
Abstract
Tuberous Sclerosis Complex (TSC) is an autosomal dominant disorder characterized by widespread hamartomas and prominent neurological involvement. It results from pathogenic variants in the TSC1 or TSC2 genes, leading to hyperactivation of the mTOR pathway and consequent dysregulation of cell growth. These tumor suppressor genes encode hamartin and tuberin, proteins critical for regulating cell proliferation, neuronal excitability and synaptogenesis. In this retrospective study, we analyzed clinical, genetic and radiological features of 81 TSC patients from Sicily, focusing on genotype-phenotype correlations and intergroup comparisons. Pathogenic TSC2 variants were more common than pathogenic TSC1 variants (61.7% vs. 38.3%). Patients with pathogenic TSC2 variants tended to exhibit a higher frequency of weekly seizures, a higher prevalence of infantile spasms and hypsarrhythmia compared to those with pathogenic TSC1 variants, consistent with a more severe phenotype. Interestingly, TSC1 patients exhibited a higher incidence of radial bands, while TSC2 patients harbored a larger average size of tubers and subependymal nodules. Cognitive and behavioral disorders were similarly distributed, although TSC1 patients had higher rates of normal or borderline cognitive function, while TSC2 patients had more severe neuropsychiatric profiles compared to TSC1. To our knowledge, this is the first comprehensive TSC1 and TSC2 mutational analysis and genotype-phenotype correlation study carried out in a large cohort of Sicilian patients affected by TSC. Our findings contribute to regional and global data on TSC, emphasizing the utility of genotype-informed management strategies.
Similar content being viewed by others


Introduction
Tuberous Sclerosis Complex (TSC) (OMIM #191100; #613254) is a rare genetic disorder affecting approximately 1:6,000–1:10,000 live births1. First described by de Bourneville, TSC manifests with a wide clinical spectrum, including cutaneous, cardiac, renal, and neurological involvement, alongside neuropsychiatric manifestations known as TAND (Tuberous Sclerosis-Associated Neuropsychiatric Disorders)2,3. The disease is caused by pathogenic heterozygous variants in either the TSC1 gene (OMIM *605284) on chromosome 9 or the TSC2 gene (OMIM *191092) on chromosome 164,5. About two-thirds of cases result from de novo pathogenic variants, especially in TSC2 gene, while familial cases show a more balanced distribution of variants between the two genes. Notably, a subset of patients who meet clinical diagnostic criteria for TSC do not have identifiable pathogenic variants in TSC1 or TSC2 through conventional genetic testing, with somatic mosaicism and intronic variants implicated in these cases6,7,8.
The hamartin-tuberin complex, encoded by TSC1 and TSC2, acts as a negative regulator of the mechanistic target of rapamycin (mTOR) pathway. Dysfunction in this complex due to inactivating mutations leads to constitutive mTOR activation, resulting in cellular hyperproliferation and aberrant growth9. Clinical diagnostic criteria for TSC, first established in 2012 and revised in 2021, now encompass 11 major and seven minor features10. Histopathologically, TSC is marked by benign tumors (hamartomas) affecting multiple organs and cortical brain lesions termed “tubers”, which disrupt synaptic networks and contribute to its neurological manifestations. Despite nearly complete penetrance, the clinical presentation of TSC is highly variable. Neurological symptoms such as epilepsy, cognitive impairment, and autism spectrum disorder (ASD) are predominant and significantly impact patients’ quality of life11,12. Recent studies, including data from the TOSCA registry, consistently show that pathogenic TSC2 variants are associated with a more severe clinical phenotype than pathogenic TSC1 variants10,13.
However, it is now well recognized that much of the clinical heterogeneity in TSC is likely attributable to the stochastic nature of postzygotic “second-hit” mutations, which are required for the development of most—if not all—organ-specific manifestations. These somatic events vary widely in timing, location, and extent, contributing significantly to the phenotypic variability observed among individuals with the same germline mutation. As such, while several genotype–phenotype studies have attempted to link specific germline variants to clinical features, the strength and consistency of these associations remain limited across diverse cohorts6,7,8,13,14,15,16,17,18.
This study analyzes the genotype-phenotype relationship in a Sicilian cohort, and evaluates the alignment of these findings with global research, with particular regard to epilepsy and neurological comorbidities.
Patients and methods
Patients and clinical assessment
This study included 81 patients (mean age 26 years, range 2–71) from four Italian centers (Unit of Pediatric Clinic and Unit of Rare Diseases of the Nervous System in Pediatric Age – “Policlinico-San Marco” University Hospital, Catania; Clinical and Instrumental Neurology and Neurophysiopathology Unit – I.R.C.C.S. Oasi Troina; Regional Reference Center for Rare Genetic and Chromosomal Diseases – Maternal and Child Department, “Villa Sofia-Cervello” Hospital, Palermo; Child Neuropsychiatry Unit, Santa Marta and Santa Venera Hospital, Acireale). Patients were required to present with a definite clinical diagnosis of TSC and a confirmed genetic alteration in TSC1 or TSC2, including pathogenic and likely pathogenic exonic variants. Patients harboring intronic variants with unknown or unpredictable effects at the protein level were included only if they had a definitive diagnosis of TSC, established according to the revised 2021 diagnostic criteria9. Retrospective analysis used data from existing medical records, supplemented by additional studies and follow-up during the period of the study. Collected data covered clinical, genetic, and radiological information. Regarding the epilepsy phenotype, for each patient, data included age at onset (in months), seizure type, frequency (seizures per week, averaged over the most recent 12-month period), EEG findings, and treatment history. Seizure outcome was categorized based on response to treatment as follows: remission (> 75% reduction in seizure frequency), good control (50–75%), partial control (25–50%), and drug-resistant epilepsy (< 25%). Data were obtained through clinical records and follow-up evaluations. Classifications were made in accordance with standard clinical definitions. All patients underwent an EEG, a brain MRI and a neuropsychiatric evaluation in the last three years. Clinical investigation included a review of medical history and a careful physical examination by clinicians experienced in the manifestations of TSC. Brain MRI reports were collected and analyzed to identify specific patterns associated with cortical tubers, subependymal nodules (SENs), sub-ependymal giant cell astrocytoma (SEGA), and radial bands. Details on clinical signs and symptoms were obtained from the physicians who were sent a standardized clinical evaluation form. Family members were consulted when necessary. In addition, a range of standardized and validated psychological measures were used to assess intellectual level, autistic disorder, anxiety, depression, and behavioral disorders. All manifestations were graded according to a binary scale (present or absent), but several features were graded in severity using numbers and words (characteristics of seizure, drug resistance, severity of intellectual disability, for example). Although complete phenotype information was collected, neurological and neuropsychiatric characteristics were the primary focus of analysis (Supplementary Tables 1 and 2). All TSC patients included in the study provided informed consent and had molecular diagnostic analysis for variants in TSC1 or TSC2. The study protocol was approved by the Ethical Committee of the University of Catania, Italy. All methods were performed in accordance with the relevant guidelines and regulations.
Mutational analysis
Genetic analysis was conducted to identify pathogenic variants in TSC1 and TSC2 through combination of multiplex ligation-dependent probe amplification (MLPA), denaturing high-performance liquid chromatography (DHPLC), and next-generation sequencing (NGS).
MLPA (MRC-Holland, SALSA P124/P046) was used as the primary method to detect large deletions or duplications in TSC1 and TSC2, as NGS is not optimized for the reliable identification of copy number variants. DHPLC was initially employed as a screening method to identify potential sequence variants, which were subsequently confirmed by Sanger sequencing (list of primers Supplementary Table 3).
Next-generation sequencing (NGS) was the preferred approach for patients born after 2016, enabling comprehensive mutation screening. Targeted NGS was carried out using a custom gene panel that included the entire coding regions and exon-intron boundaries of TSC1 (9q34.13) and TSC2 (16p13.3). The target regions encompassed all exons and at least 20 base pairs of flanking intronic sequences to facilitate the detection of splicing-affecting variants. Sequencing was performed on the Illumina MiSeq or NextSeq platform, achieving a mean coverage of > 200× and ensuring a minimum of 30× for reliable variant calling.
Raw sequence reads were processed using BWA-MEM for alignment to the hg19 or hg38 reference genome, and variant calling was performed using the GATK HaplotypeCaller. Identified variants were annotated using ANNOVAR, with pathogenicity assessed based on data from ClinVar, gnomAD, and the ACMG guidelines. When indicated, MLPA was also used to complement NGS in detecting copy number changes not adequately captured by sequencing data.
Patients in whom no pathogenic variants were identified in TSC1 or TSC2, and individuals who did not meet the revised 2021 clinical diagnostic criteria, were excluded from the study.
Neuropsychological evaluation
Neuropsychiatric characteristics and cognitive impairment were evaluated using standardized neuropsychological tests administered by trained clinicians. Cognitive functioning was assessed using the Wechsler Preschool and Primary Scale of Intelligence (WPPSI-III or WPPSI-IV) for children aged 2 years to 7 years, and the Wechsler Intelligence Scale for Children (WISC-IV) or the Wechsler Adult Intelligence Scale (WAIS-IV), depending on patient age. Full-scale IQ (FSIQ) was classified according to standard cut-off values: Normal cognitive function: FSIQ ≥ 85; Borderline cognitive function: FSIQ 70–84; Mild intellectual disability (ID): FSIQ 55–69; Moderate ID: FSIQ 40–54; Severe ID: FSIQ < 40. For neuropsychiatric comorbidities, the presence of autism spectrum disorder (ASD) was determined using the Autism Diagnostic Observation Schedule-2 (ADOS-2) and the Autism Diagnostic Interview-Revised (ADI-R), with classification based on DSM-5 criteria. Attention-deficit/hyperactivity disorder (ADHD) was diagnosed using the Conners Rating Scale and DSM-5 clinical criteria. Behavioral disorders, including oppositional defiant disorder (ODD) and anxiety disorders, were assessed using the Child Behavior Checklist (CBCL) and clinical interviews. For sleep disorders, a structured sleep history was taken, and the Pittsburgh Sleep Quality Index (PSQI) and the Sleep Disturbance Scale for Children (SDSC) were used to classify sleep disturbances.
Specific Learning Difficulties (SLD), including dyslexia, dyscalculia, and dysgraphia, were assessed using standardized academic achievement tests appropriate for age and national guidelines. The following assessments were employed: Reading and dyslexia: Batteria per la Dislessia e Disortografia Evolutiva (DDE-2) for Italian-speaking children. Mathematics and dyscalculia: Test AC-MT (Test di Abilità di Calcolo e Matematica) for evaluating mathematical abilities. Writing and dysgraphia: BVSCO-2 (Batteria per la Valutazione della Scrittura e della Competenza Ortografica) for assessing handwriting and spelling difficulties.
All assessments were conducted at specialized centers with expertise in TSC, ensuring standardized evaluation across the cohort.
Statistical analysis
Quantitative data were analyzed using the Student’s t-test, while categorical variables were assessed using the Chi-square test. Statistical significance was determined for variables including age, age at epilepsy onset, and frequencies of neurological manifestations. Percentages and proportions were calculated for categorical data, with significance assessed through a 2 × 2 contingency table analysis.
Results
TSC1 patients
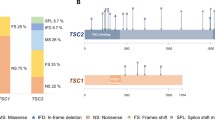
The TSC1 group included 31 patients (15 males, 16 females), with a mean age of 25.1 years (range: 2–60 years). Of these, 45.1% had inherited variants, while 54.9% were sporadic or de novo. Pathogenic TSC1 variants were widely distributed throughout the gene, involving in particular exons 4, 5, 6, 10, 15, 17, 18, 21 and 22. One patient carried an intronic variant (intron 1). Variants were intragenic deletions (48.4%), nonsense (38.7%), missense (9.6%), and in one patient a duplication (3.2%) (Supplementary Table 1). Among these patients, six came from the same family and were affected by the already reported c.1498 C > T, p.(Arg500Ter) pathogenic variant (Fig. 1a).

Genealogic tree of the three large families reported in the present study. (A) A family affected by mutation c.1498T > C of TSC1; (B) A family showing c.1096 G > T mutation of TSC2; (C) A large family affected by mutation c.3693_3696delGTCT of TSC2. Patients affected by TSC are marked in black.
TSC1 patients were affected by different types of seizures, differing in EEG findings, clinical course, age of onset and type of seizures. Epileptic seizures were present in 64.5% of patients, with a mean onset at 35.4 months. Seizure types were both generalized and focal seizures and some patients experienced multiple seizure types throughout life. More in detail, two patients (6.4%) presented infantile spasms, 12 (38.7%) had focal seizures (three with impaired awareness), six (19.3%) patients experienced generalized tonic clonic seizures, four (12.9%) suffered absence, one (3.2%) presented gelastic seizures, one (3.2%) reported seizures with fever, three (9.6%) suffered hypertonic seizures and one (3.2%) experienced atonic seizures. Their frequency ranged from 1 per lifetime to 28 per week (mean: 10.4 weeks, SD: 9.2). Severity and duration of seizures varied among patients and in the same patient. EEG abnormalities included spikes, waves, and hypsarrhythmia. Particularly, 11 patients (35.5%) had normal EEG, one (3.2%) presented hypsarrhythmia, 13 (41.9%) were reported to have spikes and waves (unilateral or bilateral), 11 (35.5%) had synchronous or asynchronous focal spikes, one (3.2%) had “arceau-like” pattern, four (12.9%) presented a theta wave pattern. Epilepsy course varied from spontaneous remission to drug resistance, with valproic acid and carbamazepine being the most common treatments: ten out of 20 patients (50%) achieved remission (complete response), while one (5%) had a good response, two (10%) a partial response, and the other and seven other (35%) showed a drug-resistant epilepsy.
Cognitive impairment levels of TSC1 patients ranged from mild to severe: 51.6% had normal cognitive functions, 9.7% had cognitive function near lower limit (borderline), 19.3% presented mild intellectual disability, 3.2% suffered from moderate intellectual disability and 16.1% had severe cognitive deficit. Autism spectrum disorder (ASD) was observed in four patients (12.9%), with only one patient showing Level 2 autism. Behavioral disorders (e.g., ADHD, anxious-depressive syndrome, borderline personality disorder) affected eight patients (25.8%), while 12 (38.7%) experienced learning disabilities. Sleep disorders were reported in only three patients (9.7%). Brain abnormalities of TSC1 group included cortical tubers (67.8% with > 5 tubers, mean size 8 mm) and subependymal nodules (51.6% with > 5 SENs, variable in size). Subependymal giant cell astrocytomas (SEGAs) were rare, found in only two patients (6.5%). White matter abnormalities and cerebral cysts were uncommon in our group of TSC1 patients, only one patient (3.2%) carrying the nonsense mutation c.2293 C > T, had many cerebral cysts.
Some pathogenic variants were associated with specific clinical patterns. The mutation c.1888_1891delAAAG, identified in two patients, was associated in both patients with numerous bilateral tubers and SENs, while epilepsy and neuropsychiatric disorders were heterogeneous, suggesting a potential role of other modifying factors. The pathogenic variants c.1004del and c.2111_2112del were linked to minimal neurological symptoms and a good quality of life, indicating a milder impact. The c.1498T mutation (associated to a common specific EEG pattern and many brain hamartomas) was found in 6 patients of the same family (Fig. 1a) and five of them showed the same EEG pattern (sharp theta waves and spikes in the left temporal lobe or bilateral). Notably, in this family, not all the patient showed epilepsy, and some of those affected were partial responders to drugs, while other drug-resistant. Cognitive functions and behavioral disorders were variable, with different grade of intellectual disability (absent to moderate/severe).
TSC2 patients
This study analyzed 50 patients with pathogenic TSC2 variants, comprising 21 males (42%) and 29 females (58%), with a mean age of 27.1 years (SD = 18.6, range 2–71 years). Variants were classified as familial (23 cases, 46%) or sporadic or de novo (27 cases, 54%) and showed extensive distribution across exons and included deletion (60%), nonsense (26%), missense (10%), and splicing mutations (6%). Furthermore, four intragenic deletions were detected in this group (one spanning from intron 1 to exon 8), as well as somatic mosaicism for the TSC2 c.3598 C > T (p.Arg1200Trp) variant, identified with a variant allele fraction of 11.4%. Among these patients, two large families were included: one (five members) harbored the c.1096G > T; (p.Glu366*) mutation (Fig. 1b). The other one comprised 16 members (among the 50 known members of this family), and presented the already reported c.3693_3696del (p.Ser1232Thrfs*92) mutation (Fig. 1c).
Of the 50 patients, 36 (72%) experienced seizures, with a mean onset age of 29.45 months (SD = 66.3, range one month to 23 years). Seizure types varied, with 32% experiencing infantile spasms, 30% focal seizures (16% with impaired awareness) and 22% generalized tonic-clonic seizures. The frequency and severity of seizures varied widely, ranging from two lifetime episodes to as many as 42 episodes per week (mean: 18.3; SD: 24.5). EEG patterns were varied, including theta waves, spikes, hypsarrhythmia, and focal spikes. Eighteen patients (36%) had normal EEG results, while focal and generalized spikes were more frequently observed. EEG patterns fluctuated over time within individual patients. The clinical course of epilepsy in TSC2 group of patients was variable, going from remission to multidrug resistance. Different antiseizures medications were used, including valproic acid, vigabatrin, carbamazepine, ACTH, topiramate, phenobarbital, levetiracetam, oxcarbazepine, lamotrigine, everolimus. Among them the most used were valproic acid and carbamazepine. TSC2 patients had been divided according to drug response into complete responder, (13 out of 36 patients, 36.1%), good responders (four patients, 11%), partial responders (six patients, 16.6%) and non-responders (nine patients, 25%).
Cognitive impairment was prevalent among TSC2 patients, with 24%, 10%, and 24% exhibiting mild, moderate, and severe intellectual disability, respectively, while 42% displaying normal cognitive function. ASD was diagnosed in 12% of patients, while 48% had learning disabilities. Behavioral issues were present in 30% of patients, predominantly ADHD (20%), irritability (10%), and oppositional behavior (10%). It should be noted that many patients presented more than one behavior disorder. Sleep disorders were rare, with only a few cases of insomnia or enuresis reported. MRI analyses revealed that 10% of TSC2 patients had no detectable cortical tubers, while 74% had multiple tubers (> 5). The average tuber sizes was 17 mm (SD = 18.3 mm), with some patients presenting cerebellar or cystic tubers. Subependymal nodules (SENs) were identified in 86% of patients, with dimensions varying widely (average size = 9.4 mm; SD = 0.9 mm). SEGAs were less common, found in 14% of patients. The average size of SEGAs was 13 mm (SD = 15 mm), ranging from 1 cm to 1.5 cm. In our cohort SEGAs were associated with different genotypes, in particular the pathogenic variants c.3094 C > T, c.5201_5216dup and c.1283_1285del. One patient harboring 2 SEGAs carried a large intragenic deletion (spanning from exon 3 to 9). Radial bands were noted in 12% of cases, mostly in parietal and temporal regions. Most patients (74%) had no white matter abnormalities, the remaining cases ranged from multiple to unspecified abnormalities. Only 3 patients (6%) had brain cysts and 2 (4%) had atrophy or hypoplasia of the corpus callosum.
Regarding the specific pathogenic variant c.3693_3696del, (p.Ser1232Thrfs*92), it was found in one of the biggest family of TSC patients in Italy. The family is made up of at least 43 members (Fig. 1c), of whom at least 25 members are affected by tuberous sclerosis. Complete information about genotype and phenotype of all the members was impossible to collect for many reasons (for example, some of them died without certain diagnosis, others refused clinical and genetic investigations, data were stored in different hospitals): for this reason, a total of 16 family members have been included in this study. Although they all carry the same mutation, an intrafamilial variable clinical presentation was observed, going from mild cutaneous phenotype to severe mental retardation and drug-resistant epilepsy. Nine patients from this family presented epilepsy. The median age of onset of epilepsy was 6 years old, ranging from birth to 23 years. Types of seizures and frequency were variable, but the most frequent were infantile spasms, tonic clonic and focal crises with impaired awareness. Regarding the EEG, the most frequent patterns were hypsarrhythmia and focal spikes (with secondary generalization) in the left frontal temporal lobes: it must be underlined that all the normal EEGs found in TSC2 epileptic patients were observed in this family. Drug response was different among the members of this family, and the biggest group is made up of good/partial responders, although there were four cases of remission. All the levels of cognitive impairment severity were present in this family, although the most frequent was mild intellectual disability. ASD was rare and none of the individuals met the full diagnostic criteria for confirmed diagnosis, only some of them had a subclinical features of ASD with limited social difficulties. Learning disability was present mainly in patients with moderate-severe intellectual disability, who were the minority of the family. The most frequent behavior disorders were ADHD and oppositional disorder. Sleep disorders were basically absent. Many bilateral tubers and SENs were reported in all the members of the family, while none of them presented SEGAs. Cerebral cysts and white matter abnormalities were absent, while some of them presented radial bands. To sum up the common characteristics of this family members were multiple tubers and SENs, absence of SEGAs, attention deficit disorder, mild developmental delay (that could explain why they managed to reproduce), partial epilepsy control, infantile spasms and focal impaired awareness seizures.
The pathogenic variant c.1096G > T (p.Glu366Ter) was present in another family (patients 69,70,71,72,73) (respectively a father and his two sons and two daughters). The types of seizures described in this family were focal with impaired awareness and infantile spasms. All patients also shared severe intellectual and learning disability. ASD, behavior and sleep disorders were absent. Brain MRI reports revealed the presence of few to many tubers and SEN, but no-one showed SEGA.
TSC1 vs. TSC2 patients
The study cohort included 81 patients, divided into TSC1 and TSC2 groups. Tables 1, 2 and 3 summarize comparisons between the two groups and the statistical data.
The proportion of TSC2 patients (61.7%) was higher than TSC1 ones (38.3%). There was no statistically significant difference between the two groups with respect to sex (TSC1: male 48.4%-female 51.6%; TSC2: male 42%-female 58%) or mean age. In our cohort, the distribution of familial versus sporadic or de novo pathogenic variants was comparable between the TSC1 and TSC2 groups. There was no statistically significant difference for types of mutations, which included intragenic deletions, duplications, missense and nonsense mutations.
TSC2 patients exhibited a greater variability in clinical manifestations, seizure types, cognitive outcomes, and behavioral disorders. There was no statistically significant difference in the overall prevalence of epilepsy between the two groups (72% in the TSC2 group vs. 64.5% in TSC1), but infantile spasms were significantly more frequently in TSC2 patients (p = 0.016). For other seizure types (focal, focal dyscognitive, tonic-clonic, atonic, hypertonic, myoclonic, gelastic) no statistically significant differences were observed. EEG findings were comparable between TSC1 and TSC2 patients, with a similar proportion showing normal patterns. Pathological findings – such as focal or generalized spike-and-wave discharges and hypsarrhythmia – were also observed at similar rates in both groups, with no significant differences in their prevalence.
Both groups showed similar responses to antiseizure medications, with no significant difference in rates of remission, partial or good response, or drug-resistance. Cognitive and behavioral disorders were similarly distributed. Importantly, seizure remission was associated with a higher likelihood of normal cognitive function. Among the TSC1 patients with seizure remission, 70% had normal cognition (90% if those with mild impairment were included). Among TSC2 patients with remission, 28.6% had normal cognitive profile, rising to 78.6% when including those with mild intellectual disability.
Tubers, SENs, and radial bands occurred at comparable frequencies, although TSC2 patients slightly more likely to have multiple and larger tubers (p < 0.01) and larger SENs (p < 0.01), while TSC1 patients were more likely to present radial (p < 0.05) and multiple radial bands (p < 0.01).
Discussion
This study presents a comprehensive genotype–phenotype analysis of a Sicilian cohort of patients with Tuberous Sclerosis Complex (TSC), offering a broad overview of the clinical and genetic landscape of TSC in the region. The cohort includes 81 patients, representing approximately 15% of the estimated TSC population in Sicily, based on a prevalence of 1 in 8,000 individuals. This substantial proportion lends statistical and clinical weight to our findings. Nevertheless, challenges in diagnosis persist, particularly in cases with milder manifestations or limited healthcare access. Patients with subtle dermatological signs or well-controlled epilepsy may remain undiagnosed, highlighting the importance of increasing awareness and access to genetic testing across all provinces.
The distribution of mutations was consistent with global data, with a predominance of TSC2 pathogenic variants (approximately two-thirds) and fewer TSC1 pathogenic variants (approximately one-third)14. Gender distribution and mean age were similar between the two groups, echoing previous studies15,16. Interestingly, our cohort showed a higher proportion of familial cases (45.6%) than reported in the literature, which often shows a 2:1 ratio of de novo to familial mutations17,18,19,20. This discrepancy may reflect more thorough family screening in our clinical setting or regional variations in reproductive behavior and healthcare practices. No statistically significant difference was observed between TSC1 and TSC2 regarding inheritance pattern, contrasting with prior findings that more sporadic cases are associated with TSC2 mutations15,21,22,23.
Regarding the distribution of variant types, we found an equal spread of intragenic deletions, duplications, missense, and nonsense mutations across both genes. This contrasts with previous reports that TSC1 variants are more frequently nonsense or deletions, while TSC2 variants are more commonly frameshift or nonsense mutations16,24,25. These differences may be due to sample size, ethnic background, or referral bias. Nevertheless, other reports have also shown balanced variant types in TSC2, supporting the heterogeneity we observed23.
Consistent with previous literature, TSC2 patients were more likely to experience seizures than TSC1 patients, although the difference did not reach statistical significance (72% vs. 64.5%)15. Notably, infantile spasms occurred significantly more frequently in the TSC2 group (p = 0.016), in line with previous studies reporting rates of 19% in TSC1 and 41% in TSC2 patients22,23,24. The age of epilepsy onset was comparable between groups, although TSC2 patients tended to have a higher seizure burden. Drug responsiveness was also similar between groups, though seizure remission was more common in TSC1 patients (50% vs. 36.1%), while TSC2 patients had a higher rate of partial responders (27.6% vs. 15%). These findings partly contradict studies showing that TSC2 mutations are associated with more severe and drug-resistant epilepsy26,27,28,29,−30.
EEG findings were broadly similar between groups. However, hypsarrhythmia was more common in TSC2 patients, albeit without statistical significance. This may reflect more extensive cortical dysplasia in TSC2, which aligns with theories of greater mTOR pathway dysregulation in these patients.
Intellectual disability rates did not significantly differ between TSC1 and TSC2 patients. Severe intellectual disability was more frequent in TSC2 patients, as documented in previous studies31,32,33,34. Some pathogenic variants in both genes were associated with variable cognitive outcomes and these differences may relate to the functional domain affected. For example, truncating mutations in the tuberin-interacting domain of TSC1 and the hamartin-binding domain of TSC2 are often associated with more severe phenotypes27,30,35,36,37,38,39,40,41,42,43,44.
Seizure control correlated with cognitive outcomes. Among TSC1 patients achieving seizure remission, 70% had no intellectual disability, rising to 90% when including those with mild impairment. In contrast, only 28.6% of seizure-free TSC2 patients had normal cognition, increasing to 78.6% when mild disability was included. Among drug-resistant patients, severe intellectual disability was present in 57.1% of TSC1 and 66.6% of TSC2 individuals. These results are in line with evidence that early seizure control is a key determinant of neurodevelopment in TSC44,45,46,47.
No significant differences were observed in ASD, ADHD, learning disabilities, or behavioral disorders. This contrasts with prior studies suggesting higher rates of ASD and behavioral issues in TSC2 patients47. Our findings may reflect the small number of patients with these specific comorbidities in our cohort.
Neuroimaging findings were generally consistent between groups, with a few exceptions. While cortical tuber prevalence was similar, TSC1 patients were more likely to have smaller tubers (p < 0.01). SENs, SEGAs, and white matter abnormalities occurred with equal frequency. However, TSC1 patients showed a significantly higher prevalence of radial bands (p = 0.011), and were more likely to exhibit more than two bands (p = 0.028). This is a novel observation and may warrant further investigation in larger studies.
Although the prevailing view is that TSC2 mutations confer a more severe clinical phenotype, our data demonstrate considerable overlap between TSC1 and TSC2 patients. Several TSC1 individuals exhibited severe neurocutaneous manifestations, and some TSC2 patients had mild disease. Therefore, while genotype is informative, it should not be the sole factor guiding clinical counseling or prognostication.
Genotype–phenotype correlations remain challenging due to phenotypic variability, age-dependent manifestation, and the influence of mosaicism or unknown modifiers contributing to postzygotic “second-hit” mutations, which are required for the development of several organ-specific manifestations6,7,8,13,1,15,16,17,18. Nonetheless, our findings emphasize the clinical relevance of integrating genetic data into personalized care strategies. The four familial observations have highlighted the complexity and intra-familial variability of TSC, underscoring the need for further research to clarify possible mutation-specific patterns.
The association between TSC2 mutations and a higher epilepsy burden, particularly infantile spasms and EEG abnormalities, supports the hypothesis that TSC2 dysfunction results in greater mTOR hyperactivation and neural excitability27. These findings highlight the need for early genotype-informed treatment strategies, including the use of vigabatrin, cannabidiol, or mTOR inhibitors48,49,50,51,52,53,54. EEG findings such as hypsarrhythmia and focal spikes, especially in TSC2 patients, may reflect perituberal cortical dysfunction not visible on conventional MRI49,53.
The neuropsychiatric profile of TSC2 patients, particularly the higher prevalence of ADHD and cognitive impairment, may reflect greater disruption of fronto-striatal and fronto-temporal circuits34,35. Our findings confirm earlier reports linking pathogenic TSC2 variants to ASD severity, reinforcing the importance of early neurodevelopmental assessment and intervention32,33,34,35,47.
Conclusions
This study represents the first comprehensive TSC mutational analysis and genotype-phenotype correlation conducted in a cohort of Sicilian patients with TSC. Our findings reinforce the established association of pathogenic TSC2 variants with more severe neurological manifestations, including infantile spasms and related EEG abnormalities. Conversely, patients harboring pathogenic TSC1 variants demonstrated a higher prevalence of radial bands, and milder cognitive involvement, emphasizing the nuanced heterogeneity of TSC.
Our findings highlight the broad variability of TSC manifestations, even among members of the same families, and underscore the need for personalized management approaches. Our results support the importance of early genetic testing to guide treatment strategies, particularly for TSC2 patients who may benefit from mTOR inhibitor therapy and proactive epilepsy management. In this context, additional or alternative genetic testing methods should be employed to effectively detect somatic mosaicism through minimally invasive approaches, such as liquid biopsy, thereby enhancing the accuracy of genotype–phenotype correlations.
Larger, multicenter, and longitudinal studies are essential to confirm these correlations, refine genotype-specific therapeutic guidelines, and assess the long-term outcomes of early interventions. By leveraging genetic insights, clinicians can further advance precision medicine in TSC, improving both clinical outcomes and quality of life for affected individuals.
Data availability
The datasets generated and/or analyzed during the current study are available in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/) and can be retrieved using the following submission IDs: 15233733; 15233796; 15233928; 15233938; 15233951; 15233966; 15233980; 15233928; 15234011; 15234019; 15234029; 15234041; 15234045; 15234142; 15234050; 15234063; 15233951; 15234070; 15234109 15234148; 15234153; 15234159; 15234185; 15234192; 15234203; 15237360; 15234242; 15234212; 15234220; 15234224; 15234375; 15234222; 15234237; 15234362; 15237397; 15234207; 15234240; 15237421, which will be linked to public variant accession numbers upon final processing. The data can be accessed upon reasonable request from the corresponding author.
References
Osborne, J. P. et al. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 615, 125–127 (1991).
Gómez, M. R. et al. History of the tuberous sclerosis complex. Brain Dev. 17 Suppl, 55–57 (1995).
Brigo, F. et al. First descriptions of tuberous sclerosis by Désiré-Magloire Bourneville (1840–1909). Neuropathology 38, 577–582 (2018).
Huschner, F. et al. Molecular EPISTOP, a comprehensive multi-omic analysis of blood from tuberous sclerosis complex infants age birth to two years. Nat. Commun. 14(1), 7664 (2023).
Kandt, R. S. et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat. Genet. 2, 37–41 (1992).
West, H. D. et al. Targeted genomic sequencing of TSC1 and TSC2 reveals causal variants in individuals for whom previous genetic testing for tuberous sclerosis complex was normal. Hum. Mutat. 2023, 4899372 (2023).
Tyburczy, M. E. et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 11, e1005637 (2015).
Ye, Z. et al. Mosaicism in tuberous sclerosis complex: Lowering the threshold for clinical reporting. Hum. Mutat. 43, 1956–1969 (2022).
Inoki, K. et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell. Biol. 4, 648–657 (2002).
Northrup, H. et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr. Neurol. 123, 50–66 (2021).
Curatolo, P. et al. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 14, 733–745 (2015).
Pinto, A. L. R. et al. Neurological manifestations of tuberous sclerosis complex: The importance of early diagnosis. Arq. Neuropsiquiatr. 80, 983–984 (2022).
Nabbout, R. et al. Historical patterns of diagnosis, treatments, and outcome of epilepsy associated with tuberous sclerosis complex: Results from TOSCA registry. Front. Neurol. 12, 697467 (2021).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80 (2001).
Northrup, H., Koenig, M. K., Pearson, D. A., Au, K. S. & Tuberous sclerosis complex. In GeneReviews® (eds Adam, M. P., Feldman, J., Mirzaa, G. M., Pagon, R. A., Wallace, S. E. & Amemiya, A.) (University of Washington, 1999).
Peron, A. et al. Genetics, genomics, and genotype-phenotype correlations of TSC: Insights for clinical practice. Am. J. Med. Genet. C Semin Med. Genet. 178, 281–290 (2018).
Tyburczy, M. E. et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum. Mol. Genet. 23, 2023–2029 (2014).
Lam, H. C., Nijmeh, J. & Henske, E. P. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J. Pathol. 241, 219–225 (2017).
Curatolo, P. et al. Tuberous sclerosis. Handb. Clin. Neurol. 111, 323–331 (2013).
Langkau, N. et al. TSC1 and TSC2 mutations in tuberous sclerosis, the associated phenotypes and a model to explain observed TSC1/TSC2 frequency ratios. Eur. J. Pediatr. 161, 393–402 (2002).
Sancak, O. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur. J. Hum. Genet. 13, 731–741 (2005).
Au, K. S. et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the united States. Genet. Med. 9, 88–100 (2007).
Jones, A. C. et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 64, 1305–1315 (1999).
Rosset, C. et al. TSC1 and TSC2 gene mutations and their implications for treatment in tuberous sclerosis complex: A review. Genet. Mol. Biol. 40, 69–79 (2017).
Kothare, S. V. et al. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia 55, 1025–1029 (2014).
Di Napoli, C. et al. TSC1 and TSC2: Tuberous sclerosis complex and its related epilepsy phenotype. J. Pediatr. Neurol. https://doi.org/10.1055/s-0041-1727142 (2021).
Zeng, L-H. et al. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 20, 445–454 (2011).
Nabbout, R., Kuchenbuch, M., Chiron, C. & Curatolo, P. Pharmacotherapy for seizures in tuberous sclerosis complex. CNS Drugs 35(9), 965–983 (2021).
Nabavi Nouri, M. et al. Epilepsy management in tuberous sclerosis complex: Existing and evolving therapies and future considerations. Pediatr. Neurol. 126, 11–19 (2022).
Henske, E. P. et al. Tuberous sclerosis complex. Nat. Rev. Dis. Prim. 2, 16035 (2016).
Feliciano, D. M. et al. The neurodevelopmental pathogenesis of tuberous sclerosis complex (TSC). Front. Neuroanat. 14 (2020).
Bolton, P. F. et al. Intellectual abilities in tuberous sclerosis complex: Risk factors and correlates from the tuberous sclerosis 2000 study. Psychol. Med. 45, 2321–2331 (2015).
Joinson, C. et al. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol. Med. 33, 335–344 (2003).
de Vries, P. J. et al. Tuberous sclerosis complex-associated neuropsychiatric disorders (TAND): New findings on age, sex, and genotype in relation to intellectual phenotype. Front. Neurol. 11, 603 (2020).
van Eeghen, A. M. et al. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur. J. Hum. Genet. 20, 510–515 (2012).
Ekong, R. et al. Variants within TSC2 exons 25 and 31 are very unlikely to cause clinically diagnosable tuberous sclerosis. Hum. Mutat. 37(4), 364–370 (2016).
Farach, L. S. et al. TSC2 c.1864C > T variant associated with mild cases of tuberous sclerosis complex. Am. J. Med. Genet. A 173(3), 771–775 (2017).
Jansen, A. C. et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann. Neurol. 60(5), 528–539 (2006).
van Eeghen, A. M. et al. Central TSC2 missense mutations are associated with a reduced risk of infantile spasms. Epilepsy Res. 103(1), 83–87 (2013).
O’Connor, S. E. et al. A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology 61(3), 409–412 (2003).
Wentink, M. et al. Functional characterization of the TSC2 c.3598C > T (p.R1200W) missense mutation that co-segregates with tuberous sclerosis complex in mildly affected kindreds. Clin. Genet. 81(5), 453–461 (2012).
Khare, L. et al. A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. J. Med. Genet. 38(5), 347–349 (2001).
Camposano, S. et al. Distinct clinical characteristics of tuberous sclerosis complex patients with no mutation identified. Ann. Hum. Genet. 73(2), 141–146 (2009).
Farach, L. S. et al. Drug-Resistant epilepsy in tuberous sclerosis complex is associated with TSC2 genotype: More findings from the preventing epilepsy using vigatrin (PREVeNT) trial. Pediatr. Neurol. 159, 62–71 (2024).
De Ridder, J. et al. Early epileptiform EEG activity in infants with tuberous sclerosis complex predicts epilepsy and neurodevelopmental outcomes. Epilepsia 62, 1208–1219 (2021).
Curatolo, P. et al. Advances in the genetics and neuropathology of tuberous sclerosis complex: Edging closer to targeted therapy. Lancet Neurol. 21, 843–856 (2022).
Moavero, R. et al. Early clinical predictors of autism spectrum disorder in infants with tuberous sclerosis complex: Results from the EPISTOP study. J. Clin. Med. 8(6), 788 (2019).
Krueger, D. A. et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 4(12), 877–887 (2017).
Hwang, S-K. et al. Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Mol. Brain 9, 56 (2016).
Maász, A. et al. Three-year follow-up after intrauterine mTOR inhibitor administration for fetus with TSC-Associated rhabdomyoma. Int. J. Mol. Sci. 24(16), 12886 (2023).
Samanta, D. Evolving treatment strategies for early-life seizures in tuberous sclerosis complex: A review and treatment algorithm. Epilepsy Behav. 161, 110123 (2024).
Chu-Shore, C. J., Major, P., Camposano, S., Muzykewicz, D. & Thiele, E. A. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 51(7), 1236–1241 (2010).
Coban, G. et al. Synthetic MRI in children with tuberous sclerosis complex. Insights Imaging 13(1), 115 (2022).
Rosengren, T. et al. Mutational analysis of TSC1 and TSC2 in Danish patients with tuberous sclerosis complex. Sci. Rep. 10(1), 9909 (2020).
Acknowledgements
The Authors wish to thank the “Associazione Sclerosi Tuberosa” (AST) for its support; The Authors also wish to thank the Health Operational Plan (FSC 2014-2020) “Trajectory 4-Biotechnology, Bioinformatics and Pharmaceutical Development” entitled “PHARMA-HUB”.
Author information
Authors and Affiliations
Contributions
A.D.P. wrote the first draft of the manuscript; C.D., E.D. and S.S. performed the literature research; M.P., F.C., R.S., M.V., A.Z. and M.B. followed-up the patients; M.E. and M.L.B. validated the results; A.P. & M.R. critically revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Approvement of the Ethical committee of the University has been obtained, as well as consent to participate from patients and/or parents or legal guardians of minor patients.
Consent for publication
Written informed consent for publication of this study was obtained from all adult patients and the parents or legal guardians of minor participants.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Praticò, A.D., Di Napoli, C., Salafia, S. et al. Genetic screening of tuberous sclerosis complex in Sicily with a focus on neurological manifestations. Sci Rep 15, 20347 (2025). https://doi.org/10.1038/s41598-025-04718-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04718-6
