
Abstract
Vitamin B12 (VB12) is essential for human health, and its deficiency can lead to various health issues, including anemia and neurological problems. Additionally, the presence of antibiotic resistance genes in various environments raises concerns about the spread of antibiotic resistance. This study aims to explore potential connections between these two functional gene categories in freshwater environments, which are closely intertwined with human health. We conducted a comprehensive metagenomic sequencing analysis across 23 sampling points from five different eutrophic urban lakes. The results highlight the dominance of the precorrin-2 synthesis pathway in the microbial synthesis of VB12 within urban lakes. Eutrophication may potentially enhance the precorrin-2 synthesis pathway while inhibiting others. The risk of antibiotic resistance is reduced in the anaerobic pathway of VB12 synthesis, while the abundance of metal resistance genes is increased. Binning analysis reveals that 26 metagenome-assembled genomes (MAGs) actively participate in VB12 synthesis, with at least 4 MAGs showing resistance during the VB12 synthesis process and demonstrating pathogenicity. These findings provide critical insights into the VB12 synthesis process and its implications for human health in terms of resistance risks.
Similar content being viewed by others


Introduction
Cobalamin, commonly known as vitamin B12 (VB12), is an essential micronutrient for nearly all living organisms in Earth’s biosphere1,2. Beyond its critical role in natural ecosystems, VB12 serves as a vital cofactor in key metabolic processes, including DNA synthesis and nervous system function, particularly in mammalian hosts such as the human gut microbiome3. However, most animals lack the biosynthetic capacity to produce VB12 de novo4, necessitating its acquisition either from dietary sources or through microbial synthesis—primarily by select bacteria and archaea, provided sufficient cobalt is bioavailable5.
The microbial synthesis of VB12 is a highly complex, enzymatically driven process that proceeds via two distinct carbon feedstock pathways: five-carbon glutamate or four-carbon glycine6,7. This biosynthesis involves a series of condensation, dehydrogenation, and amination reactions, culminating in the formation of precorrin-2—a pivotal intermediate generated through either aerobic or anaerobic routes6,7. Subsequent enzymatic steps, including ring closure, deacetylation, and cobalt incorporation mediated by methyltransferases, yield cob(II)alamin, which is further metabolized into active VB12 via the adenosylcobalamin (AdoCbi) pathway6,7. Additionally, alternative routes such as the pseudocobalamin salvage pathway contribute to VB12 production8. Notably, VB12 deficiency in humans can disrupt methyl group metabolism, leading to severe neurological and hematological disorders such as methylmalonic aciduria and hyperhomocysteinemia9.
Concurrently, the global rise in antibiotic resistance represents one of the most pressing public health crises of the 21st century10. The proliferation of antibiotic resistance genes (ARGs) in environmental reservoirs—driven by both natural bacterial evolution and anthropogenic activities (e.g., clinical misuse, agricultural overuse)12,13,14,15—has accelerated the emergence of multidrug-resistant pathogens10,11. Environmental microbiota serve as dynamic hubs for ARG dissemination, facilitating horizontal gene transfer (HGT) among diverse bacterial taxa16,17. This genetic mobility, compounded by selective pressures from antibiotic pollution, threatens to render once-treatable infections increasingly recalcitrant to therapy18.
Despite the dual significance of VB12 biosynthesis and ARGs in microbial ecology and human health, their potential interplay remains unexplored. Given that both processes are predominantly mediated by prokaryotes, we hypothesize that ecological drivers (e.g., nutrient availability) and genetic linkages may concurrently shape their distributions. To test this, we employ urban lakes—anthropogenically influenced ecosystems—as model systems to address four key questions via metagenomics and geochemical analyses: (1) Distribution dynamics of VB12 synthesis genes across urban lakes of varying trophic states; (2) Environmental drivers modulating VB12-associated gene abundance; (3) Potential linkages between VB12 biosynthesis capacity and antibiotic resistome; (4) Genomic profiling of VB12-producing strains with ARG transmission potential. This research aspires to illuminate the intricate relationships among VB12 synthesis, microbial communities, and antibiotic resistance within urban lakes, offering valuable insights for comprehending and managing the repercussions of these processes on both human health and the environment.
Materials and methods
Study sites
In this study, a total of 23 surface water samples were systematically collected from five representative urban lakes in Wuhan, China. The two largest urban lakes in China, Tangxunhu (TXH) and Donghu (DH), characterized as mesoeutrophic, boast expansive water areas of 47.6 km2 and 33.9 km2respectively. Yanxihu (YXH), identified as a eutrophic lake, encompasses a water area of 11.8 km2. In contrast, Nanhu (NH) and Shahu (SH), both hypereutrophic lakes, are situated amidst hospitals, sewage treatment plants, universities, and a densely populated area, with water areas measuring 7.6 km2 and 3.1 km2respectively19,20. Sampling points were systematically distributed across each lake at intervals of approximately 1–5 km, with the spacing adjusted based on lake size to ensure representative coverage. Notably, Lake SH—being relatively small—was assigned only three sampling points, while all other lakes were sampled at five predetermined locations. Detailed information can be found in Fig. S1A.
Sampling, shotgun metagenomic sequencing and measurement of environmental factors
Total DNA was extracted using the DNeasy PowerWater Kit (QIAGEN, Hilden, Germany), The concentration and purity of the isolated DNA were assessed with a Qubit 3.0 fluorometer (Thermo Fisher Scientific, USA). Subsequently, a paired-end sequencing library (150 bp read length) was prepared and subjected to high-throughput sequencing on an Illumina Hiseq platform (Illumina, San Diego, CA) by Biomarker Technologies Corporation (Beijing, China) as previously study21. A multi-parameter probe (WTW 3430) was used to record in situ measurements of dissolved oxygen (DO), electrical conductivity (EC), pH. Laboratory analyses were conducted for nitrate, nitrite, ammonia, orthophosphate, dissolved organic carbon (DOC), and Chl a using standardized methods21. The detailed physiochemical parameters can be found in the supplementary material provided by Mao et al., in 2023.
Metagenome analysis
Raw metagenome sequencing reads underwent filtration to eliminate low-quality reads using FASTP22and were subsequently de novo assembled using MEGAHIT23with k-mer size ranging from 21 to 149. Assembled contigs longer than 500 bp were processed for open reading frame (ORF) prediction via Prodigal24and a non-redundant gene catalog was generated using CD-HIT (98% identity, 90% coverage)25. Gene abundance was normalized to transcripts per million (TPM) to enable cross-sample comparisons. This approach minimizes the impact of variations in total read count and gene length26. Functional annotation of predicted genes against the VB12Path database employed DIAMOND (with an E-value of ≤ 1e-5). All samples were rarified to the identical sequencing depth (33,055,308 sequences per sample) through random resampling. ARGs and metal resistance genes (MRGs) were annotated using the deepARG27 and metal resistance gene database28, respectively. The predicted ORFs were initially translated into amino acid sequences using the EMBOSS29. Subsequently, BLASTp was employed for annotation against the aforementioned databases, utilizing the same alignment thresholds as Mao et al., in 2023. Finally, MetaCompare was employed to evaluate the resistome risk by estimating the coexistence of ARGs, mobile genetic elements (MGEs), and human pathogens30.
Metagenomic Binning and metagenome-assembled genomes (MAGs) annotation
Genome assembly and binning procedures were executed using the MetaWRAP pipeline31. First, quality-filtered sequences were assembled into contigs with MEGAHIT23. Next, genome binning was performed on the assembled contigs with MetaBAT232, and the resulting bins were refined using the Bin_refinement module. These bins were then dereplicated with dRep33 at a 99% average nucleotide identity (ANI) threshold for strain-level clustering. The quality of the dereplicated bins was evaluated with CheckM34 based on completeness and contamination; only bins meeting the criteria of > 50% completeness and < 10% contamination were retained35. Taxonomic classification of the obtained MAGs was performed using GTDB-Tk36. Gene prediction for the MAGs was conducted with Prodigal, followed by functional annotation against the METABOLIC, VB12Path, KEGG, COG, pathogen-host interaction (PHI), CRISPR-Cas, mobileOG-db, and repeat VB12Path databases37,38,39,40.
Statistical analysis
The data were first subjected to logarithmic transformation, followed by Z-score normalization. Next, analysis of variance (ANOVA) was used to assess the statistical significance of differences in mean gene abundance among samples. Data were visualized using online tools (https://www.bioincloud.tech). Principal coordinate analysis (PCoA) was performed with the “amplicon” R package to evaluate β-diversity patterns of functional genes across lakes. Additionally, permutational multivariate analysis of variance (PERMANOVA, adonis) tested the significance of inter-group differences. To examine associations, Spearman rank correlation analysis was applied to (1) environmental factors vs. VB12 functional genes and (2) VB12 genes vs. resistance genes (ARGs/MRGs). Univariate linear regression further modeled relationships between VB12 genes and resistance risk. For visualization, phylogenetic trees were refined using iTOL (https://itol.embl.de/tree), and genome circular maps were generated with CGView (https://paulstothard.github.io/cgview).
Results and discussion
Spatial distribution variations of VB12 functional genes and environmental drivers
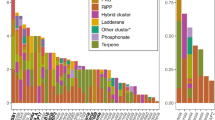
The recently developed VB12Path database provides a robust metagenomic platform to investigate cobalamin (VB12) biosynthesis across environmental and human microbiomes7. In our study, five functional genes critical for VB12 synthesis were identified across 23 sampling sites (Fig. 1A), annotated as: cobalt-precorrin-8 methylmutase (cbiC), uroporphyrinogen-III c-methyltransferase (cobA), precorrin-8X methylmutase (cobH), siroheme synthase (cysG), and glutamate-1-semialdehyde 2,1-aminomutase (hemL). These genes are involved in three pathways related to VB12 synthesis, namely anaerobic (cbiC), precorrin-2 synthesis (cysG, cobA, hemL), and the aerobic (cobH) pathway. Among them, the functional genes hemL and cobA, involved in the precorrin-2 synthesis pathway, dominated the gene pool (collectively 97.0% relative abundance), with hemL alone exceeding 70% in all lakes (Fig. 1A). This aligns with reports by Zhou et al. (2021), who observed high hemL abundance in marine and gut microbiomes7,8. Given its role in initiating VB12 synthesis7, hemL’s prevalence suggests a bottleneck effect in the pathway, where its activity may gatekeep downstream VB12 production.

Differential distribution characteristics of VB12 synthesis genes in different lakes. (A) Abundance distribution patterns of identified VB12 synthesis genes in the five lakes. (B) Variations in the distribution of VB12 synthesis genes among the five lakes (Data were standardized using Z-scores, the letters (a, b, ab, bc) denote homogeneous subsets based on Duncan’s multiple range test, where groups sharing no common letters are significantly different (p < 0.05).
Differential analysis revealed significant inter-lake variability (p < 0.01) for all genes except cysG (Fig. 1B; Tables S1–S2). Consistent with this, principal coordinates analysis (PCoA) based on the Bray-Curtis matrix further confirmed the spatial distribution differences of these functional genes (p < 0.01; Fig. 2A). Distinct separation was observed among NH, TXH, YXH, and SH lakes. The ADONIS test confirmed statistically significant differences between them (p < 0.05). These pronounced variations in functional genes were consistent with the significant distribution patterns of microbial communities21,41. Given the significant relationships between microbial communities and nutrients, we speculate that microbial VB12 synthesis in lakes may be associated with their eutrophication status. Therefore, a correlation analysis was conducted between all VB12 functional genes and environmental factors. CobA exhibited strong positive correlations with nitrate/nitrite (p < 0.05; Fig. 2B, Table S3), implicating nitrogen availability in precorrin-2 pathway activation. HemL showed inverse relationships with most measured factors (p < 0.05; Fig. 2B), suggesting suppression under eutrophic conditions. The previous research also has indicated that a deficiency in VB12 is associated with dietary, environmental, and genetic factors42. Nitrate nitrogen, nitrite nitrogen, and chlorophyll a—established indicators of aquatic eutrophication43,44, demonstrated significant correlations with VB12 synthesis-related genes in this study, suggesting their potential dichotomous regulation of VB12 biosynthesis. Stimulation of cobA by nitrogen influx, potentially accelerating early pathway steps. Concurrent repression of hemL, which dominates the gene pool (79.8%). We hypothesize that this paradox—nutrient enrichment favoring pathway initiation while limiting its keystone enzyme—could constrain net VB12 output. VB12 is essential for one-carbon metabolism and epigenetic regulation45. Its synthesis limitation may impair cellular reprogramming and tissue repair, particularly in ecosystems where eutrophication alters microbial niche partitioning. Future work should quantify VB12 titers directly to test whether gene abundance patterns translate to functional bottlenecks.

Distribution patterns and driving factors of functional genes among different lakes. (A) Principal coordinates analysis (PCoA) of VB12 functional genes based on the Bray-Curtis distances. The significance of the differential distribution was statistically tested by adonis. (B) Association analysis between VB12 functional genes and environmental factors (Spearman’s, ***p < 0.001; **p < 0.01; *p < 0.05).
Association between VB12 biosynthesis pathways and antimicrobial resistance: mechanistic insights and ecological implications
VB12 functional genes and ARGs represent inherent genomic features of prokaryotic organisms that have evolved through complex evolutionary processes. These genetic elements can confer either beneficial or detrimental traits to human health through diverse mechanisms46,47,48. In this study, we observed positive correlation between prokaryotic diversity and the abundance of four VB12 biosynthesis genes aligns with theoretical expectations (Fig. S1C). In addition, horizontal gene transfer (HGT) serves as a key evolutionary mechanism facilitating gene flux across diverse microorganisms and plays a critical role in the rapid dissemination of genetic traits within microbial communities49. Understanding potential associations between different gene categories is therefore essential for unveiling functional interactions within microbial systems. In this study, Spearman rank correlation was employed to investigate potential associations between VB12 functional genes and resistance genes (Fig. 3). The results indicated a significant negative correlation between the CbiC involved in the anaerobic pathway and ARGs (p < 0.05; Fig. 3A, Table S4). Furthermore, the CobH and CobA, participating in the aerobic pathway and precorrin-2 synthesis pathway, respectively, demonstrated a significant positive correlation with ARGs (p < 0.05; Fig. 3A, Table S4). This suggested a higher likelihood of co-occurrence between ARGs and CobH/CobA. Despite hemL exhibiting the highest abundance among all genes, no significant associations with any ARGs were observed. MGEs play a crucial role in the dissemination of ARGs in urban lakes21. The significant relationship observed in this study between ARGs and genes related to VB12 synthesis may suggest that MGEs, as crucial mediators of HGT50, may exhibit selectivity during the process of gene movement51.
It was noteworthy that the results of the correlation analysis revealed a significant positive correlation between all VB12 functional genes and diverse MRGs (p < 0.05, Fig. 3A, Table S5). Particularly, CbiC, CobH, and CobA exhibited a noteworthy positive correlation with the majority of MRGs (p < 0.05, Fig. 3A, Table S5), indicating that VB12-producing microbes may simultaneously evolve metal detoxification strategies. The metallation process during VB12 synthesis could structurally facilitate metal ion binding/sequestration52. This aligns with cellular-level evidence showing VB12 enzymes participate in metal coordination52. Furthermore, the results of the univariate linear regression analysis between VB12 functional genes and resistance risk indicated a significant negative correlation between CbiC and resistance risk (Fig. 3B, Fig. S1B). This suggests that the anaerobic pathway might compete for cellular resources with resistance mechanisms, thereby mitigating antimicrobial resistance development.

Correlation analysis between genes related to VB12 synthesis and resistance genes, as well as resistance risk. (A) The potential associations between VB12 functional genes and antibiotic resistance genes (ARGs) as well as metal resistance genes (MRGs) (Spearman’s, ***p < 0.001; **p < 0.01; *p < 0.05). (B) Regression analysis between VB12 functional genes and resistance risk.
Evolutionary analysis of MAGs co-harboring VB12 functional genes and resistance genes
The emergence of metagenomic binning techniques is paramount for the identification of genes with specific functions, such as resistance genes and pathway-related metabolic genes53. Based on these methodological advances, we conducted binning analysis across all samples, identifying 26 medium-to-high-quality MAGs (contamination rate less than 10%, completeness greater than 50%) associated with VB12 synthesis. These MAGs spanned 6 phyla, 9 classes, 13 orders, 15 families, and 21 genera (Fig. 4, Table S6), revealing distinct phylogenetic patterns in VB12 synthesis pathways. Specifically, 13 MAGs were assigned to Bacteroidota, involved in the salvage pathway, while 6 belonged to Proteobacteria, participated solely in precorrin-2 synthesis (Fig. 4, Table S6). This indicates distinct roles for Bacteroidota and Proteobacteria in the VB12 synthesis process. 4 MAGs associated with Cyanobacteria uniquely harbored three VB12 functional genes, engaging in both aerobic (cobB/cobM) and precorrin-2 pathways (hemC). This phylogenetic divergence aligns with existing literature. Proteobacteria are known to express vitamin biosynthesis genes54, while both Bacteroidota and Proteobacteria play established roles in biochar-driven composting55. Notably, cyanobacterial VB12 synthesis has been previously documented in planktonic species56, corroborating our findings.
All 26 MAGs carried ARGs (1–16 per MAG), with 18 concurrently hosting metal resistance genes (MRGs; 1–6 per MAG). Interestingly, MAGs involved in the precorrin-2 synthesis pathway carried more MRGs, while those participating in the salvage pathway exhibited more ARGs (Fig. 4, Table S6). This dichotomy suggests divergent evolutionary pressures on resistance gene co-occurrence: the precorrin-2 pathway may favor MRG retention, whereas the salvage pathway appears more permissive to ARGs. These observations support our earlier conclusion that VB12 synthesis potentially mitigates ARG-associated resistance risk while promoting MRG coexistence.
Functional characteristics of microbial taxa with VB12 synthesis potential
Microbial functional traits are of paramount importance for human societal development57. To characterize functional profiles of VB12-synthesizing microorganisms carrying resistance determinants, we selected MAGs involved in various VB12 synthesis pathways, carrying more than 10 ARGs, and simultaneously harboring MRGs for further functional analysis. The results reveal that selected MAGs possessed the potential for horizontal gene transfer. Each MAG carried 12–15 MGEs, contributing significantly to the horizontal transfer of genes (Fig. 5, Fig. S2, S3, Table S7). Notably, these MAGs carried an average of 175 pathogen-host interaction (PHI) genes—critical mediators of human disease transmission58. When PHI genes undergo MGE-mediated transfer to human-associated microbes, they may potentiate disease emergence59. This risk is particularly concerning for ARGs, as their HGT could exacerbate antibiotic resistance in clinical settings60.

Evolutionary relationship between MAGs with the potential for resistance and VB12 synthesis. Assign names to the MAGs based on the annotated genera, and then color them at the phylum level. The figure illustrates the phylum, class, order, and family to which the MAGs belong. The height of the right column indicates the number of resistance genes carried by each MAG.
In addition, understanding the CRISPR-Cas system is essential for gaining profound insights into how bacteria recognize and eliminate invading viral genomes, contributing to the development of innovative antiviral strategies61,62. Our investigation revealed that these MAGs carry 2–8 Cas proteins, predominantly consisting of Cas3 and Cas4 (Fig. 5, Fig. S2, S3, Table S7). Cas3 protein, with DNA cleavage and helicase activities, functions as a core enzyme in the Type I CRISPR-Cas system63, while Cas4 protein is involved in integrating foreign DNA sequences into bacterial DNA64,65. Bin.709 also found to host a Cas2 protein site (Fig. 6, Table S7), a core protein in the CRISPR system responsible for the initial phase of integrating foreign fragments66,67,68. These findings offer crucial insights for developing efficient and precise gene-editing tools.
Furthermore, functional annotation of the aforementioned MAGs was carried out using the COG and KEGG databases69 (Fig. 6, Fig. S4 – S9, Table S7). The results demonstrated a high degree of consistency between the annotations from both databases, with most genes being associated with metabolic pathways. This observation suggested that metabolic functions likely played a more critical role in these MAGs compared to other functional categories. Microbial metabolic functions encompass the degradation, synthesis, and transformation of organic compounds, as well as participation in biochemical processes linked to cellular energy and material metabolism40,70. Consequently, microorganisms harboring a greater abundance of metabolic genes may have contributed significantly to environmental nutrient cycling, energy flow, and ecological equilibrium. These findings implied that such microorganisms could have possessed adaptive advantages in their respective habitats71, enabling more efficient utilization and conversion of available nutrients. Additionally, KEGG annotation revealed that each MAG contained genes associated with infectious diseases and antimicrobial resistance (Fig. 6, Fig. S4–S9, Table S7), which corroborated earlier functional annotations from individual databases.

The genomic features of the metagenomic-assembled genome bin.709. In the figure, the characteristics of mobile genetic elements (MGEs) and the distribution of CRISPR-Cas sites are also shown separately.

The KEGG and COG functional annotation results for the metagenomic-assembled genome bin.709. The size of the circles and the height of the bars represent the number of genes, with different colors indicating various functional classifications.
Conclusion
Firstly, this study employed metagenomic approaches to investigate the distribution patterns and environmental drivers of microbial genes associated with vitamin B12 (VB12) biosynthesis in urban lake surface waters. Our analysis revealed that hemL was the predominant gene among all VB12 synthesis-related genes examined. Notably, we observed substantial spatial heterogeneity in the distribution of microbial VB12 synthesis genes across different urban lakes, with eutrophication levels (particularly nitrate nitrogen concentrations) showing a negative correlation with hemL gene abundance (Fig. S10). However, it should be noted that the VB12 synthesis-related genes detected in this study were identified through genomic prediction approaches. Further experimental validation via PCR and quantitative real-time PCR (qPCR) remains necessary to confirm these findings.
Secondly, by employing specific, meticulously curated functional gene databases alongside widely used public databases, this study conducted a more precise and comprehensive analysis of the genomic metabolic characteristics of microbes with the potential for synthesizing VB12. It was discovered that 26 metagenome-assembled genomes (MAGs) had the capability to synthesize VB12 and concurrently harbored ARGs. Among these, 4 MAGs simultaneously carried ARGs and MRGs, while also demonstrating the ability to synthesize VB12. These MAGs not only possessed functional elements conducive to horizontal gene transfer but also exhibited pathogenicity. Freshwater systems are increasingly recognized as critical reservoirs for antibiotic-resistant bacteria (ARB) and ARGs, posing substantial public and veterinary health risks21,72,73. Indirect transmission further escalates risks via contaminated aquatic food chains (e.g., raw shellfish) and urban agricultural irrigation, which can introduce ARB into the food supply. Compounding these threats, eutrophic waters serve as hotspots for horizontal gene transfer, accelerating the spread of resistance mechanisms to clinically relevant pathogens74. Addressing this multifaceted challenge demands a “One Health” approach75, integrating advanced wastewater treatment technologies, systematic ARB/ARG monitoring, and public awareness campaigns to curb antibiotic misuse. Without coordinated intervention, these urban aquatic reservoirs will continue to fuel the global antimicrobial resistance crisis, undermining both human and animal health.
These findings provide crucial evidence for future research exploring the link between the VB12 synthesis metabolic mechanism and human resistance risks.
Data availability
All metagenomic raw data used in this study have been deposited in the National Center for Biotechnology Information with the primary accession code PRJNA887701, detailed information on NCBI sequence submissions in Table S8.
References
King, A. L. et al. CO2 and vitamin B12 interactions determine bioactive trace metal requirements of a Subarctic Pacific diatom. ISME J. 5, 1388–1396. https://doi.org/10.1038/ismej.2010.211 (2011).
Sonnenburg, E. D. & Sonnenburg, J. L. Gut microbes take their vitamins. Cell. Host Microbe. 15, 5–6. https://doi.org/10.1016/j.chom.2013.12.011 (2014).
Degnan, P. H., Taga, M. E. & Goodman, A. L. Vitamin B12 as a modulator of gut microbial ecology. Cell. Metab. 20, 769–778. https://doi.org/10.1016/j.cmet.2014.10.002 (2014).
LeBlanc, J. G. et al. B-group vitamin production by lactic acid bacteria–current knowledge and potential applications. J. Appl. Microbiol. 111 https://doi.org/10.1111/j.1365-2672.2011.05157.x (2011). 1297 – 309.
Franco-Lopez, J. et al. Correlations between the composition of the bovine microbiota and vitamin B(12) abundance. mSystems 5, 10–1128. https://doi.org/10.1128/mSystems.00107-20 (2020).
Fang, H., Kang, J. & Zhang, D. Microbial production of vitamin B(12): a review and future perspectives. Microb. Cell. Fact. 16, 15. https://doi.org/10.1186/s12934-017-0631-y (2017).
Zhou, J. et al. VB12Path for accurate metagenomic profiling of microbially driven cobalamin synthesis pathways. mSystems 6, e0049721. https://doi.org/10.1128/mSystems.00497-21 (2021).
Zhang, X., Tan, Z. & Jiao, J. Lactobacillus plantarum or Macleaya cordata affects the biosynthesis of vitamin B12 by ileal microbiota of weaned goats: a metagenomics study. Acta Microbiol. Sinica. 63, 4218–4231. https://doi.org/10.13343/j.cnki.wsxb.20230184 (2023).
Akduman, N. et al. Bacterial vitamin B(12) production enhances nematode predatory behavior. ISME J. 14, 1494–1507. https://doi.org/10.1038/s41396-020-0626-2 (2020).
Samreen, Ahmad, I., Malak, H. A. & Abulreesh, H. H. Environmental antimicrobial resistance and its drivers: a potential threat to public health. J. Glob Antimicrob. Resist. 27, 101–111. https://doi.org/10.1016/j.jgar.2021.08.001 (2021).
Das, M. K., Das, S. & Srivastava, P. K. An overview on the prevalence and potential impact of antimicrobials and antimicrobial resistance in the aquatic environment of India. Environ. Monit. Assess. 195, 1015. https://doi.org/10.1007/s10661-023-11569-z (2023).
Heuer, H., Schmitt, H. & Smalla, K. Antibiotic resistance gene spread due to manure application on agricultural fields. Curr. Opin. Microbiol. 14, 236–243. https://doi.org/10.1016/j.mib.2011.04.009 (2011).
Lu, Y. et al. Long-term biogas slurry application increased antibiotics accumulation and antibiotic resistance genes (ARGs) spread in agricultural soils with different properties. Sci. Total Environ. 759, 143473. https://doi.org/10.1016/j.scitotenv.2020.143473 (2021).
Pu, C. et al. Impact of direct application of biogas slurry and residue in fields: in situ analysis of antibiotic resistance genes from pig manure to fields. J. Hazard. Mater. 344, 441–449. https://doi.org/10.1016/j.jhazmat.2017.10.031 (2018).
Behling, A. H. et al. Addressing antibiotic resistance: computational answers to a biological problem? Curr. Opin. Microbiol. 74, 102305. https://doi.org/10.1016/j.mib.2023.102305 (2023).
Zainab, S. M., Junaid, M., Xu, N. & Malik, R. N. Antibiotics and antibiotic resistant genes (ARGs) in groundwater: A global review on dissemination, sources, interactions, environmental and human health risks. Water Res. 187, 116455. https://doi.org/10.1016/j.watres.2020.116455 (2020).
Jiang, Q., Feng, M., Ye, C. & Yu, X. Effects and relevant mechanisms of non-antibiotic factors on the horizontal transfer of antibiotic resistance genes in water environments: A review. Sci. Total Environ. 806, 150568. https://doi.org/10.1016/j.scitotenv.2021.150568 (2022).
Romandini, A. et al. Antibiotic resistance in pediatric infections: global emerging threats, predicting the near future. Antibiotics 10, 393. https://doi.org/10.3390/antibiotics10040393 (2021).
Xiao, X. et al. Pollution characteristics of antibiotics and antibiotic resistance genes in urban lakes of Wuhan. Environ. Sci. Technol. 42, 9–16. https://doi.org/10.19672/j.cnki.1003-6504.2019.03.002 (2019).
Yang, X., Xiong, B. & Yang, M. Relationships among heavy metals and organic matter in sediment cores from lake nanhu, an urban lake in wuhan, China. J. Freshw. Ecol. 25, 243–249. https://doi.org/10.1080/02705060.2010.9665074 (2010).
Mao, C. et al. Microbial communities, resistance genes, and resistome risks in urban lakes of different trophic states: internal links and external influences. J. Hazard. Mater. Adv. 100233. https://doi.org/10.1016/j.hazadv.2023.100233 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, D. et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 1–11. https://doi.org/10.1186/1471-2105-11-119 (2010).
Fu, L. et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. (2012). https://doi.org/10.1093/bioinformatics/bts565 Bioinformatics.
Seyler, L. M., Trembath-Reichert, E., Tully, B. J. & Huber, J. A. Time-series transcriptomics from cold, oxic subseafloor crustal fluids reveals a motile, mixotrophic microbial community. ISME J. 15, 1192–1206. https://doi.org/10.1038/s41396-020-00843-4 (2021).
Arango-Argoty, G. et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 1–5. https://doi.org/10.1186/s40168-018-0401-z (2018).
Pal, C. et al. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, D737–D743. https://doi.org/10.1093/nar/gkt1252 (2014).
Staden, R., Judge, D. P. & Bonfield, J. K. Analyzing Sequences Using the Staden Package and EMBOSS, in Introduction To Bioinformaticsp. 393–410 (Springer, 2003).
Oh, M. et al. MetaCompare: a computational pipeline for prioritizing environmental resistome risk. FEMS Microbiol. Ecol. 94, fiy079. https://doi.org/10.1093/femsec/fiy079 (2018).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 1–13. https://doi.org/10.1186/s40168-018-0541-1 (2018).
Kang, D. D. et al. MetaBAT 2: an adaptive Binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. https://doi.org/10.7717/peerj.7359 (2019).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868. https://doi.org/10.1038/ismej.2017.126 (2017).
Parks, D. H. et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. https://doi.org/10.1101/gr.186072.114 (2015).
Luo, Z. et al. Depth-dependent variability of biological nitrogen fixation and diazotrophic communities in Mangrove sediments. Microbiome 9, 212. https://doi.org/10.1186/s40168-021-01164-0 (2021).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. https://doi.org/10.1093/bioinformatics/btac672 (2022).
Guo, C. et al. GcPathogen: a comprehensive genomic resource of human pathogens for public health. Nucleic Acids Res. 52, D714–d723. https://doi.org/10.1093/nar/gkad875 (2024).
Grant, J. R. et al. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–w492. https://doi.org/10.1093/nar/gkad326 (2023).
Brown, C. L. et al. mobileOG-db: a manually curated database of protein families mediating the life cycle of bacterial mobile genetic elements. BioRxiv https://doi.org/10.1101/2021.08.27.457951 (2021).
Zhou, Z. et al. METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10, 33. https://doi.org/10.1186/s40168-021-01213-8 (2022).
Xiao, F. et al. The impact of anthropogenic disturbance on bacterioplankton communities during the construction of Donghu tunnel (Wuhan, China). Micro Ecol. 77, 277–287. https://doi.org/10.1007/s00248-018-1222-0 (2019).
Surendran, S. et al. An update on vitamin B12-related gene polymorphisms and B12 status. Genes Nutr. 13, 2. https://doi.org/10.1186/s12263-018-0591-9 (2018).
Wang, J., Fu, Z., Qiao, H. & Liu, F. Assessment of eutrophication and water quality in the estuarine area of lake wuli, lake taihu, China. Sci. Total Environ. 650, 1392–1402. https://doi.org/10.1016/j.scitotenv.2018.09.137 (2019).
Morsy, A. et al. Evaluation of the water quality and the eutrophication risk in mediterranean sea area: A case study of the Port said harbour, Egypt. Environ. Challenges. 7, 100484. https://doi.org/10.1016/j.envc.2022.100484 (2022).
Kovatcheva, M. et al. Vitamin B(12) is a limiting factor for induced cellular plasticity and tissue repair. Nat. Metab. 5, 1911–1930. https://doi.org/10.1038/s42255-023-00916-6 (2023).
Doxey, A. C. et al. Aquatic metagenomes implicate Thaumarchaeota in global cobalamin production. ISME J. 9, 461–471. https://doi.org/10.1038/ismej.2014.142 (2015).
Sultana, S. et al. Vitamin B12 is not shared by all marine prototrophic bacteria with their environment. ISME J. 17, 836–845. https://doi.org/10.1038/s41396-023-01391-3 (2023).
Ondon, B. S., Li, S., Zhou, Q. & Li, F. Sources of antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARGs) in the soil: A review of the spreading mechanism and human health risks. Rev. Environ. Contam. Toxicol. 256, 121–153. https://doi.org/10.1007/398_2020_60 (2021).
Sieber, K. B., Bromley, R. E. & Dunning Hotopp, J. C. Lateral gene transfer between prokaryotes and eukaryotes. Exp. Cell. Res. 358, 421–426. https://doi.org/10.1016/j.yexcr.2017.02.009 (2017).
Horne, T., Orr, V. T. & Hall, J. P. How do interactions between mobile genetic elements affect horizontal gene transfer? Curr. Opin. Microbiol. 73, 102282. https://doi.org/10.1016/j.mib.2023.102282 (2023).
Craig, N. L. Target site selection in transposition. Annu. Rev. Biochem. 66, 437–474. https://doi.org/10.1146/annurev.biochem.66.1.437 (1997).
Osman, D. et al. The requirement for Cobalt in vitamin B(12): A paradigm for protein metalation. Biochim. Biophys. Acta Mol. Cell. Res. 1868, 118896. https://doi.org/10.1016/j.bbamcr.2020.118896 (2021).
Ma, J. et al. Horizontal transfer potential of antibiotic resistance genes in wastewater treatment plants unraveled by microfluidic-based mini-metagenomics. J. Hazard. Mater. 465, 133493. https://doi.org/10.1016/j.jhazmat.2024.133493 (2024).
Kong, L. et al. Cross-feeding between filamentous Cyanobacteria and symbiotic bacteria favors rapid photogranulation. Environ. Sci. Technol. 57, 16953–16963. https://doi.org/10.1021/acs.est.3c04867 (2023).
Duan, Y. et al. Bacterial community dynamics and co-occurrence network patterns during different stages of biochar-driven composting. Bioresour Technol. 384, 129358. https://doi.org/10.1016/j.biortech.2023.129358 (2023).
Grossman, A. & Nutrient Acquisition The generation of bioactive vitamin B12 by microalgae. Curr. Biol. 26, R319–R321. https://doi.org/10.1016/j.cub.2016.02.047 (2016).
Dominguez-Bello, M. G., Godoy-Vitorino, F., Knight, R. & Blaser, M. J. Role of the Microbiome in human development. Gut 68, 1108–1114. https://doi.org/10.1136/gutjnl-2018-317503 (2019).
Durmuş, S., Çakır, T., Özgür, A. & Guthke, R. A review on computational systems biology of pathogen-host interactions. Front. Microbiol. 6, 235. https://doi.org/10.3389/fmicb.2015.00235 (2015).
Richardson, E. J. et al. Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2, 1468–1478. https://doi.org/10.1038/s41559-018-0617-0 (2018).
Lerminiaux, N. A. & Cameron, A. D. S. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 65, 34–44. https://doi.org/10.1139/cjm-2018-0275 (2019).
Brezgin, S. et al. Immunity and viral infections: modulating antiviral response via CRISPR-Cas systems. Viruses 13, 1373. https://doi.org/10.3390/v13071373 (2021).
Wang, H., La Russa, M. & Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264. https://doi.org/10.1146/annurev-biochem-060815-014607 (2016).
Gong, B. et al. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc. Natl. Acad. Sci. U S A. 111, 16359–16364. https://doi.org/10.1073/pnas.1410806111 (2014).
Hu, C. et al. Mechanism for Cas4-assisted directional spacer acquisition in CRISPR-Cas. Nature 598, 515–520. https://doi.org/10.1038/s41586-021-03951-z (2021).
Yang, H. & Patel, D. J. New CRISPR-Cas systems discovered. Cell. Res. 27, 313–314. https://doi.org/10.1038/cr.2017.21 (2017).
Nuñez, J. K. et al. Foreign DNA capture during CRISPR-Cas adaptive immunity. Nature 527, 535–538. https://doi.org/10.1038/nature15760 (2015).
Jackson, S. A. et al. CRISPR-Cas: adapting to change. Science 356, eaal5056. https://doi.org/10.1126/science.aal5056 (2017).
Santiago-Frangos, A. et al. Structure reveals why genome folding is necessary for site-specific integration of foreign DNA into CRISPR arrays. Nat. Struct. Mol. Biol. 30, 1675–1685. https://doi.org/10.1038/s41594-023-01097-2 (2023).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Davenport, R. et al. Decomposition decreases molecular diversity and ecosystem similarity of soil organic matter. Proc. Natl. Acad. Sci. U S A 120, e2303335120. (2023). https://doi.org/10.1073/pnas.2303335120
Bang, C. et al. Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology 127, 1–19. https://doi.org/10.1016/j.zool.2018.02.004 (2018).
72 Yang, Y. et al. Antibiotic resistance genes in surface water of eutrophic urban lakes are related to heavy metals, antibiotics, lake morphology and anthropic impact. Ecotoxicology 26, 831–840. https://doi.org/10.1007/s10646-017-1814-3 (2017).
Wang, M. et al. Impacts of net cages on pollutant accumulation and its consequence on antibiotic resistance genes (ARGs) dissemination in freshwater ecosystems: insights for sustainable urban water management. Environ. Int. 183, 108357. https://doi.org/10.1016/j.envint.2023.108357 (2024).
EFSA Panel on Biological Hazards (BIOHAZ). Role played by the environment in the emergence and spread of antimicrobial resistance (AMR) through the food chain. Efsa J. 19, e06651. https://doi.org/10.2903/j.efsa.2021.6651 (2021).
Who, O. One health. World Health Organ. 736. (2017).
Acknowledgements
We appreciate Jie Huang (Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan) for their diligent sampling efforts and providing open-access data. This manuscript is supported by National Natural Science Foundation of China (Juan Ren, 82473252, 81772793); the Scientific and Technological Research Foundation of Shaan’xi Province, Key Research and Development Project, General project (Juan Ren, 2023-YBSF-666); the Shaan’xi Provincial Key Research and Development Plan Project (Key support projects Category A) (Juan Ren, 2021A011); the Basic and Clinical Integration Innovation Project of Xi’an Jiao tong University (Juan Ren, YXJLRH2022006); the Nanjing Tianqing Scientific Research Fund of First Affliated Hospital of Xi’an Jiaotong University (Juan Ren, TQ202205), and the China Scholarship Council (Chengzhi Mao, No. 202204910442).
Author information
Authors and Affiliations
Contributions
Ning Lan: Writing–original draft, Conceptualization, Visualization. Min Chen: Writing–review & editing. Xuan Wang: Writing–review & editing. Shuheng Bai: Writing–review & editing. Chengzhi Mao: Writing–original draft, Conceptualization, Methodology, Formal analysis, Investigation, Writing–review & editing, Funding acquisition. Juan Ren: Writing–review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lan, N., Mao, C., Chen, M. et al. Metagenomic insights into resistance trends related to microbial VB12 synthesis in eutrophic urban lakes. Sci Rep 15, 20599 (2025). https://doi.org/10.1038/s41598-025-06125-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06125-3
