
Abstract
The main focused of this work is the designing of isoquinoline-based derivatives through structural modeling of synthesized compound (BPDI). Two distinct series of derivatives were developed: MPBID1–MPBID6, in which electron-withdrawing groups were introduced at unfused phenyl ring on isoquinoline core and MPBID1′–MPBID6′, where substitutions were made at unfused phenyl ring on isoquinoline core. Quantum chemical calculations were employed by DFT/TD-DFT at M06/6-311G(d, p) functional. Frontier molecular orbitals (FMOs), natural bonding orbital (NBO), non-linear optics (NLO), density of states (DOS), global reactivity parameters (GRPs), transition density matrix (TDM) and UV-Visible analyses of designed compounds were performed to understand their NLO responses. FMO results are supported by TDM analysis showing that all the designed compounds have smaller energy gap values than the reference compound. Among all the designed compounds, MPBID4′ is the most suitable candidate for NLO study because of the lowest energy gap with a larger bathochromic redshift. NBO study has confirmed the stability of compounds. Dipole moment, average hyperpolarizability, first hyperpolarizability and second hyperpolarizability values of designed compounds were also better than that of reference compound. This study reveals that structural tailoring performs a key role in the development of attractive and best NLO materials for optoelectronic devices.
Similar content being viewed by others
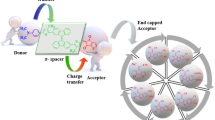


Introduction
In recent years, non-linear optics (NLO) have gained considerable importance due to their various applications in optical communications, quantum optics1information technology, biomedical imaging2material sciences3optoelectronic devices4,5,6 optical data storage, dynamic image processing, telecommunications7, sensing, optical computing, and many more1. Furthermore, NLO has emerged as a prominent field of research in various disciplines of solid-state physics3medicine8nuclear research9 and chemical dynamics10. Researchers in both theoretical and experimental domains are increasingly focusing on NLO materials because of their critical role in advancing modern technologies11. The growing demand for high-performance NLO materials has led scientists to explore both organic and inorganic domains7,12. These materials have much more attention due to their low cost, ease of design and small dielectric constant13. Initially, NLO research was mainly focused on inorganic compounds that were used by scientists for NLO applications14. Alkali metal atoms like silicones were used for NLO materials as they show several properties like low toxicity, thermal stability, a potential source of electrons15 and high efficiency. Unfortunately, there were certain drawbacks associated with inorganic compounds including high cost, hardness, non-tunable energy levels and manufacturing difficulty16.
Over time, organic compounds gained substantial consideration over inorganic compounds due to their tremendous benefits like greater damage threshold, higher photovoltaic coefficient, speedy response time, low development cost, versatility of design, tunable bands and low dielectric coefficient17,18,19,20,21. Synthetic organic compounds are considered more important due to their simple reaction chemistry and tolerance of structural modeling for better NLO response22,23. Different organic compound classes like fullerene, non-fullerene, polymers, and dyes have been studied in the literature to explore better NLO responses24. The properties of organic NLO compounds improved through electronic charge transfer from one fragment to another fragment with the aid of π-linker25. This charge transfer (ICT) increases with the structural tailoring of end-capped acceptors in various organic compounds. These NLO related properties arise from ICT generating push-pull mechanism that can minimize energy gap thus leading to the generation of high performing NLO material26,27,28.
Among various classes of organic chromophores, donor–π–acceptor (D–π–A) systems have emerged as promising candidates for NLO applications owing to their strong intramolecular charge transfer (ICT) characteristics, structural tunability, and high polarizability29.
Isoquinoline, a nitrogen-containing hetero aromatic compound, represents a versatile electron-accepting unit due to its extended conjugation and electron-deficient nature. Incorporating isoquinoline moieties into organic chromophores can enhance the molecular hyperpolarizability (β), facilitate ICT processes, and stabilize charge-separated states, making them ideal components in the design of efficient NLO materials. Moreover, the rigid and planar structure of isoquinoline contributes to improved thermal and photochemical stability, which is critical for practical applications30.
Recent studies have highlighted the potential of isoquinoline-functionalized compounds in organic electronics, but their role in NLO applications remains relatively underexplored. The present work aims to investigate the structure–property relationships of novel isoquinoline-based chromophores. Herein, a synthesized compound 5,6-diphenylbenzol[4,5]imidazole[2,1-a]isoquinoline (DPBI)31 has been taken as parent compound and designed into the reference compound MPBIR by structural modelling;30one of the phenyl groups of BPDI was replaced with the methyl group as shown in Fig. 1. Two new series (MPBID1-MPBID6 and MPBID1′-MPBID6′) were designed by structural modulation of reference compound (MPBIR) by introducing electron withdrawing moieties at terminal benzene rings (Fig. 2). According to the literature survey, all the designed derivatives have not been reported yet and their NLO properties have not been studied. Their optoelectronic properties were investigated through DFT/TDDFT approaches. It is expected that this investigation might serve as a source for the researchers to explore advance NLO materials with outstanding characteristics.
Computational study
All the theoretical calculations of designed derivatives were accomplished at M06/6-311G(d, p) functional by employing Gaussian 16 program package32. At first, the geometrical optimization was performed to get true minima geometries. The absence of imaginary frequency supported the successful optimization of entitled chromophores. These optimized structures were then utilized to performed further analyses like absorption spectra, frontier molecular orbital (FMOs), density of states (DOS), global reactivity parameters (GRPs), transition density matrix (TDM), natural bond orbitals (NBO) and NLO investigations at the above-mentioned functional.
In order to check the effect of different media on absorption spectra of entitled compounds, UV-Visible analysis was accomplished at gaseous phase and in chloroform media. The conductor-like polarizable continuum model (CPCM)33 was utilized to explore the effect of chloroform solvent on UV-Vis properties of entitled compounds. For the interpretation of data different software like GaussSum34Multiwfn version 3.835, PyMOlyze version 2.034, Avogadro version 1.2.0n36Gauss-view version 5.037, and Chemcraft38 were utilized.
Result and discussion
The synthesized parent chromophore 5,6-diphenylbenzol[4,5]imidazo[2,1-a]isoquinoline (DPBI) is designed into the reference chromophore (MPBIR) by replacing one of the phenyl group with methyl group to reduce steric hindrance and lesser the computational cost. After this structural modification, two series of new compounds (MPBID1-MPBID6) and (MPBID1′-MPBID6′) are designed by introducing electron with drawing moieties at two different position on benzene ring of isoquinoline core. The first series MPBID1-MPBID6 was designed by introducing -F, -Cl, -Br, -NO2 moieties at the benzene ring that is fused with isoquinoline and the second series MPBID1′-MPBID6′ was designed by introducing these moieties at phenyl ring which is unfused with core as shown in Fig. 2. In order to investigate the effect of these electron with drawing moieties on isoquinoline core we divided the compounds in to three fragments as shown in Fig. 2 illustrated with red gray and white color balls. The IUPAC names of these designed compounds are presented in Table S1. Furthermore, the ChemDraw structure are displayed in Figures S1 and S2. Different analyses like electronic properties, first hyperpolarizability, second hyperpolarizability, energy gap, NBO and UV-visible spectra of the designed compounds have been performed by DFT computations. The cartesian coordinates of designed compounds are given in Tables S2-S13.

Structural modification of Parent chromophore DPBI into reference compound MPBIR.

Sketch map of designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′).
Frontier molecular orbital (FMO) analysis
FMO investigation is a powerful tool to examine different electronic transitions, light absorbance, molecular reactivity, optical behavior and chemical stabilities of the compound10,39,40. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) plays an important role in determining the optical and electronic properties of the compounds4114. HOMO is regarded as an electron donator while LUMO is regarded as an electron acceptor42,43. The energy difference is known as the energy gap (∆E), which is also defined as ELUMO-EHOMO. The ∆E of the designed compounds obtained by FMO is directly related to the chemical and kinetic stability of the molecules44. If the molecule has a higher ∆E value then it is considered as less reactive, hard molecule. While the molecule with a lower ∆E value shows a higher degree of softness and greater reactivity45,46. Thus, the molecules with lower ∆E possess higher ICT and offer the best NLO response47,48. The FMO results of MPBID1-MPBID6 and MPBID1′-MPBID6′ are shown in Tables 1 and 2. The other molecular orbital energy values of entitled compounds EHOMO−1, ELUMO+1, EHOMO−2, and ELUMO+1 are provided in Tables S14 and S15 and their structures are displayed in Figures S3 and S4.
The calculated values of HOMO and LUMO energy of compounds MPBIR and MPBID1-MPBID6 along with their energy gap values are tabulated in Table 1. The EHOMO, ELUMO and energy gap values of the MPBIR were examined as -5.762, -1.938 and 3.824 eV, respectively. The EHOMO values of the MPBID1-MPBID6 were − 5.811, -5.859, -5.858, -6.225, -6.004 and − 6.093 eV, respectively. While the ELUMO values of MPBID1-MPBID6 were noted as -2.061, -2.096, -2.083, -3.146, -2.027 and − 2.312 eV respectively. The energy gap values of MPBID1-MPBID6 were investigated as 3.750, 3.763, 3.775, 3.079, 3.977 and 3.781 eV, respectively. Interestingly, all the designed compounds have lower ∆E than the reference compound except MPBID5. These reduced energy gap values are due to the introduction of electron withdrawing groups (-F, -Cl, -Br and -NO2, etc.) in fragment 3. The energy gap of the designed molecules reduces in the following order: MPBID5 > MPBIR > MPBID6 > MPBID3 > MPBID2 > MPBID1 > MPBID4. The highest energy gap value among all the designed molecules was found in MPBID5 (5-methyl-6-(4-(trifluoromethyl)phenyl)benzo[4,5]imidazo[2,1-a]isoquinoline) because of the presence of -CF3 unit which is less electronegative than -NO2 unit. The lowest energy gap value (3.079 eV) was found in the MPBID4 molecule because of the replacement of the -Br group with the -NO2 group which is a strong electronegative unit with greater negative inductive effect (-I) on fragment 3. It shows that structural modeling of the acceptor moieties has a comparable influence on the energy gap of the molecules. Figure 3 showed the electron density on HOMO/LUMO of designed compounds.
Table 2 discloses the HOMO/LUMO values of reference and designed compounds (MPBIR and MPBID1′-MPBID6′) along with their energy gap values. The ELUMO values of MPBIR and MPBID1′-MPBID6′ were − 1.938, -1.966, -2.071, -2.077, -3.304, -2.239 and − 2.456 eV respectively. While 5.797, -5.826, -5.823, -6.046, -5.904 and − 5.968 eV were the EHOMO values of derivatives MPBID1′-MPBID6, respectively. The 3.831, 3.755, 3.746, 2.742, 3.665 and 3.512 eV were the energy gap values of MPBID1′-MPBID6, respectively. The presence of a strong electron withdrawing unit (-NO2) in MPBID4′ enhanced the π-conjugation and resonance of the molecule leading to a lower energy gap value (2.742 eV). The energy gap entitled chromophore diminished in the following order: MPBID1′> MPBIR′> MPBID2′> MPBID3′> MPBID6′> MPBID5′> MPBID4′. Except MPBID1′ all other derivatives showed reduced energy gap when electron withdrawing moieties are introduced on fused benzene ring of isoquinoline core might be due to greater resonance phenomena. Figures 3 and 4 show that the electronic charge density is distributed on the entire molecule in both HOMO and LUMO which illustrated good ICT in these derivatives.

Molecular orbital of MPBIR and MPBID1- MPBID6 illustrating charge densities.

Molecular orbital of MPBID1′-MPBID6 illustrating charge densities drawn with the help of Avogadro software, Version 1.2.0. (http://Avogadro.cc/). All out put files of entitled compounds were accomplished by Gaussian 16 version D.01 (https://gaussian.com/g16citation/).
The FMO analysis of these designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′) are compared with each other in other to find out which end capped changes have better electronic results. When compared the energy gap of these designed compounds then it shows that almost all the values are comparably same except MPBID4 and MPBID4′. There is a great difference in their energy gap values MPBID4 (3.079 eV), and MPBID4′ (2.742 eV). Both these compounds have same nitro group (-NO2) but the difference in their energy gap values is due to the location of attachment of end capped unit. In MPBID4 the -NO2 group is attached to fragment 3 which is not fused with fragment 2 (central core). While in MPBID4′ the -NO2 group is attached to fragment 1 which is fused with fragment 2 having a better resonance than the other one. This resonance property makes the energy gap of MPBID4′ lower (2.742 eV), making it a better option as compared to MPBID4 (3.079 eV). Optimized frequency structures of reference and all designed compounds are displayed in Figures S5 and S6.
Global reactivity parameters
The FMO (Egap=ELUMO−EHOMO) study is a important factor for the estimation of global reactivity parameters (GRPs) such as electron affinity (EA), electronegativity (X)49global electrophilicity index (ω), ionization potential (IP), chemical potential (µ), global softness and global hardness (η)50,51,52. These parameters were estimated using Koopmans’s theorem53. The electron donating and electron accepting capabilities of the compounds are determined by IP and EA that are the energy mandatory to extract the electron from the HOMO orbital10. The electrophilic strength of the compounds can be measured by using this parameter. The energy gap (∆E) is directly proportional to the hardness and stability of the compounds while inversely proportional to reactivity and softness. Thus, the molecules with smaller energy gaps are soft molecules that are more reactive, easily tunable and less stable as compared to the compounds having larger energy gap values. These soft molecules with lower energy gaps are considered as better competitor showing the best NLO response44,54. Equations for the calculation of global reactivity parameters are displayed in Table S15. The calculated GRP values of designed compounds are represented in Tables 3 and 4.
The data from the above Table 3 showed that GRP results for the MPBID1-MPBID6 were closely related to energy gap values. The ionization potential values for reference and designed compounds were ranged from 5.762 to 6.093 eV, respectively. All the designed compounds have greater IP and EA values than the reference compound MPBIR. The global hardness values of the reference and designed compounds were 1.912, 1.875, 1.882, 1.888, 1.539, 1.989 and 1.891 eV, respectively. The MPBID5 has the highest η value (1.989 eV) means that it is a hard molecule with more stability and less reactivity than all other designed compounds. MPBID4 has the lowest η value (1.539 eV) along with the highest global softness value (0.325 eV − 1) and shows the highest reactivity and a larger rate of polarizability holding efficient NLO response. The increasing order of global softness (σ) was investigated as MPBID5 < MPBIDR < MPBID6 = MPBID3 < MPBID2 < MPBID1 < MPBID4.
Table 4 demonstrated the GRP values for the MPBID1′-MPBID6′ which showed that GRP results for MPBID1′-MPBID6′ were closely related to energy gap values. The global hardness and chemical potential are directly related to energy gap values i.e. lesser the global hardness of the molecule, the lower its energy gap value55. The ionization potential values for designed compounds were investigated as 5.797, 5.826, 5.823, 6.046, 5.904, 6.004 and 5.968 eV respectively. All the designed compounds have greater IP, EA and electronegativity values than reference compounds. The 0.261, 0.266, 0.265, 0.365, 0.272 and 0.284 eV− 1 were the global softness values of the designed compounds MPBID1′-MPBID6′, respectively. MPBID4′ has the highest global softness value (0.365 eV− 1) means that it is a soft molecule with less stability and more reactivity and a larger rate of polarizability than all other designed compounds. The increasing order of global softness (σ) was found as MPBID1′< MPBID3′< MPBID2′< MPBID5′< MPBID6′< MPBID4′.
When compared the GRPs of MPBID4 and MPBID4′, both have the highest global softness (σ) values. In MPBID4, the -NO2 group is attached to fragment 3 while in MPBID4′ -NO2 group is attached to fragment 1. But when both of these compounds were compared with each other MPBID4′ showed a slightly better result (σ = 0.365 eV− 1) than that of MPBID4 (σ = 0.325 eV− 1 ) because it is directly attached to fragment 2 having a better resonance effect. Overall, this investigation found that these designed compounds have greater charge transfer capability from HOMO to LUMO resulting in good NLO response.
UV-visible analysis
UV-Visible analysis is performed at M06/6-311G(d, p) level in both gaseous and chloroform phase, in order to understand the absorption peak of designed compounds. UV-Visible analysis is performed to demonstrate the type of transitions, optical properties and photophysical properties of the designed compounds. Table S17 states all of the UV-visible parameters like excitation energy (E), oscillator strength (fos) and maximum absorption (λmax)of the designed compounds56. The absorption spectra of designed compounds (MPBID1-MPBID6) in both gaseous and chloroform phase is displayed in Fig. 5.

UV-Visible spectra of designed compounds (MPBID1-MPBID6) in gaseous and solvent phase. These spectra were drawn by utilizing the Origin 8.5 software (https://www.originlab.com/).
Table S16 demonstrates the absorption values of reference and designed compounds in both the gaseous and solvent phase. Due to the solvent effect, the λmax of the designed compounds observed more bathochromic shift in the chloroform phase as compared to the gaseous phase. Chloroform is utilized as a solvent due to its moderate polarity, enabling effective dissolution of various organic compounds. It influences molecular orbital parameters by lowering HOMO and LUMO energy levels, thereby reducing the HOMO-LUMO gap. The λmax of the reference compound was investigated as 319 nm with 3.888 eV excitation energy. The highest value of λmax (319 nm) was observed in MPBID4 with 0.049 oscillator strength in gaseous state. In solvent phase, the exciton energy values of MPBID1-MPBID6 were 3.860, 3.828, 3,823, 3.815, 4.046 and 3.992 eV, respectively. The exciton energy is directly related to charge transfer thus the above mention values show that the absorption spectra of the designed compound are better than the reference compound. The 321, 324, 324, 325, 306 and 311 nm were the λmax values of MPBID1-MPBID6, respectively. The lowest value of λmax (306 nm) was observed in MPBID5 with 0.412 oscillator strength which may be due to the presence of -CF3 which is a less electronegative unit. The highest value of λmax (325 nm) was observed in MPBID4 with 0.636 oscillator strength. The lower excitation energy (3.815 eV) that defines the higher charge transfer ability was seen in MPBID4 due to the presence of a strong electron withdrawing moiety (-NO2). A higher value of λmax means a lower energy gap as wavelength and energy gap are inversely proportional to each other. The increasing order of λmax of the designed compounds was MPBID4 > MPBID3 > MPBID2 > MPBID1 > MPBIDR > MPBID6 > MPBIR5.
Table S31 demonstrated that the compounds MPBID1′-MPBID6′ have comparable results with respect to each other in both the gaseous and solvent phases. The highest λmax value (319 nm) in the gaseous state was found in compound MPBID4′ with 0.049 oscillator strength. In the solvent phase, the λmax of MPBIDR and MPBID1′-MPBID6′ were investigated as 319, 318, 323, 324, 338, 328 and 336 nm, respectively. MPBID4′ has the highest λmax value in both gaseous (319 nm) and solvent phase (338 nm) among all the designed compounds. This is due to the presence of the strongest electron withdrawing group -NO2 at fragment 1 which create a strong push-pull architecture. The increasing order of λmax of the designed compounds is MPBID4′> MPBID6′> MPBIR5′ MPBID3′> MPBID2′> MPBIDR > MPBID1′. The absorption spectra of designed compounds (MPBID1′-MPBID6′) in both gaseous and chloroform phase are displayed in Fig. 6. All other data related to UV-visible analysis of designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′) is provided in supplementary tables (Tables S16-S45).

UV-Visible spectra of designed compounds (MPBID1′-MPBID6′) in gaseous and solvent phase. These spectra were drawn by utilizing the Origin 8.5 software (https://www.originlab.com/).
Natural bond orbital analysis (NBO)
NBO analysis is a powerful tool that is used to investigate the transfer of charge between empty and filled orbitals34,57,58. Some properties like intramolecular and intermolecular transitions, charge distribution, type of bonding and interactions in designed compounds are also described by NBO analysis59,60. Delocalization of charge density and their transfer from donor to acceptor region is also explored by NBO analysis61,62. These calculations are used to evaluate intramolecular and intermolecular interactions like hydrogen bonding, conjugated transitions, orbitals interactions and orbital hybridization63 which results in system stabilization. The interactions of donors and acceptors are the main reason to yield stabilization energies64. NBO analysis results of the designed compounds are tabulated in Tables 5 and 6. NBO analysis of designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′) performed at M06/6-311G (d, p) are tabulated in Tables S43-S55. Some properties like intramolecular and intermolecular transitions, charge distribution, type of bonding and interactions in designed compounds are also described by NBO analysis59,60. Delocalization of charge density and their transfer from donor to acceptor region is also explore by NBO analysis61,62. These calculations are used to evaluate intramolecular and intermolecular interactions like hydrogen bonding, conjugated transitions, orbitals interactions and orbital hybridization63 which result in system stabilization. The interactions of donor and acceptors are the main reason to yield stabilization energies64. NBO analysis results of the designed compounds are tabulated in Tables 5 and 6. NBO analysis of designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′) performed at M06/6-311G d, p) are tabulated in Tables S46-S58.
Usually, four types of accepted transitions take place i.e. π→ π*, σ→ σ*, LP → π* and LP → σ*. Other transitions are also present but they are forbidden and thus are neglected in NBO analysis. Among all the aromatic interactions that occur π → π* stands out since it exhibits higher stabilization energy values. The transitions of LP → π* and LP → σ* show slightly stronger behavior than σ→ σ* transitions which act as the least dominant transitions among all. The detection of hyperconjugation and charge transfer in designed compounds depends upon π→ π* transitions. For π→ π* transitions in MPBIR, the maximum energy value of π(C13-C32) → π*(N2-C12) transition was observed as 26.4 kcal/mol while the minimum value of stabilization energy of π(N2-C12) → π*(N2-C12) was observed as 0.54 kcal/mol. Contrastingly, The stabilization energy of 7.81 kcal/mol for σ(N2-C11) → σ*(C12-C13) makes it the most stable transition in σ→ σ* transitions whereas σ(C9-H10) → σ*(N2-C11) shows the lowest stability with 0.51 kcal/mol. Moreover, in the case of LP to π* transitions, LP (1) (N1) → π*(N2-C12) has greater stabilization energy of 47.32 kcal/mol as compared to LP→ σ* transitions with 11.71 kcal/mol of stabilization energy.
In the same manner, π→ π* transitions of MPBID1-MPBID6 were π(C13-C21) → π*(N2-C12), π(C13-C21) → π*(N2-C12), π(C13-C21) → π*(N2-C12), π(C18-C19) → π*(N40-O42), π(C13-C21) → π*(N2-C12) and π(C13-C21) → π*(N2-C12), with maximum stabilization energy of 26.8,26.18, 26.13, 27.34, 25.09 and 24.81 kcal/mol respectively. Other π→ π* transitions were π(N2-C12) → π*(N2-C12), π(N2-C12) → π*(N2-C12), π(N2-C12) → π*(N2-C12), π(N40-O42) → π*(C18-C19), π(N2-C12) → π*(C13-C21) and π(N2-C12) → π*(C13-C21) with minimum stabilization energy values of 0.57, 0.53, 0.52, 3.98, 9.5 and 9.78 kcal/mol respectively.
Similarly, σ→ σ* transitions of MPBID1-MPBID6 were σ(N2-C11) → σ*(C12-C13), σ(N2-C11) → σ*(C12-C13), σ(N2-C11) → σ*(C12-C13), σ(N2-C11) → σ*(C12-C13), σ(N2-C11) → σ*(C12-C13) and σ(C18- C40) → σ*(C40-N41) with highest stabilization energy of 7.88, 7.89, 7.89,7.9, 7.9 and 8.86 kcal/mol respectively. The σ→ σ* transitions with lowest stabilization energy of 0.51, 0.51, 0.51, 0.51, 0.51 and 0.51 kcal/mol were σ(C9-H10) → σ*(N2-C11), σ(C9-H10) → σ*(N2-C11), σ(C9-H10) → σ*(N2-C11), σ(C9-H10) → σ*(N2-C11), σ(C9-H10) → σ*(N2-C11) and σ(C9-H10) → σ*(N2-C11) respectively.
LP transitions with σ* and π* were also noticed in designed compounds (MPBID1-MPBID6). LP to π* transitions were LP(1)(N1) → π* (N2-C12), LP(1)(N1) → π* (N2-C12), LP(1)(N1) → π* (N2-C12), LP(3)(O41) → π* (N40-O42), LP(1)(N1) → π* (N2-C12) and LP(1)(N1) → π* (N2-C12) with highest stabilization energy of 47.02, 47.01, 47.01, 175.92, 46.94 and 46.83kcal/mol respectively. LP to σ* transitions were LP(1)(N2)→σ*(N1-C12), LP(2)(O42)→σ*(N40-O41), LP(1)(N2)→σ*(N1-C12), LP(2)(O42)→ σ*(N40-O41), LP(1)(N2)→σ*(N1-C12) and LP(1)(N41)→σ*(C18-C40) with highest stabilization energy of 11.79, 20.62, 11.78, 20.62, 11.77 and 12.06 kcal/mol respectively.
In the same way, as in possibility 1, stabilization energy in all types of transitions are noted in designed compounds. Table 6 shows the NBO analysis results of the MPBID1′-MPBID6′. The most stable π → π* transition of 26.32 kcal/mol arises from the bonding pattern of π(C13-C22) to π*(N2-C12) within MPBID1 molecule. The π → π* transitions receiving the least stabilization energy stand at 0.5 kcal/mol due to the activation of π(C29-C30) to π*(C29-C30). The highest stabilization energy of 7.82 kcal/mol was observed in MPBID1′ when σ(N2-C11) → σ*(C12-C13) occurred and the lowest stabilization energy was 0.5 kcal/mol for the σ(C9-H10)→ σ*(N2-C11).
In MPBID2′ the highest π→ π* stabilizing energy was observed in the transition π(C13-C22) → π*(N2-C12) which reached 26.28 kcal/mol yet the π(N2-C12) → π*(N2-C12) transition proved least stable with an energy of 0.53 kcal/mol. The most stable σ→ σ* transition in MPBID2′ occurred for σ(N2-C11) → σ*(C12-C13) at 7.83 kcal/mol but σ(C9-H10) → σ*(N2-C11) showed the most unstable transition at 0.51 kcal/mol. The stabilization energy in LP (1) (N1) → π* (N2-C12) reaches a maximum value of 47.08 kcal/mol while LP (1) (N2) →σ*(N1-C12) exhibits the minimum energy value of 11.76 kcal/mol.
Analysis of MPBID3′ revealed that the π(C13-C22) → π*(C14-C16) transition exhibited 19.63 kcal/mol as its maximum stabilization energy but π(N2-C12) → π*(N2-C12) transition displayed only 0.53 kcal/mol. MPBID3′ exhibited its highest π→ σ* transition stabilization of 7.82 kcal/mol for σ(N2-C11) → σ*(C12-C13) but σ(C9-H10) → σ*(N2-C11) showed the least energy value at 0.5 kcal/mol. The stabilization energy is at its maximum when LP (1) (N1) transfers to π* (N2-C12) at 47.07 kcal/mol yet LP (1) (N2) →σ*(N1-C12) has the lowest value at 11.74 kcal/mol among all LP to π* transitions.
The largest observed value for π→ π* transitions with 20.9 kcal/mol stabilization energy appeared in π(C7-C9) → π*(C4-C5) whereas the smallest value of 0.53 kcal/mol stabilization energy occurred from π(N2-C12) → π*(N2-C12). MPBID4′ demonstrates σ(N2-C11) → σ*(C12-C13) transitions that have 7.84 kcal/mol stabilization energy as its highest value and σ(C9-H10) → σ*(N2-C11) transitions that show 0.5 kcal/mol stabilization energy as the minimum value. The highest stabilization energy of 178.34 kcal/mol exists in LP (3) (O41) → π* (N40-O42) but LP (2) (O41) → σ*(N40-O42) exhibits the lowermost stabilization energy of 20.62 kcal/mol.
The π→ π* transitions of MPBID5′ were found to reach their maximum stabilization point of 26.24 kcal/mol when analyzing the π(C13-C22) → π*(N2-C12) connection and the minimum stabilization occurred at 0.53 kcal/mol through the π(N2-C12) → π*(N2-C12) bond. The highest stabilization energy value of 46.84 kcal/mol belongs to LP (1) (N1) → π* (N2-C12), whereas LP (3) (F42) →σ*(C40-F43) demonstrates the lowest stabilization energy at 10.9 kcal/mol.
From the above-mentioned data, it is believed that among all designed compounds, MPBID4 and MPBID4′ have the highest NBO values with greater stabilization energy and charge transfer properties. Thus, NBO analysis showed that the enhancement in hyperconjugation and greater ICT value plays an important role in stabilizing the designed compounds.
Density of state (DOS)
DOS analysis supports the delocalization of electrons in HOMO and LUMO orbitals65. DOS result supported the result of FMO diagrams and is used for the determination of electronic properties65. DOS analysis was performed at M06/6-311G (d, p) functional. Calculated DOS percentage values can confirm that the electronic charge distribution patterns are due to the attachment of different acceptor moieties66. In DOS pictographs, left side values show the valance band (HOMOs) while the right side values show the conduction band (LUMOs) along x-axis on DOS maps35. For a better explanation of the DOS study, compounds were divided into 3 fragments. These 3 fragments (fragment 3, fragment 2 and fragment 1) are illustrated by red, green and blue colors in the pictographs, respectively. Figure 7 is the graphical representation of the DOS of MPBID1-MPBID6.
Table S59 demonstrated the DOS analysis of MPBIR and MPBID1-MPBID6. For the reference compound fragments 1,2 and 3 contributed 51.0, 48.9 and 0.2% to LUMO while contributed 18.8, 80.2 and 1.0% to HOMO, respectively. Fragment 1 contributed 49.2, 55.6, 55.9, 87.0, 61.5 and 69.9% to LUMO and 19.3, 19.3, 19.8, 15.0, 15.9 and 16.1% to HOMO in MPBID1-MPBID6, respectively. Fragment 2 contributed 48.9, 44.4, 44.0, 13.0, 38.5 and 30.1% to LUMO and 79.9, 79.9, 79.4, 84.1, 83.2 and 83.0% to HOMO, respectively. In the same manner, fragment 3 contributed 1.9, 0,1, 0.1, 0.0, 0.0% and 0.0% to LUMO and 0.8, 0.8, 0.8, 0.9, 0.9 and 0.9% to HOMO in MPBID1-MPBID6, respectively. Thus, major charge distribution contribution of HOMO was present in fragment 2 and for LUMO major contribution of charge distribution was present in fragment 1. As shown in DOS pictographs, for reference compound, the highest density of the HOMO was present in fragment 2 and the highest density of LUMO was present in fragment 3. For the designed compounds, fragment 2 exhibited a maximum charge cloud on the HOMO at -6 eV while fragment 1 showed the greatest peak of charge density on the LUMO at -2 eV. Thus, the DOS pictograph shows the transfer of charge from fragment 2 to fragment 1 in all MPBID1-MPBID6. In MPBID4, the highest charge density (84.1%) on HOMO was occupied by fragment 2, and the highest charge density (87.0%) on LUMO was occupied by fragment 1. All this charge distribution factor reveals that MPBID4 transfers the charge proficiently among all other designed compounds.

DOS of reference and designed compounds (MPBID1-MPBID6) drawn by utilizing PyMOlyze 1.1 version (https://sourceforge.net/projects/pymolyze/).
For the compounds MPBID1′-MPBID6′, DOS values were demonstrated in Table S60. Fragment 1 contributed 50.7, 50.7, 50.7, 0.2, 49.4 and 0.5% to LUMO and 18.8, 18.7, 18.8, 18.9, 18.8 and 18.8% to HOMO in chromophores MPBID1′-MPBID6′, respectively. Fragment 2 contributed 49.1, 48.7, 49.2, 1.7, 48.2 and 3.1% to LUMO and 80.3, 78.1, 80.3, 79.8, 80.2 and 80.2% to HOMO, respectively. Fragment 3 contributed 0.2, 0.6, 0.1, 98.1, 2.3 and 96.5% to LUMO and 0.9, 2.4, 0.9, 1.3, 1.0 and 1.0% to HOMO in MPBID1′-MPBID6′, respectively. The chromophores are divided into 3 fragments. These 3 fragments (fragment 3, fragment 2 and fragment 1) were shown by blue, green and red lines in the pictographs, respectively. Figure 8 is the graphical representation of the DOS of compounds MPBID1′-MPBID6′. For the compounds MPBID1′-MPBID6′, fragment 2 exhibited a maximum electronic cloud on the HOMO at -6 eV while fragment 1 showed the greatest peak of on the LUMO at -3 eV. Thus, the DOS pictograph showed the transfer of charge from fragment 2 to fragment 1 in MPBID1′-MPBID6′. Thus, in all designed compounds DOS calculation showed that charge was efficiently transferred from HOMO to LUMO.

DOS of designed compounds (MPBID1′-MPBID6′) drawn by utilizing PyMOlyze 1.1 version (https://sourceforge.net/projects/pymolyze/).
Transition density matrix (TDM)
Transition density matrix (TDM) is employed for the explanation of various transition processes like intramolecular charge transfer movement in reference and designed compounds. The behavior of transitions from the ground state (S0) to the excited state (S1), electron hole localization and electron excitations can also be calculated by TDM analysis65,67. TDM plots of the designed compounds were computed using the M06 functional with the 6-311G(d, p) basis set. The intensity scale which signifies the charge transfer density from lowest to highest value is displayed on the right side of TDM maps. Hydrogen atoms were excluded from the TDM study due to their minimal contribution to electronic transitions. For TDM analysis, the designed compounds were divided into three fragments: fragment 1, fragment 2, and fragment 3. These fragments are represented by black, green, and red lines, respectively. The TDM spectra of the reference and designed compounds are shown in Fig. 9.

TDM heat maps of reference and designed compounds (MPBID1-MPBID6). These were drawn with the help of Multiwfn 3.7software (http://sobereva.com/multiwfn/). All out put files of designed compounds were accomplished by Gaussian 16 version D.01 (https://gaussian.com/g16citation/).
In above Fig. 9, the TDM heat maps display the nature of transitions of the MPBIR and MPBID1-MPBID6. It can be shown that the charge was transferred diagonally from fragment 2 to fragment 1. The majority of charge coherence was seen in fragment 1 and fragment 2 while no charge was observed in fragment 3. The green and red spots signify higher electronic clouds in TDM heat maps. MPBID4 shows excellent charge transfer at fragment 1, this behavior was due to the presence of a highly electron deficient group (-NO).
To understand the charge transfer of MPBID1′-MPBID6′, their fragments were also divided into 3 portions i.e. fragment 1, fragment 2 and fragment 3 characterized by black, green and red lines (Fig. 10). There was efficient charge transfer coherence in compounds MPBID1′-MPBID3′ but MPBID4′-MPBID6′ compounds showed the higher electronic cloud expressed by red and green spots. In these designed compounds, the electronic charge coherence was observed both diagonally and off diagonally all over the TDM map. From the TDM map, it has been clear that all the designed compounds have a continuous flow of charge mobility inside the molecule. Thus, these designed compounds are found to be unique and effective with great potential to be used in the development of NLO materials.

TDM heat maps of designed compounds (MPBID1′-MPBID6′). These were drawn with the help of Multiwfn 3.7software (http://sobereva.com/multiwfn/). All out put files of designed compounds were accomplished by Gaussian 16 version D.01 (https://gaussian.com/g16citation/).
Exciton binding energy (E b)
Exciton binding energy (Eb) is another important method that is used for the analysis of optoelectronic properties of compounds68. Coulombic forces of interaction between hole and electron can also be calculated by binding energy. The strength of Eb and coulombic interactions shows direct correlation but forms inverse correlation with the exciton dissociation process69,70. Less binding energy values along with lower columbic interactions results in higher exciton dissociation rate. The binding energy exists as the numeric difference between electronic energy (EH−L) and optical energy gap (Eopt)71. Whereas, electronic energy is the HOMO-LUMO energy gap and Eopt is the first smallest excitation energy. The theoretical estimation of Eb of the designed compounds is calculated by the given Eq. 1.
.
Eb is exciton binding energy, EH−L is energy energy gap and Eopt is the smallest quantity of energy required for first excitation from the ground state (S0) to the excited state (S1)72.
Table 7 illustrates the binding energy values of compounds MPBID1-MPBID6. The Eb values of reference and designed compounds are 0.875, 0.877, 0.878, 0.877, 0.704, 0.752 and 0.704eV respectively. These results indicate that the designed derivatives have lower binding energy values than the reference compound. Decreasing order of binding energy values is MPBID2 > MPBID1 = MPBID3 > MPBIDR > MPBID5 > MPBID4 = MPBID6. The lowest Eb value was found in MPBID4 and MPBID6 (Eb = 0.704eV) which means these compounds have a higher degree of maximum charge dissociation potential in contrast to other chromophores.
According to data mentioned in Table 8, the Eb values of MPBIDR and MPBID1′-MPBID6′ were 0.875, 0.876, 0.868, 0.866, 0.669, 0.854 and 0.813eV respectively. All entitled compounds have smaller value of binding energy as compared to reference compounds except MPBID1′. The decreasing order of binding energy of the designed compound is detected as MPBID1′> MPBIDR′> MPBID2′> MPBID3′> MPBID5′> MPBID6′> MPBID4′. MPBID4′ has the lowest binding energy value of 0.669eV among all other designed compounds.
As we compare the binding energy values of both possibilities of designed compounds (MPBID1-MPBID6 and MPBID1′-MPBID6′) has been shown that almost all the values were comparably the same except for MPBID4 and MPBID4′. The Eb value of MPBID4 is 0.704eV whereas the Eb value of MPBID4′ is 0.669eV which means MPBID4′ compound shows the smallest Eb value with the greatest charge dissociation potential among all the designed compounds. Thus, MPBID4′ is a potential candidate for non-linear optic materials. The compounds that have binding energy values lower than 1.9eV are considered perfect photonic materials with significant NLO response73. Interestingly, all our designed compounds have Eb values lower than 1.9eV, which means they may have promising optical activity and might be used for various NLO applications.
Molecular electrostatic potential (MEP)
The MEP surface uses a color band to show the positive, negative, and neutral electrostatic potentials23. Whereas blue indicates the probability of nucleophilic attack, red indicates the possibility of electrophilic attack. The order in which the electrostatic potential increases is red < orange < yellow < green < blue.74 As shown in FiguresS7 and S8, MEP analysis was carried out at the M06/6-311G (d, p) theoretical level to predict the nucleophilic and electrophilic sites of the suggested compounds in order to assess their reactivity. The oxygen and nitrogen atoms are surrounded by the most important negative potential zone (red). Therefore, among all the derivatives, oxygen atoms are the most favorable site for electrophilic assault. However, the blue color primarily indicates the presence of hydrogen and carbon atoms, suggesting that the nucleophile is likely to bind at these sites. Yellow indicates the positions of the midway potential, which is situated above the aromatic ring and lies between the extremes (red and blue) and the average (green).
Non-linear optics (NLO)
NLO study develops rapidly due to its applications in different fields like medical, electronics, telecom, optic memory systems and optic transmission modulation10. In comparison of inorganic molecules, organic compounds exhibit higher capacity for developing strong NLO materials75. Thus, organic compounds revealed significant applications in the field of electrochemical sensors, optical devices76photonic devices77photonic materials78biomedicines and signal manipulation79. In organic compounds, NLO response is established due to the pull and push mechanism of compounds depending on the nature of the donor, acceptor and pi-spacer. According to the literature, polarizability is influenced by the energy gap (∆E). Compounds with smaller energy gaps possessed high polarizability and hyperpolarizability values. Dipole moment (µtotal)80polarizability (<α> )81, first-order hyperpolarizability (βtotal)81 and second-order hyperpolarizability (γtotal)81 are NLO parameters that are attained by M06 functional with 311G(d, p) basis set. These NLO parameters for MPBID1-MPBID6 were calculated using Eqs. 2–5.
As tabulated in Table 9, the µtotal of the MPBIDR was 4.964D while dipole moment values of MPBID1-MPBID6 were 4.9709, 5.1204, 5.0709, 8.1239, 5.6057 and 7.3023D respectively. Interestingly, all the designed compounds have larger dipole moment values as compared to the reference compound. The enhancement in the value of dipole moment may be due to the introduction of different electron withdrawing groups at acceptor moieties. In the designed compounds, the highest dipole moment value was found in MPBID4 with 8.1239D while the lowest dipole moment value was found in MPBID1 with 4.9709D value. The decreasing order of dipole moment was MPBID4 > MPBID6 > MPBID5 > MPBID2 > MPBID3 > MPBID1 > MPBIRR. A greater dipole moment(µtotal) value leads to greater ICT capability of the compound. As MPBID4 has the highest dipole moment value (8.1239D) showed a greater electron transportation rate among all other designed compounds.
Electrical characteristics of the organic compounds are determined by polarizability values (<α> ). The linear polarizability value of the MPBIDR was 5.09 × 10− 23 esu. while the polarizability values of the MPBID1-MPBID6 were 5.08 × 10− 23, 5.42 × 10− 23, 5.57 × 10− 23, 5.46 × 10− 23, 5.17 × 10− 23 and 5.43 × 10− 23 esu. respectively. All the designed compounds have comparable < α > values with reference compounds. The decreasing order of linear polarizability was MPBID3 > MPBID4 > MPBID6 > MPBID2 > MPBID5 > MPBIDR > MPBIR1. The βtotal value of reference and designed compounds were 1.48 × 10− 29, 1.15 × 10− 29, 1.35 × 10− 29, 1.26 × 10− 29, 1.10 × 10− 29, 3.30 × 10− 29 and 5.09 × 10− 29 esu. respectively. The decreasing order of βtotal value was MPBID4 > MPBID6 > MPBID5 > MPBIDR > MPBID2 > MPBID3 > MPBIR1.
The γtotal values of the reference and designed compounds were 1.15 × 10− 34,1.13 × 10− 34, 1.40 × 10− 34, 1.51 × 10− 34, 3.08 × 10− 34, 1.03 × 10− 34 and 1.60 × 10− 34 esu. respectively. All the designed compounds have greater γtotal values than that of the reference compound. The decreasing order of γtotal values was MPBID4 > MPBID6 > MPBID3 > MPBID2 > MPBIDR > MPBID1 > MPBIR5.
According to data illustrated in Table 10, dipole moment values of the reference and designed compounds were 4.9643, 4.2449, 4.1417, 4.1583, 5.3302, 4.4368 and 5.252D respectively. Interestingly, all the designed compounds have larger dipole moment values as compared to the reference compound. The decreasing order of dipole moment was MPBID4′ > MPBID6′ > MPBIDR′ > MPBID5′ > MPBID1′ > MPBID3′ > MPBID2′. The highest dipole moment value was found in MPBID4′ with 5.3302D while the lowest dipole moment value was found in MPBID2′ with 4.1417D value. < α > values of MPIBR and MPBID1′-MPBID6′ were 5.09 × 10− 23, 5.08 × 10− 23, 5.41 × 10− 23, 5.56 × 10− 23, 5.22 × 10− 23, 5.32 × 10− 23 and 5.53 × 10− 23 esu. respectively. Increasing order of < α > values of designed compounds was MPBIR3′> MPBID6′> MPBID2′> MPBID5′> MPBID4′> MPBIDR′> MPBID1′. The βtotal value of reference and designed compounds were 1.48 × 10− 29, 1.69 × 10− 29, 1.67 × 10− 29, 1.77 × 10− 29, 1.55 × 10− 29, 2.06 × 10− 29 and 3.57 × 10− 29 esu. respectively. Increasing order of βtotal values of designed compounds was as follows: MPBIR6′> MPBID5′> MPBID3′> MPBID1′> MPBID2′> MPBID4′> MPBIDR′. All the designed compounds have greater βtotal values than the reference compound. The γtotal values of the reference and designed compounds were 1.15 × 10− 34, 1.14 × 10− 34, 1.52 × 10− 34, 1.67 × 10− 34, 8.85 × 10− 35, 1.43 × 10− 34 and 2.13 × 10− 34 esu. respectively. The results are further compared with paranitroanaline as paranitroanaline is considered as a standard compound (βtotal = 6.46 × 10− 30 e.s.u.) for exploration of hyperpolarizability properties71. All the designed compounds have greater hyperpolarizability values than paranitroanaline. This paranitroanaline related inspection revealed that all the designed compounds are most efficient for use in NLO materials.
To evaluate the efficiency of the designed chromophores, a comparative analysis was conducted with 6-aminoquinoline (6AQ) 82, a well-known benchmark molecule in NLO studies. The β_total value of 6AQ is reported to be 6.46 × 10⁻³⁰ esu. Remarkably, all designed compounds exhibit significantly enhanced first-order hyperpolarizability values in comparison. Among them, MPBID4 (11.0 × 10⁻²⁹ esu) and MPBID6′ (3.57 × 10⁻²⁹ esu) showed the most prominent improvement, surpassing the reference by nearly one order of magnitude. This substantial enhancement can be attributed to the extended π-conjugation and optimized donor–π–acceptor architecture, which facilitate stronger intramolecular charge transfer. These findings confirm that the structural modifications applied in the present study have effectively amplified the NLO response and demonstrate the potential of the designed molecules as superior candidates for nonlinear optical applications compared to conventional systems like 6AQ. Tables S61-S68 (supplementary information) provide the major contributing tensor values of designed compounds.
Conclusion
In this study, a series of organic molecules (MPBID1–MPBID6 and MPBID1′–MPBID6′) were designed by structural modification of a reference compound with various electron-withdrawing end-capped acceptor units. MPBID1′–MPBID6′ featured substitutions at fragment 3, while MPBID1–MPBID6 were modified at fragment 1. The inclusion of electron-withdrawing groups (-F, -Cl, -Br, -NO₂, -CF₃) significantly enhanced the optoelectronic properties of these compounds compared to the reference molecule, MPBIDR. Quantum chemical analyses revealed that these designed molecules exhibited smaller energy gap values, improved charge transfer rates (evident from FMO analysis, DOS, and TDM heat maps), and higher softness (GRP data). The compounds also demonstrated prominent absorption in the visible region with low transition energy. Notably, MPBID4 (3.079 eV) and MPBID4′ (2.742 eV) showed the smallest energy energy gaps and maximum redshifts (325.00 nm and 337.83 nm, respectively), attributed to the strong electron-withdrawing effects of the nitro (-NO₂) group. Furthermore, MPBID4′ outperformed MPBID4 due to the direct fusion of the nitro group at fragment 3 with the reference core, enabling enhanced resonance effects. Moreover, the NLO analysis revealed that all designed compounds exhibited high values for total dipole moment (µtotal), average polarizability (<α> ), first hyperpolarizability (βtotal), and second hyperpolarizability (γtotal), confirming superior nonlinear optical characteristics. Among them, MPBID4′ emerged as the most promising candidate due to its unique NLO properties. In conclusion, this computational study highlights that the designed molecules possess excellent optoelectronic and NLO properties, indicating their potential utility in future applications across nonlinear optics and related fields.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Lakhera, S., Rana, M. & Dhuliya, V. Enhancement of the electronic and optical properties of superalkali metal adsorbed Al10N10 nanocage. Mater. Sci. Semiconduct. Process. 185, 108882 (2025).
Garmire, E. Nonlinear optics in daily life. J. O. e. Optics Express, 21, 30532-30544 (2013).
Khalid, M. et al. Promising impact of push–pull configuration into designed octacyclic naphthalene-based organic scaffolds for nonlinear optical amplitudes: A quantum chemical approach. Sci. Rep. 13, 20104 (2023).
Peng, Z. & Yu, L. J. M. Second-order nonlinear optical polyimide with high-temperature stability. Macromolecules, 27, 2638–2640 (1994).
Tsutsumi, N., Morishima, M. & Sakai, W. J. M. Nonlinear optical (NLO) polymers. 3. NLO polyimide with dipole moments aligned transverse to the Imide linkage. Macromolecule, 31, 7764–7769 (1998).
Marinescu, M. et al. Synthesis, characterization and antimicrobial activities of some schiff bases with Non-Linear optical applications. Chem. Proceed. 7, 2022007040 (2022).
Lakhera, S. et al. Enhanced nonlinear optical and optical limiting responses of 7-diethylamino-4-methyl coumarin functionalized with silver nanoparticles: A combined experimental and DFT study. J. Photochem. Photobiol., A. 457, 115910 (2024).
Zyss, J. & Ledoux, I. Nonlinear optics in multipolar media: theory and experiments. Chemical reviews, 94, 77-105 (1994).
Sreedharan, R., Ravi, S., Raghi, K. R., Kumar, T. M. & Naseema, K. Growth, linear-nonlinear optical studies and quantum chemistry formalism on an organic NLO crystal for opto-electronic applications: experimental and theoretical approach. SN Appl. Sci. 2, 1–18 (2020).
Ahmad, M. S. et al. Synthesis and XRD, FT-IR vibrational, UV–vis, and nonlinear optical exploration of novel tetra substituted imidazole derivatives: A synergistic experimental-computational analysis. Journal of Physics and Chemistry of Solids, 115, 265–276 (2018).
Akram, M. et al. A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties. Journal of Molecular Structure, 1160, 129–141 (2018).
Franken, P., Hill, A. E., Peters, C. & Weinreich, G. Generation of optical harmonics. Physical review letters, 7, 118 (1961).
Ivanova, B. & Spiteller, M. Possible application of the organic barbiturates as NLO materials. Crystal growth & design, 10, 2470-2474 (2010).
Lakhera, S., Devlal, K. & Rana, M. Utilization of Methyclothiazide adsorbed with malonamide for quantum chemical applications: A DFT and DFT-D2/D3 studies. Optik 295, 171485 (2023).
Yaqoob, J. et al. Optimized nonlinear optical (NLO) response of silicon carbide nanosheet by alkali metals doping: A DFT insight. The European Physical Journal Plus,137, 233 (2022).
Yan, C. et al. Non-fullerene acceptors for organic solar cells. Nature Reviews Materials, 3, 1–19 (2018).
Wang, Y., Tam, W., Stevenson, S., Clement, R. & Calabrese, J. J. C. p. L. New organic non-linear optical materials of Stilbene and diphenylacetylene derivatives. Chemical Physics Letters, 148, 136–141 (1988).
Tam, W., Wang, Y., Calabrese, J. C. & Clement, R. A. Organometallics and organics for nonlinear optics: new material for second harmonic generation. In Nonlinear Optical Properties of Organic Materials. Vol. 971. 107-112 . (SPIE, 1988).
Liu, L., Young, J., Smeu, M. & Halasyamani, P. S. Ba4B8TeO19: A UV nonlinear optical material. Inorg. Chem. 57, 4771–4776 (2018).
Yamashita, S. A tutorial on nonlinear photonic applications of carbon nanotube and graphene. J. Lightwave Technol. 30, 427–447 (2011).
Guo, L., Guo, Z. & Li, X. Design and preparation of side chain electro-optic polymeric materials based on novel organic second order nonlinear optical chromophores with double carboxyl groups. J. Mater. Sci. Mater. Electron. 29, 2577–2584 (2018).
Khan, M. U. et al. Quantum chemical designing of Indolo [3, 2, 1-jk] carbazole-based dyes for highly efficient nonlinear optical properties. Chem. Phys. Lett. 719, 59–66 (2019).
Lakhera, S., Rana, M. & Devlal, K. Comprehensive quantum chemical study of the associative complex of para-aminobenzoic acid and 7-diethylamino 4-methyl coumarin by adsorption and aromatic bridges. J. Mol. Model. 30, 37 (2024).
Khalid, M. et al. Theoretical approach towards benzodithiophene-based chromophores with extended acceptors for prediction of efficient nonlinear optical behaviour. Arab. J. Sci. Eng. 49, 339–359 (2024).
Kumar, P. R., Ravindrachary, V., Janardhana, K. & Poojary, B. Structural and optical properties of a new chalcone single crystal. J. Cryst. Growth. 354, 182–187 (2012).
Janjua, M. R. S. A. Quantum mechanical design of efficient second-order nonlinear optical materials based on heteroaromatic imido-substituted hexamolybdates: First theoretical framework of POM-based heterocyclic aromatic rings. Inorg. Chem. 51, 11306–11314 (2012).
Janjua, M. R. S. A. et al. Solvent-dependent non-linear optical properties of 5, 5′-disubstituted-2, 2′-bipyridine complexes of ruthenium (II): a quantum chemical perspective. Aust. J. Chem. 68, 1502–1507 (2015).
Janjua, M. R. S. A. et al. First principle study of electronic and non-linear optical (NLO) properties of triphenylamine dyes: interactive design computation of new NLO compounds. Aust. J. Chem. 69, 467–472 (2015).
Etabti, H. et al. Computational investigation of the stability and efficiency of Indenoindene-based A-π-D-π-A type compounds in organic photovoltaics cells. Phys. Chem. Res. 12, 1019–1035 (2024).
Mir, M. A. DFT analysis for structure, vibrational assignments, and molecular properties of medicinally important isoquinoline. J. Dispers. Sci. Technol. 1–11 (2024).
Obata, A., Sasagawa, A., Yamazaki, K., Ano, Y. & Chatani, N. J. C. Nickel-catalyzed oxidative C–H/N–H annulation of N-heteroaromatic compounds with alkynes. Chem. Sci. 10, 3242–3248 (2019).
Frisch, M. et al. Gaussian 16 Revision C. 01, 2016. 1, 572 (2016).
Barone, V. & Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A. 102, 1995–2001 (1998).
O’boyle, N. M., Tenderholt, A. L. & Langner, K. M. J. Cclib: a library for package-independent C.mputational C.emistry algorithms. J. Comput. Chem. 29, 839–845 (2008).
Lu, T. & Chen, F. J. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Hanwell, M. D. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1–17 (2012).
Dennington, R., Keith, T., Millam, J. & Wallingford, J. I. GaussView 5.0. Gaussian 20 (2008).
Zhurko, G. & Zhurko, D. ChemCraft, version 1.6. (2009).
Uzun, S., Esen, Z., Koç, E., Usta, N. C. & Ceylan, M. J. Experimental and density functional theory (MEP, FMO, NLO, Fukui functions) and antibacterial activity studies on 2-amino-4-(4-nitrophenyl)-5, 6-dihydrobenzo [h] quinoline-3-carbonitrile. J. Mol. Struct. 1178, 450–457 (2019).
Khalid, M., Lodhi, H. M., Khan, M. U. & Imran, M. Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: A DFT approach. RSC Adv. 11, 14237–14250 (2021).
Qin, C. & Clark, A. E. J. C. P. L. DFT characterization of the optical and redox P.operties of natural P.gments relevant to dye-sensitized solar cells. Chem. Phys. Lett. 438, 26–30 (2007).
Rasool, F. et al. Heterocyclic donor moiety effect on optical nonlinearity behavior of chrysene-based chromophores with push–pull configuration via the quantum chemical approach. ACS Omega. 9, 3596–3608 (2024).
Amiri, S. S., Makarem, S., Ahmar, H. & Ashenagar, S. J. Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1, 3, 4-oxadiazol-2-yl) thio) benzene-1, 2-diol. J. Mol. Struct. 1119, 18–24 (2016).
Ferdowsi, P. et al. Molecular design of efficient organic D–A––A dye featuring triphenylamine as donor fragment for application in dye-Sensitized solar cells. ChemSusChem 11, 494–502 (2018).
Khan, M. U. et al. Theoretical investigation of nonlinear optical behavior for rod and T-Shaped phenothiazine based D-π-A organic compounds and their derivatives. J. Saudi Chem. Soc. 25, 101339 (2021).
Parr, R. G., Szentpály, L. & Liu, S. J. Electrophilicity index. 121, 1922-1924 (1999).
Dai, X., Dong, B., Ren, M. & Lin, W. J. Unique D–π–A–π–D type fluorescent probes for the two-photon imaging of intracellular viscosity. J. Mater. Chem. B. 6, 381–385 (2018).
Yamada, S., Bessho, J., Nakasato, H., Tsutsumi, O. Color tuning donor–acceptor-type Azobenzene dyes by controlling the molecular geometry of the donor moiety. Dyes Pigm. 150, 89–96 (2018).
Fukui, K. Role of frontier orbitals in chemical reactions. Science 218, 747-754 (1982).
Lesar, A. & Milošev, I. Density functional study of the corrosion Inhibition properties of 1, 2, 4-triazole and its amino derivatives. Chem. Phys. Lett. 483, 198–203 (2009).
Kovačević, N. & Kokalj, A. J. C. Analysis of molecular electronic structure of imidazole-and benzimidazole-based inhibitors: a simple recipe for qualitative Estimation of chemical hardness. Corros. Sci. 53, 909–921 (2011).
Chattaraj, P. K., Duley, S. & Domingo, L. R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 10, 2855–2861 (2012).
Koopmans, T. J. p. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. 1, 104-113 (1934).
Khalid, M. et al. First principles study of electronic and nonlinear optical properties of A–D–π–A and D–A–D–π–A configured compounds containing novel quinoline-carbazole derivatives. 10, 22273–22283 (2020).
Rashid, M. et al. Multicomponent synthesis of pyrido [2, 3-b] pyrazine derivatives: electrochemical DNA sensing, nonlinear optical properties and biological activity. Physica 13, 32160–32174 (2023).
Mahmood, A., Khan, S. U. D. & ur Rehman, F. J. Assessing the quantum mechanical level of theory for prediction of uv/visible absorption spectra of some aminoazobenzene dyes. J. Saudi Chem. Soc. 19, 436–441 (2015).
Karakas, A., Elmali, A., Unver, H. Linear optical transmission measurements and computational study of linear polarizabilities, first hyperpolarizabilities of a dinuclear iron (III) complex. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 68, 567–572 (2007).
Rafiq, M. et al. Synthesis, XRD, spectral (IR, UV–Vis, NMR) characterization and quantum chemical exploration of benzoimidazole-based hydrazones: A synergistic experimental‐computational analysis. Appl. Organomet. Chem. 33, e5182 (2019).
Weinhold, F. & Landis, C. R. Practice. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2, 91–104 (2001).
Dhamodharan, P., Sathya, K., Dhandapani, M. J. J. & o., M. S. Physico-chemical characterization, density functional theory (DFT) studies and Hirshfeld surface analysis of a new organic optical material: 1H-benzo [d] imidazol-3-ium-2, 4, 6-trinitrobenzene-1, 3 Bis (olate). J. Mol. Struct. 1146, 782–792 (2017).
Nan, M. I. et al. Mono-and di-substituted pyrene-based donor-π-acceptor systems with phenyl and Thienyl π-conjugating bridges. Dyes Pigm. 181, 108527 (2020).
Khalid, M. et al. Non-covalent interactions abetted supramolecular arrangements of N-Substituted benzylidene acetohydrazide to direct its solid-state network. J. Mol. Struct. 1230, 129827 (2021).
Reed, A. E., Weinhold, F., Weiss, R. & Macheleid, J. J. T. Nature of the contact ion pair trichloromethyl-chloride (CCl3+ Cl-). A theoretical study. J. Phys. Chem. 89, 2688–2694 (1985).
Khalid, M. et al. Frontier molecular, natural bond orbital, UV-Vis spectral stduy, solvent influence on geometric parameters, vibrational frequencies and solvation energies of 8-Hydroxyquinoline. Int. J. Pharm. Sci. Res. 8, 457–469 (2017).
Khalid, M. et al. Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 25, 101305 (2021).
Ans, M. et al. Designing three-dimensional (3D) non‐fullerene small molecule acceptors with efficient photovoltaic parameters. ChemistrySelect 3, 12797–12804 (2018).
Shafiq, I. et al. The impact of structural modifications into benzodithiophene compounds on electronic and optical properties for organic solar cells. Mater. Chem. Phys. 308, 128154 (2023).
ul Ain, Q. et al. Designing of benzodithiophene acridine based donor materials with favorable photovoltaic parameters for efficient organic solar cell. Comput. Theor. Chem. 1200, 113238 (2021).
Khan, M. U. et al. Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high‐efficiency organic solar cells. Int. J. Quantum Chem. 120, e26377 (2020).
Ans, M., Iqbal, J., Ayub, K., Ali, E. & Eliasson, B. Spirobifluorene based small molecules as an alternative to traditional fullerene acceptors for organic solar cells. Mater. Sci. Semiconduct. Process. 94, 97–106 (2019).
Kanis, D. R., Ratner, M. A. & Marks, T. J. J. C. R. Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 94, 195–242 (1994).
Kim, B. G., Zhen, C. G., Jeong, E. J., Kieffer, J. & Kim, J. Organic dye design tools for efficient photocurrent generation in dye-sensitized solar cells: exciton binding energy and electron acceptors. Adv. Funct. Mater. 22, 1606–1612 (2012).
Khalid, M. et al. Exploration of efficient electron acceptors for organic solar cells: rational design of Indacenodithiophene based non-fullerene compounds. Sci. Rep. 11, 19931 (2021).
Mahalakshmi, G., Balachandran, V., NBO, HOMO & LUMO analysis and vibrational spectra (FTIR and FT Raman) of 1-amino 4-methylpiperazine using ab initio HF and DFT methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 135, 321–334 (2015).
Bi, W. et al. A switchable NLO Organic-Inorganic compound based on conformationally chiral disulfide molecules and Bi (III) I5 Iodobismuthate networks. Adv. Mater. 20, 1013–1017 (2008).
Adant, C., Dupuis, M., Bredas, J. Ab initio study of the nonlinear optical properties of urea: electron correlation and dispersion effects. Int. J. Quantum Chem. 56, 497–507 (1995).
Morks, M. Magnesium phosphate treatment for steel. Mater. Lett. 58, 3316–3319 (2004).
Iwan, A. & Sek, D. Polymers with triphenylamine units: photonic and electroactive materials. Prog. Polym. Sci. 36, 1277–1325 (2011).
Margar, S. N. & Sekar, N. Nonlinear optical properties of curcumin: solvatochromism-based approach and computational study. Mol. Phys. 114, 1867–1879 (2016).
Kromann, J. C., Steinmann, C. & Jensen, J. H. Improving solvation energy predictions using the SMD solvation method and semiempirical electronic structure methods. J. Chem. Phys. 149 (2018).
Ullah, F., Ayub, K. & Mahmood, T. Remarkable second and third order nonlinear optical properties of organometallic C 6 Li 6-M 3 O electrides. New J. Chem. 44, 9822–9829 (2020).
Pandey, N., Mehata, M. S., Pant, S. & Tewari, N. Structural, electronic and NLO properties of 6-aminoquinoline: a DFT/TD-DFT study. J. Fluoresc. 31, 1719–1729 (2021).
Acknowledgements
The work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP 2502).
Author information
Authors and Affiliations
Contributions
Iqra Shafiq: Conceptualization, Data curation; formal analysis, writing, reviewing and editing. visualization. Nadeem Raza: Conceptualization; methodology; Formal Analysis; Data curation; visualization; Funding acquisition. Saba Dildar: Data curation; formal analysis, writing & reviewing original draft. Saifullah Bullo: Methodology; Investigation; software; project administration; resources; validation; supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shafiq, I., Raza, N., Dildar, S. et al. Unraveling the NLO potential of isoquinoline functionalized chromophores via molecular modeling using DFT/TD-DFT approaches. Sci Rep 15, 23236 (2025). https://doi.org/10.1038/s41598-025-04911-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04911-7
