleavage occurs, which cleavages a single-strand DNA reporter24. Cas12 exhibits trans-cleavage activity on a variety of reporter probes, including those modified with fluorescence, biotin, or other detectable molecules. It is employed to develop pathogen detection systems that use blue LEDs or robust equipment such as a UV light transilluminator25,26,27,28.
In light of the previously discussed challenges in diagnosis like costs and the need for a simple screening method, the objective of this study is to develop a fluorescence-based detection prototype for T. cruzi employing CRISPR/LbCas12a system, applicable to insect vectors and parasite reservoirs to establish a highly specific, sensitive, rapid, programmable, cost-effective, and portable detection system that any person with a short training can use for the detection of T. cruzi.
Methods
Bioinformatic analysis
To achieve molecular detection of T. cruzi, we targeted conserved DNA regions to enable specific detection of the parasite. A thorough literature review led to selection of three genes: SR18S, H2 A, and Cytb29,30,31. Representative sequences from the NCBI database of each gene were downloaded and aligned using the SNAPGENE software (Fig. 1A). Consequently, we designed primers for the SR18 s and H2 A gene, while primers for the Cytb gene were adapted from previously reported for T. cruzi detection31. We designed guide RNAs (gRNAs) for each gene with 24-pb length and a PAM sequence TTTV in the CHOPCHOP v2 software32. These gRNAs were chosen based on high specificity for T. cruzi, efficiency scores, and zero off-target effects in other genome regions. Then, we filtered the designs in BLAST and selected the most optimal for each gene. In like manner, designed primers were analyzed in BLAST to select the most specific design with 100% homology for T. cruzi. Each primer was compared and aligned against negative controls with Rhodnius prolixus, Leishmania infantum, Trypanosoma theileri, and Lutzomyia sp as shown in Annex Table 1.

Schematic of the CRISPR/LbCas12a assay workflow for detection of T. cruzi in reservoirs and vectors of the parasite. A Bioinformatic analysis. BLAST analysis and literature review of target regions from T. cruzi. B Sampling individuals and DNA extraction. Isolation of DNA of T. cruzi from collected samples from D. marsupialis and intestine of R. pallescens. C PCR or RPA amplification. PCR amplification in thermocycler or isothermal amplification with RPA. D gRNAs validation. In amplified DNA, the gRNA guides the LbCas12a protein cleavage (Cis cleavage). Then, the protein cleaves the fluorescent single-stranded DNA reporter due to its Trans activity. The reporter is excited at 480 nm and emits fluorescence at 530 nm E Fluorescentdetection in the spectrophotometer. The emitted fluorescence is measured in a spectrophotometer over two hours at 37 °C. F Fluorescent detection under UV light in a transilluminator. Visualization of positive samples under UV light on a transilluminator. G Fluorescent detection cleavage under blue light fluorescent emission prototype “TropD-Detector. Employing blue LED light, the reporter is excited, and the emitted fluorescence is registered and detected in a conventional cellphone. Figure created with BioRender.com.
The three analyzed genes were tested on species used as non-target controls. Ten possible designs were obtained within the amplified region for each gene, with potential binding sites containing PAM (TTTV) motifs. One gRNA was selected for each gene, prioritizing high specificity and 0% off-target score, as shown in Table 1.
DNA isolation from T. cruzi from R. pallescens intestine
The collection of R. pallescens specimens was conducted between November 2021 and February 2022 by personnel from the Medical Entomology Laboratory in local neighborhoods of Bucaramanga, Colombia. The sampling was carried out with Angulo trap33, in three neighborhoods with forest fragments. A total of 26 specimens were collected: twelve from the “Pan de Azúcar” neighborhood, eight from “Limoncito,” and six from “Bucarica.” DNA extraction was done with Qiagen DNeasy Blood and Tissue protocol (Qiagen, Valencia, CA, USA) from dissected hindgut samples and stored at − 20 °C.
Blood sample collection and DNA extraction of D. marsupialis
Collected samples from D. marsupialis were obtained from specimens treated at the Wildlife Rescue and Care Center of the metropolitan area of Bucaramanga. Initially, these animals were weighed employing a digital scale with a 20 kg capacity (Vibra, Terrace, USA®). A veterinarian anesthetized each specimen using xylazine (doses of 2.2 mg/kg) and ketamine hydrochloride (doses of 5–7.5 mg/kg). Ultimately, 0.5–1 mL blood samples were isolated from the caudal vein for each animal, employing 3 mL syringes and 23G x 1” needles, and stored in microtubes with EDTA. DNA extraction from D. marsupialis blood samples was conducted with the Corpogen extraction Kit, following the manufacturer’s protocol (CorpoGen, Bogotá, Colombia®). Briefly, 250 µL of blood with EDTA was extracted, with the final elution carried out in 100 µL of elution buffer. Finally, the quality and quantity of the extracted DNA were assessed using a NanoDrop 2000 (Thermo Fisher Scientific, Massachusetts, USA) The collection of blood samples from D. marsupialis was conducted as part of the research project titled “Epidemiological characterization of Trypanosoma cruzi and Leishmania spp. infection in D. marsupialis (common opossum) from the Metropolitan Area of Bucaramanga, Colombia,” led by the Universidad Cooperativa de Colombia. This study was approved by the Scientific Research Ethics Committee, which issued Bioethical Concept No. BIO563, as recorded in Minutes No. 2/2023, dated October 25, 2023.
For wild insects, Decree 1376 of June 27, 2013, was taken into account, which regulates the collection permit for specimens of wild species of biological diversity for non-commercial scientific research purposes. The Medical Entomology Laboratory, part of CINTROP-UIS, operates under Framework Permit for Specimen Collection No. IDB0398, issued by the National Authority of Environmental Licenses. The collection of R. pallescens was conducted following laboratory animal handling protocols and approved by the Scientific Research Ethics Committee (CEINCI) Comité de ética en investigación científica CEINCI” of the Industrial University of Santander (acronym in Spanish CEINCI), as recorded in Minutes No. 15, dated August 27, 2021. Nucleotides used in this study were sourced from the project “Producción de nucleótidos a partir de biomasa residual de la agroindustria para diagnóstico por biología molecular en el departamento de Santander,” also approved by CEINCI under Minutes No. 20, dated May 20, 2022. Further- more, the study was carried out in compliance with the arrive guidelines and following the 1989 Colombian Law 84 (Chapter IV, Art. 23–26) and Resolution 8430 (1993, Title IV, Art. 83–93) that regulates animal research in Colombia.
Determination of parasite DNA concentration by qPCR
To estimate the DNA concentration in our samples, we used a reference sample with a previously determined parasite DNA concentration34. Based on this reference, we performed a relative quantification using TaqMan qPCR, following the standard protocol for this method. qPCR reactions were carried out in a QuantStudio 1 thermocycler, using a total reaction volume of 20 µL, containing 10 µL of Luna Universal Probe qPCR Master Mix (New England Biolabs, NEB), 0.8 µL of each primer (10 µM) and 0.4 µL of TaqMan probe (10 µM). Quantification was performed using the cycle threshold (Ct) method, interpolating the values against a standard curve generated from serial dilutions of DNA with a known concentration (Annex Fig. 2).

TropD-Detector device. A The fluorescent emission prototype utilizes blue light at 480 nm to excite the sample containing the CRISPR/LbCas12a system and the ssDNA fluorescence reporter with RPA. The emitted fluorescence passes through a light filter, enabling visual detection using a smartphone. Positive samples display a bright green color, while the negative control exhibits a green color with a transparent background. B Fluorescence emission from positive samples of T. cruzi extracted from culture, D. marsupialis and the intestine of R. pallescens. C Visual cleavage detection of diluted DNA amplified from T. cruzi culture. D Prototype of TropDetector. The device is powered by a 9-volt battery, which supplies energy to a blue LED positioned on the right side. E Visual observation of a positive sample using a conventional mobile phone. Figure created with BioRender.com
PCR and RPA amplification
Designed primers were validated through PCR using DNA isolated from the intestine of R. pallescens infected with T. cruzi. We employed the Q5® Hot Start High-Fidelity 2× Master Mix (New England Biolabs, American), in a T100 thermocycler (Bio-RAD Inc). After validation, we amplified DNA samples of T. cruzi from the Silvio X10 strain culture and blood samples of D. marsupialis naturally infected with the parasite (Fig. 1B). The amplified products were visualized on a 2% agarose gel stained with SYBR® Safe DNA Gel Stain (Invitrogen, #S33102). Designed primers were also tested against various DNA samples used as negative controls, including Rhodnius prolixus, Leishmania infantum, Trypanosoma theileri, and Lutzomyia sp (Annex Fig. 1). RPA amplification, we employed the RPA TwistAmpbasic KIT from TwistDX Co. (Cambridge, UK). For RPA amplification, the reaction was incubated at 37 °C for 20 min with the addition of 2.5 µL of 280 mM Magnesium Acetate (MgOAc) and 2.4 µL of each primer at a concentration of 10 µM (Fig. 1C). During the incubation, the reaction was briefly agitated at the 4-minute mark. The same concentrations recommended by the supplier were used. The primers used for PCR amplification of Cytb gene were also utilized for RPA amplification in samples obtained from culture, vector, and reservoir of T. cruzi.
gRNAs validation
Designed gRNAs were synthesized commercially by IDT (Integrated DNA Technologies, Inc.) and validated through in vitro cleavage of PCR products, which were observed on agarose gel using amplified T. cruzi DNA extracted from R. pallescens. We adapted the methodology from the in vitro digestion protocol by New England Biolabs (M0653), with concentrations modified during the standardization. CRISPR reactions were performed in a 30 µL reaction mixture consisting of 6 µL of 300 nM LbCAS12a protein, 6 µL of 300 nM gRNAs, and 6 µL of NEB reaction buffer, incubated at 37° C for ten minutes. To visualize the DNA cleavage, 15 µL of the reaction mix was run on a 2% agarose gel stained with SYBR® Safe DNA Gel Stain (Invitrogen, #S33102). Full-length agarose gel images, including additional experiments that were not relevant to the main study, are provided in the Supplementary Information (Annex Fig. 3). Lanes irrelevant to the study (e.g., genomic DNA tests) are not discussed in the main text.

Dilutions observed by spectrophotometry and UV transillumination. A Raw fluorescence values from diluted T. cruzi DNA amplified Cytb gene from culture, cleaved by the CRISPR/LbCas12a system. B Normalized fluorescence data emitted by the cleavage of diluted amplified DNA from culture. C Raw fluorescence data from CRISPR/LbCas12a cleaving in the Cytb gene of T. cruzi amplified DNA diluted from the intestine of R. pallescens. D Normalized fluorescence data emitted by the cleavage of diluted amplified DNA extracted from the intestine of R. pallescens E, F. Visual cleavage detection of T. cruzi from culture and extracted from the intestine of R. pallescens under UV light using a transilluminator
Target gene sequencing
After gRNA validation, the DNA amplified was sequenced from T. cruzi genes SR18 s, H2 A, and Cytb extracted from R. pallescens intestine. Bidirectional sequencing was conducted through Macrogen’s Sanger sequencing service. The obtained sequences were aligned and compared with reference sequences obtained from the NCBI database. The results were analyzed using the SnapGene software.
Visualization of fluorescent reporter cleavage
Once it is verified that LbCas12a protein produces the cleavage in the amplified product, the ssDNA reporter is added (Fig. 1D). The fluorescent system methodology was based on Chen et al. 2018. We selected a single-stranded DNA (ssDNA) reporter probe labeled with a fluorophore and quencher (FQ) (56-FAM/TTATT/3IABkFQ), which was synthesized commercially by IDT32. It is excited about a wavelength of 480 nm and emits detectable fluorescence at 530 nm. The ssDNA reporter, gRNA, and LbCas12a protein concentrations were adapted as previously described35. The fluorescence reaction mixture was adjusted to 100 nm of ssDNA reporter, while the final concentration of gRNA and LbCas12 was set at 60 nm. An equivalent volume of protein was used in the NEB buffer; molecular water was added to achieve a final volume of 50 µL.
We quantified the emitted fluorescence using dark plates in a SYNERGY H1 spectrophotometer, taking measurements every three minutes at 37° C for 2 h, with 40-s shaking intervals between readings (Fig. 1E). The samples were subsequently visualized on a UV transilluminator, allowing visual identification of positive samples (Fig. 1F). To evaluate the sensitivity of the CRISPR/LbCas12a system, we diluted the amplified DNA obtained from PCR to final concentrations of 40 ng/uL, 20 ng/uL, 10 ng/uL, 5 ng/uL, 1 ng/uL. Those dilutions were tested through RPA amplification in the designed device. The same approach was applied to assess the sensitivity of PCR combined with CRISPR/LbCas12 system by performing serial dilutions from 1:2 to 1: 32 using T. cruzi DNA extracted from cultures of the parasite with an initial concentration of 172.8 parasite equivalents/mL. The DNA diluted was amplified through PCR using the Cytb gene and observed on a 2% agarose gel. RPA amplification was conducted in the dilution 1:32 and compared with the results obtained through PCR. Cleavage by the CRISPR system was then evaluated using the amplified DNA from these dilutions with the designed guide. Each assay was performed in triplicate.
Design an LED reading device
We designed a portable device using affordable materials (Fig. 1G). It features a darkened cavity to prevent external light. At one end, a blue LED emits light in the range of 480–485 nm is powered by a conventional 9-volt battery. A lens with a focal length of f = 5 cm focused the light onto a PCR tube sample. The emitted fluorescence from the sample is observed at a 90˚ angle relative to the incident light. A high-pass filter was used with a cutoff wavelength of 500 nm. A camera was positioned behind the filter to capture the image of the excited sample. A light trap was positioned adjacent to the sample to reduce the intensity of reflected light. Fluorescence visualization of the samples was achieved using a standard mobile phone. We validated the samples obtained from the culture, insects, and reservoir. Subsequently, we confirmed the system’s sensitivity using samples derived from the dilution of the amplified product and the dilution of the DNA provided to the PCR. All the assays were compared with the results obtained under UV light visualization, and in the spectrophotometric quantification.
Statistical analysis
Fluorescence readings from the spectrophotometer were normalized as reported36. We first employed a Kolmorgorov-Smirnov test to evaluate the normality of the replicates. Kruskal-Wallis test was used to evaluate differences in non-normal data, followed by a post hoc Dunn’s test, while a one-way ANOVA was conducted in normal data, followed by a Tukey post hoc test. The homogeneity of the replicas was applied to the normalized data in the sensitivity tests (Annex Table 2). All analyses were executed using Statistica Version 10.
Results
PCR and RPA amplification
We amplified the Cytb, SR18 s, and H2 A genes in different samples by PCR, including non-target controls (Annex Fig. 1). The PCR product was positive in the three T. cruzi samples. Selected primers from bioinformatic analysis produced a PCR product length between 600 pb and 900 pb as shown in Fig. 5A-C. In T. cruzi DNA extracted from the intestines of R. pallescens, a faint band was observed at a concentration of 18 ng/µL, whereas a strong band was observed in T. cruzi from culture at 9.5 ng/uL. Unexpectedly, bands were also observed in the other species used here, indicating non-target amplification in the PCR technique. For example, a weak band was detected in T. theileri at 203 ng/uL for both the SR18 s and H2 A genes. Similarly, in L. infantum at 9.9 ng/uL, positive amplification was observed for the SR18 s gene, while ambiguous amplification occurred for the Cytb gene. In Lutzomya sp, at 3.9 ng/uL, a weak band was detected for the SR18 s gene and ambiguous amplification for the H2 A gene. No amplification was observed in the DNA from R. pallescens at 23.9 ng/µL (Annex Fig. 1). RPA amplification. The resulting band did not display the same intensity compared with PCR and showed smearing, with multiple faint bands. This occurred because the amplified product was not subsequently purified.
gRNAs validation
The designed primers successfully amplified the target site for cleavage by CRISPR/LbCas12a system in the three target genes. Using the Cytb gRNA, two distinct bands were observed on an agarose gel, corresponding to the fragments generated at the cleavage site of the guide-protein-DNA complex. However, no cleavage was detected for the SR18S and H2 A gRNAs, as assessed by agarose gel visualization and fluorescent emission (Annex Fig. 5). In contrast, the gRNA yielded fluorescence levels exceeding 20,000 relative fluorescence units (RFU), as shown in Fig. 5D. The gRNA was validated using T. cruzi DNA extracted cultures, as well as samples from the intestine of R. pallescens, and blood samples of the opossum D. marsupailis (Fig. 5E).

Sequencing of the target site from amplified T. cruzi DNA extracted from the intestine of Rhodnius pallescens in Bucaramanga, Colombia. The green bases indicate the gRNA binding site for each gRNA target gene. The PAM site consists of TTTV nucleotides. All results can be observed in Annex Fig. 6
The amplified Cytb gene product was diluted to concentrations of 40 ng/uL, 20 ng/uL, 10 ng/uL, 5 ng/uL, and 1 ng/uL, and subsequently cleaved using the CRISPR/LbCas12a system with the corresponding gRNA. Cleavage was observed at all dilutions, as evidenced by agarose gel visualization, the larger fragment corresponded to a 481 pb band, while the shorter fragment measured 241 pb. (Fig. 5F). At the 1 ng/uL concentration, a weak band corresponds to the larger fragment of 481 pb length.

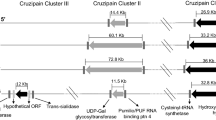
A–C Representation of the Cytb, SR18S, and H2 A genes selected for the identification of T. cruzi, along with specific primers and gRNAs design. D Fluorescence intensity graph displaying raw data for each designed gRNA. No DNA was added to the Control. The Cytb gRNA showed significant fluorescence with p < 0.05 E Comparative graph of normalized fluorescence (Raw data/Control) corresponding to three T. cruzi samples. The culture sample demonstrated a significant difference from T. cruzi extracted from the vector’s intestine (p = 0,000189) and the reservoir (p = 0,024). F Cleavage evaluation of diluted amplified products, two bands were observed with 481 pb and 241 pb. Full gel image can be observed in the Annex Fig. 5A
DNA sequencing
Sequencing of T. cruzi samples extracted from the vector’s intestine revealed point mutations in the gRNA binding region of the SR18S and H2 A genes. In the SR18S gRNA binding site, a double insertion was detected near the PAM site. Similarly, in the H2 A gRNA binding region, two base mutations and one indel were detected. In contrast, no variations were found in the Cytb gRNA binding site (Fig. 5).
Cleavage efficiency assessment
The CRISPR/LbCas12a system’s cleavage efficiency was assessed using diluted T. cruzi amplified DNA from culture by measuring fluorescence emission in a spectrophotometer. We observed fluorescence emission at all dilution levels for the cultured DNA and extracted from the vector’s intestine. The raw fluorescence data for cultured DNA (Fig. 3A) were normalized by dividing the fluorescence values for each measurement by the corresponding control without DNA (Fig. 3B). The same approach was applied to the vector’s intestine samples, as shown in Fig. 3C-D. The homogeneity of replicates for each dilution was confirmed with a Kruskal-Wallis test (p > 0.05). A transilluminator evaluated the fluorescence mix in PCR tubes under UV light. We observed fluorescence in all dilutions with a green color, clearly distinguishable from the control without DNA (Fig. 3E, F)
Fluorescence emission analysis using the “TropD-Detector” device
This simple and practical device facilitated qualitative evaluation of diluted samples, avoiding carcinogenic UV radiation, unlike the transilluminator (Fig. 2A). The emission was filtered correctly, producing a green color in the PCR tubes containing positive samples. T. cruzi samples derived from the vector’s intestine and reservoirs exhibited fluorescence at 40 ng/uL, clearly differentiable from the control (Fig. 2B). In the dilutions of amplified DNA, visible fluorescence was observed at all evaluated dilutions, except for the control tube without DNA (Fig. 2C). The designed device enabled the fluorescence excitation of the ssDNA reporter using LED light powered by a 9-volt battery (Fig. 2D). Fluorescence was detected using a conventional smartphone in a darkened room (Fig. 2E).
Analytical sensitivity testing with pre-amplified DNA Dilution
The analytical sensitivity of the CRISPR/LbCas12a system was evaluated by diluting the DNA sample prior to PCR. We realized serial dilutions ranging from 1:2 to 1:32 using T. cruzi DNA derived from culture, starting with an initial concentration of 172.8 parasite equivalents/mL. Positive bands were observed down to a concentration of 118 parasite equivalents/mL (1:16 dilution), with band intensity decreasing proportionally to the DNA concentration (Fig. 6A). Cleavage results were consistent across all visualization systems with the PCR amplifications. No amplification was detected at the 1:32 dilution, which aligned with the absence of fluorescence in both the spectrophotometer and the UV transilluminator in PCR amplifications (Fig. 6B–D), as well as with the results from the TropD-Detector device (Fig. 6E). The fluorescence data emitted by the dilution were similar to those of the control without DNA; however, significant differences were observed (Kruskal-Wallis test p < 0.05). RPA amplifications were directly confirmed in diluted DNA samples by observing fluorescence at a 1:32 dilution, which corresponded to a DNA concentration of 116 parasite equivalents/mL (Fig. 6F). These results were verified using both the designed device and a UV spectrophotometer (Figure G-H).

Detection of diluted T. cruzi DNA observed in the device, spectrophotometer, and transilluminator. A Diluted T. cruzi DNA from the culture before PCR, starting at 172.8 parasite equivalents/mL, with no band detected at the 1:32 dilution. Full gel image is shown in the Annex Fig. 5B. B Raw fluorescence data from CRISPR/LbCas12a cleavage in the Cytb gene of amplified products of the pre-PCR dilutions obtained from culture. C Normalized fluorescence data emitted by the cleavage of amplified products of the pre-PCR dilutions obtained from culture. D Visual cleavage detection of diluted DNA under UV light using a transilluminator. E Visual cleavage detection of diluted DNA prior to PCR from T. cruzi culture in the TropD-Detector device F RPA amplification in the 1:32 DNA dilution. A full gel image is shown in Fig. 5A. G Visual detection in the TropD-Detector device was obtained from culture DNA compared with diluted DNA until 116 parasite equivalents/mL were amplified with RPA. H Visual observation under UV light in a transilluminator, samples amplified with RPA
Discussion
The CRISPR/LbCas12a system, employed in this study in conjunction with the TropD-Detector device, enabled the detection of T. cruzi in samples obtained from vectors and reservoirs. Its versatility allowed the development of a portable device capable of identifying positive samples using a smartphone or other image-capturing devices. As mentioned before, the unexpected PCR amplification observed in non-target controls suggests potential cross-reactivity of the primers with non-target species rather than contamination or handling errors, highlighting the challenges of achieving absolute specificity in molecular diagnostics15,18. The methods used in this study validate the CRISPR technology as a viable alternative for parasite detection.
PCR amplification was used as a validation system for the gRNAs and for standardizing the method. However, due to differences in processing time, RPA amplification is more advantageous for the practical application of the designed device in real-world settings. RPA achieves comparable results in 20 min, versus the 90 min required for PCR (Fig. 7). Furthermore, RPA operates at 37 °C, the same incubation temperature as the Cas protein, enabling use within the same tube, as demonstrated for T. gondii37. Additionally, the RPA-CRISPR system could enhance Point-of-Care Testing (POCT) by delivering rapid results from patient samples38, offering a more practical solution that requires minimal training, less-skilled personnel, and limited infrastructure.

Workflow diagram for detection. The process begins with DNA extraction using the QIAGEN extraction kit, which requires 20 min. Next, DNA amplification is performed, either through RPA (20 min) or PCR (90 min). Traditional PCR requires gel preparation and, consequently, the samples must undergo electrophoresis. Additionally, a visualization system is necessary to observe the DNA within the gel. In some cases, sequencing may be required for further verification. Otherwise, RPA amplification proteins and Cas protein incubation occur at 37 °C, eliminating the need for temperature variation. Following amplification, a CRISPR/LbCas12a cleavage step (10 min) is performed, in which the guide RNA and Cas protein identify and cleave the target region (cis cleavage). Finally, a fluorescence signal is generated during trans cleavage by the Cas protein, taking 20 min to produce visible fluorescence on the TropD-Detector. Figure created BioRender.com
Although the gRNAs were designed through bioinformatic analysis of various available sequences in the SR18 s and H2 A genes, we observed indels and point mutations in the target sites that impeded efficient binding. These differences were located within 5 to 10 bases of the PAM site, a critical area known as the seed region in Cas proteins39. Previous studies have shown that mismatches in this sensitive region reduce the gRNA’s recognition capacity, as reported for Cas1240. In contrast, no mutations were found between the designed gRNA and the target site in the Cytb gene (Fig. 5), as confirmed by the fluorescence emitted using the Cytb gRNA (Fig. 5D), demonstrating efficient binding.
We reduced the ssDNA reporter to a final concentration of 100 nM, optimizing the number of reactions while lowering the cost per sample. This concentration aligns with the findings of Lei (2022) in detecting Toxoplasma gondii. It is half of the concentration reported by Dueñas (2022) for Leishmania sp., and is ten times lower than that mentioned by Lee 2020 in Plasmodium sp. 35,36,37.
Sensitivity testing with pre-amplified dilution with PCR enabled detection up to 118 parasite equivalents/mL. PCR amplification at 116 parasite equivalents/mL did not yield any detectable product. However, RPA amplification at the same concentration resulted in a positive signal, subsequently confirmed through the CRISPR/LbCas12a system. These results demonstrate that RPA offers a higher sensitivity compared to PCR. Various factors, such as the amplification technique, sample type, or targeted gene, can influence sensitivity. For instance, the CRISPR/Cas detection system combined with RPA amplification has been applied in zoonotic diseases caused by parasites such as human African trypanosomiasis, toxoplasmosis, or malaria, achieving a detection sensitivity as low as 0.03 parasites/µL for the SR18 s gene, as reported in two species of Plasmodium sp35,37,41. Similarly, a detection limit of 3.1 parasites/µL was achieved for Leishmania spp. using a comparable CRISPR-based approach42. In this context, the TropD-Detector device, with an estimated sensitivity of 116 parasite equivalents/mL, shows a comparable detection capacity, demonstrating that this system performs within the sensitivity range of other CRISPR-based diagnostics for protozoan parasites.
The results obtained using the TropD-Detector device were consistent with those from UV transilluminator and spectrophotometer quantification, showing a decreased fluorescence intensity with DNA decreasing concentration. The device demonstrated greater sensitivity in differentiating the 1 ng/uL concentration from the control without DNA compared to transilluminator images. This improved sensitivity is attributed to the high-pass light filter, which effectively prevents interference from the blue LED light with the fluorescence intensity emitted by the positive samples. The CRISPR-Cas system for detection using blue light for reporter excitation has been applied in previous studies. For instance, Yu et al. (2021) utilized this system to detect Cryptosporidium parvum, the parasitic agent of a disease known as cryptosporidiosis, primarily affecting the digestive system in humans. However, this method typically requires specialized equipment for sample visualization, such as the ‘Tanon 5200 Fluorescence Imager” or a portable gel imagining system43. Other researchers have employed this methodology by developing specially designed cells for sample positioning44, which is unnecessary for the TropD-Detector device, as the samples can be placed in standard PCR tubes.
A limitation of the current detection system that reduces its applicability in field settings is the absence of an integrated DNA extraction step. However, this methodology could be complemented with alternative field-adapted extraction methods. Recent research has employed DNA extraction techniques that can be effectively performed under field conditions, using simplified protocols that require only a heat source and minimal handling. For instance, the EZ-Fast method has been successfully implemented for crude DNA extraction in approximately 20 min, without requiring advanced laboratory equipment or commercial kits, making it highly suitable for low-resource environments45.
The selection of molecular targets without thorough bioinformatics validation and prior sequencing can lead to false-negative results, as observed with the SR18S and H2 A genes. This underscores the importance of verifying sequence conservation across different geographical strains through a combination of bioinformatics analysis and prior sequencing to improve assay reliability. These intra-species variations must be considered before field implementation. They can be addressed through prior characterization in the study area or by designing multiple guides targeting different sequences within the selected region to ensure reliable pathogen detection. We present a device suitable for low-infrastructure settings, with the potential to be scalable for use with human patients suspected of having CD.
Conclusion
The TropD-Detector device successfully detected T. cruzi in samples from vectors, reservoirs, and cultures of the parasite, demonstrating the adaptability and scalability of the CRISPR/Cas system for portable, precise, practical, and cost-effective methodologies. These features are critical for molecular diagnostic tests, particularly in facilitating patient screening for tropical diseases.
Limitations of the study
The device presented in this study enables the visual detection of DNA samples that have been previously extracted. Future efforts should combine this device with portable extraction methods for a fully portable technique.
Data availability
The sequence data obtained from T. cruzi extracted from Rhodnius pallescens has been submitted to the NCBI database with the following accession number: Cytb gene PV344711, H2 A gene PV344712, SR18 s gene PV124446. Alternatively, the sequences can be requested by email at jonedulu@uis.edu.co. Additionally, all raw data generated from the fluorescence spectrophotometer experiments, including the data used to produce the graphs presented in this manuscript, are provided in the supplementary materials.
References
Duque, J. et al. Vectores De Chagas Y Su Control. Hemiptera: Reduviidae Triatominae (Ediciones UIS, 2024).
Irish, A., Whitman, J. D., Clark, E. H., Marcus, R. & Bern, C. Updated estimates and mapping for prevalence of Chagas disease among adults, United States. Emerg. Infect. Dis. 28, 1313–1320 (2022).
Requena-Méndez, A. et al. Prevalence of Chagas disease in Latin-American migrants living in Europe: a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 9, e0003540 (2015).
Echeverría, L. E. et al. WHF IASC roadmap on Chagas disease. Glob Heart. 15, 26 (2020).
Monteiro, F. A. et al. Systematics, and biogeography of the triatominae, vectors of Chagas disease. Adv. Parasitol. 99, 265–344 (2018).
Briceño-León, R. La Enfermedad de Chagas En Las Américas: Una perspectiva de Ecosalud. Cad Saúde Públ.. 25, S71–S82 (2009).
Shikanai-Yasuda, M. A. & Carvalho, N. B. Oral transmission of Chagas disease. Clin. Infect. Dis. 54, 845–852 (2012).
López-García, A. & Gilabert, J. A. Oral transmission of Chagas disease from a one health approach: a systematic review. Trop. Med. Int. Health. 28, 689–698 (2023).
Ríos, J. F., Arboleda, M., Montoya, A. N., Alarcón, E. P. & Parra-Henao, G. J. Probable brote de transmisión oral de Enfermedad de Chagas En Turbo. Antioq. Biomed. 31, 185 (2011).
Olivera, M. J., Rincón Acevedo, C. Y., Olivera, A. J., Mendez-Cardona, S., & Vera Soto, M. J. Addressing Chagas disease from a one health perspective: risk factors, lessons learned and prevention of oral transmission outbreaks in Colombia. Sci. One Health. 3, 100066 (2024).
Velásquez-Ortiz, N. et al. Trypanosoma cruzi parasite burdens of several Triatomine species in Colombia. Trop. Med. Infect. Dis. 7, 445 (2022).
Cantillo-Barraza, O. et al. Eco-epidemiological study of an endemic Chagas disease region in Northern Colombia reveals the importance of Triatoma maculata (Hemiptera: Reduviidae), dogs and Didelphis marsupialis in Trypanosoma cruzi maintenance. Parasit. Vectors. 8, 482 (2015).
Abras, A. et al. Serological diagnosis of chronic Chagas disease: is it time for a change?? J. Clin. Microbiol. 54, 1566–1572 (2016).
Gomes, Y. M., Lorena, V. M. B. & Luquetti, A. O. Diagnosis of Chagas disease: what has been achieved? What remains to be done with regard to diagnosis and follow up studies? Mem. Inst. Oswaldo Cruz. 104 (Suppl 1), 115–121 (2009).
Stadhouders, R. et al. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5’ nuclease assay. J. Mol. Diagn. 12, 109–117 (2010).
Viljoen, G. J., Nel, L. H. & Crowther, J. R. Molecular Diagnostic PCR Handbook (Springer, Berlin, 2005).
Hirschhorn, J. W., Avery, A. & Schandl, C. A. Managing a PCR contamination event in a molecular pathology laboratory. In Clinical Applications of Nucleic Acid Amplification (eds Myers, M. B. & Schandl, C. A.) 15–26 (Springer US, 2023).
Seiringer, P. et al. Comparison of four PCR methods for efficient detection of trypanosoma Cruzi in routine diagnostics. Diagn. Microbiol. Infect. Dis. 88, 225–232 (2017).
Ramírez, J. D. et al. Evaluation of the analytical and diagnostic performance of a digital droplet polymerase chain reaction (ddPCR) assay to detect trypanosoma Cruzi DNA in blood samples. PLoS Negl. Trop. Dis. 12, e0007063 (2018).
de Alarcón, B. & Jackson, Y. Chagas Disease Epidemiology: From Latin America to the World. In Chagas Disease: A Neglected Tropical Disease (eds. Pinazo Delgado, M.-J. & Gascón, J.) 27–36 (Springer International Publishing, Cham, 2020).
Chen, Y. et al. Genome editing by CRISPR/Cas12 recognizing AT-Rich PAMs in Shewanella oneidensis MR-1. ACS Synth. Biol. 11, 2947–2955 (2022).
Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F. & Collins, J. J. CRISPR-based diagnostics. Nat. Biomed. Eng. 5, 643–656 (2021).
Li, Y., Li, S., Wang, J. & Liu, G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 37, 730–743 (2019).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
Bai, L. et al. Rapid, visual, and sequence-specific detection of Salmonella in egg liquid with vis-NEAA, a CRISPR/Cas12 empowered new strategy. J. Agric. Food Chem. 70, 2401–2409 (2022).
Zhou, B. et al. CRISPR/Cas12a based fluorescence-enhanced lateral flow biosensor for detection of Staphylococcus aureus. Sens. Actuators B Chem. 351, 130906 (2022).
Qian, W., Huang, J., Wang, X., Wang, T. & Li, Y. CRISPR-Cas12a combined with reverse transcription recombinase polymerase amplification for sensitive and specific detection of human Norovirus genotype GII.4. Virology 564, 26–32 (2021).
Ding, X. et al. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11, 4711 (2020).
Brisse, S., Verhoef, J. & Tibayrenc, M. Characterisation of large and small subunit rRNA and mini-exon genes further supports the distinction of six trypanosoma Cruzi lineages. Int. J. Parasitol. 31, 1218–1226 (2001).
Pavia, P. X., Vallejo, G. A., Montilla, M., Nicholls, R. S. & Puerta, C. J. Detection of trypanosoma Cruzi and trypanosoma rangeli infection in triatomine vectors by amplification of the histone H2A/SIRE and the sno-RNA-C11 genes. Rev. Inst. Med. Trop. Sao Paulo. 49, 23–30 (2007).
Brisse, S. et al. Evidence for genetic exchange and hybridization in trypanosoma Cruzi based on nucleotide sequences and molecular karyotype. Infect. Genet. Evol. 2, 173–183 (2003).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Angulo, V. M. & Esteban, L. New trap for the capture of triatomines in wild and peridomestic habitats. Biomedica 31, 264–268 (2011).
Murcia-Cueto, I. S. et al. First report of trypanosoma Cruzi infection in urban Hedgehog (Atelerix albiventris) in Colombia. Vet. Parasitol. (Amst). 55, 101116 (2024).
Lee, R. A. et al. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc. Natl. Acad. Sci. U. S. A. 117, 25722–25731 (2020).
Dueñas, E. et al. Novel CRISPR-based detection of leishmania species. Front. Microbiol. 13, 958693 (2022).
Lei, R. et al. RPA/CRISPR/Cas12a-Based On-Site and rapid nucleic acid detection of Toxoplasma gondii in the environment. ACS Synth. Biol. 11, 1772–1781 (2022).
Zakiyyah, S. N. et al. Detection of tropical diseases caused by mosquitoes using CRISPR-based biosensors. Trop. Med. Infect. Dis. 7, 309 (2022).
Moon, J. & Liu, C. Asymmetric CRISPR enabling cascade signal amplification for nucleic acid detection by competitive CrRNA. Nat. Commun. 14, 7504 (2023).
Mann, J. G. & Pitts, R. J. PrimedSherlock: a tool for rapid design of highly specific CRISPR-Cas12 CrRNAs. BMC Bioinform. 23, 428 (2022).
Sima, N. et al. SHERLOCK4HAT: a CRISPR-based tool kit for diagnosis of human African trypanosomiasis. EBioMedicine 85, 104308 (2022).
DNAzyme-Substrated, C. R. I. S. P. R. /Cas12 Assay for Label-Free Detection of Single-Celled Parasitic Infection.
Yu, F. et al. CRISPR/Cas12a-based on-site diagnostics of Cryptosporidium parvum IId-subtype-family from human and cattle fecal samples. Parasit. Vect.. 14, 208 (2021).
Fozouni, P. et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333e9 (2021).
Yamazaki, Y., Thongchankaew-Seo, U., Nagao, K., Mekata, H. & Yamazaki, W. Development and evaluation of a point-of-care test with a combination of EZ-Fast DNA extraction and real-time PCR and LAMP detection: evaluation using blood samples containing the bovine leukaemia DNA. Lett. Appl. Microbiol. 71, 560–566 (2020).
Acknowledgements
To Gustavo Adolfo Rincón and Juliana Cuadros Martínez for their collaboration in the collection of biological material and identification of the triatomines.
Funding
The authors thank the ”Sistema general de regalias” which funded the project: “Producción de nucleótidos a partir de biomasa residual de la agroindustria para diagnóstico por biología molecular en el departamento de Santander” code BPIN 2021000100331. The authors also thank the “Universidad Industrial de Santander” which funded the project “Producción de machos estériles de Aedes aegypti mediante la Técnica del Insecto Estéril guiada con precisión - TIEgp y validación de su efecto supresor contra Aedes aegypti silvestre en condiciones de laboratorio”.
Author information
Authors and Affiliations
Contributions
L.A.O.R: Performed experimental assays, formal analysis, investigation, methodology, TropD-Detector device designing and writing original draft, Writing review & editing. R.C: TropD-Detector device designing, writing – review & editing. J.J.D: Supplying DNA samples from D. marsupialis, writing – review & editing. S.C.M.S: Project administration, resources, writing – review & editing. J.E.D: Project design, formal analysis, investigation, project administration, resources, supervision, writing – original draft, writing – review & editing. All authors contributed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ortiz-Rodríguez, L.A., Cabanzo, R., Jaimes-Dueñez, J. et al. TropD-detector a CRISPR/LbCas12a-based system for rapid screening of Trypanosoma cruzi in Chagas vectors and reservoirs. Sci Rep 15, 19107 (2025). https://doi.org/10.1038/s41598-025-04017-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-04017-0
