
Abstract
Crossbreeding with Booroola or Texel sheep harboring major genes for prolificacy and muscularity enhances productivity but it may limit adaptation and survivorship in crossbred lambs. Thus, the trade-offs that impact productivity and adaptability have not been quantified or modeled and remain largely elusive at the genetic level, limiting the development of optimized breeding strategies. This study investigates the genomic architecture of purebred Moghani sheep and the first paternal backcross (PBC1) generation of crossbred lambs, including Booroola Merino × Moghani (BMM), Booroola Romney × Moghani (BRM), Texel Dalzell × Moghani (TDM), and Texel Tamlet × Moghani (TTM). Genotyping-by-sequencing (GBS) was used to assess genetic diversity, admixture patterns, and selection signatures. Structure analysis revealed complex admixture in BMM and BRM, while TDM and TTM were more homogeneous. Purebred Moghani sheep exhibited the highest genetic diversity (HO = 0.521 ± 0.10) and the lowest inbreeding (FIS = − 0.474), serving as a key genetic bridge among the groups. In contrast, BRM and TTM showed lower heterozygosity (HO = 0.410 ± 0.09 and 0.431 ± 0.10) and increased inbreeding (FROH), with extended runs of homozygosity (ROH), suggesting recent inbreeding and reduced effective population sizes. The functional annotation of ROH islands connected TDM and TTM to immune response and muscle development pathways like VEGF and insulin signaling, while BMM and BRM were linked to metabolic and reproductive pathways like central carbon metabolism in cancer (mTOR) and prolactin signaling (LHB). Taken together, these results highlight the need for improved breeding methods that prioritize trade-offs associated with reduced genetic diversity in crossbred populations. Nevertheless, given the limited genotype representation, the results should be taken with caution; so, next research should cover a larger panel of genotypes in order to have a more complete knowledge.
Similar content being viewed by others
Introduction
Sheep play a crucial role in global agriculture, supplying meat, milk, and wool for human consumption. Domesticated approximately 10,500 years ago in the Fertile Crescent, specifically between southeastern Anatolia and the Zagros Mountains in Iran, their wild ancestors still inhabit this region today1,2. Iran is home to 27 indigenous sheep breeds, presenting considerable potential to meet world protein demands and boost exports. However, many of these breeds, including the Moghani sheep, face challenges such as slow growth, low feed efficiency, poor reproductive performance, and low lean meat percentage, which hinder their economic viability3,4,5,6.
The Moghani breed is characterized by its large fat tail and low prolificacy, yet it possesses out-of-season reproductive potential, making it a valuable genetic resource. Despite this, its productivity remains limited. Early breeding attempts, such as crossing Arkhamerino with Moghani sheep between 1999 and 2001, aimed to improve wool quality and growth traits but had limited success7,8,9. To address these shortcomings, a national breeding initiative was introduced to enhance muscularity and prolificacy in Iranian Moghani sheep by incorporating genetic material from high-performance sheep breeds known for superior carcass composition (Texel: Tamlet and Dalzell) and reproductive efficiency (Booroola: Merino and Romney)10,11,12. However, crossbred lambs have shown lower adaptability and resilience to environmental challenges, leading to reduced survivability compared to pure Moghani sheep12. This reduced survivability rate poses a significant obstacle to extending crossbreeding programs aimed at enhancing productivity in Iran’s indigenous sheep breeds.
Breeding for improved economic traits in sheep is complex due to the polygenic nature of these traits, which are influenced by multiple genes and environmental factors4. Traditional breeding methods, while effective to some extent, often fail to fully exploit the genetic potential of sheep populations. Advanced genomic technologies, such as high-density SNP arrays and genome-wide selection analyses, have improved genetic variation detection in livestock. SNP genotyping and copy number variation analysis effectively identify selection signatures and key economic traits13,14. Genomic tools like GBS provide deeper insights into genetic architecture shaped by natural and artificial selection15,16,17, enabling more informed breeding decisions that enhance productivity and sustainability18,19,20.
Genomic studies focusing on ROH, which are segments of the genome that are identical by descent, have become critical for understanding inbreeding levels and population structure21. The genomic inbreeding coefficient (FROH), derived from ROH, provides an accurate measure of inbreeding, independent of allelic frequencies and pedigree data22. Longer ROH segments indicate common ancestry, while shorter segments point to more distant ancestors23,24. ROH islands, which are genomic regions under selective sweeps, offer insights into both population history and the selective pressures that shape adaptive traits18,22,25,26,27. These regions have been observed across various livestock species, including sheep19,28,29,30,31, goats32,33,34,35, pig36,37,38, horses39,40,41,42, cattle43,44,45,46, and chicken22,47,48, highlighting their significance in identifying selection signatures and understanding the evolutionary forces at play in breeding programs.
Given the productivity challenges of the Moghani breed and its crossbred lambs, it is crucial to assess both the benefits and genetic consequences of crossbreeding, including reduced adaptability and potential inbreeding depression. This study uses GBS to investigate the genomic architecture of pure and crossbred Moghani lambs, focusing on five populations: pure Moghani and four PBC1 groups, including Booroola Merino × Moghani (BMM), Booroola Romney × Moghani (BRM), Texel Dalzell × Moghani (TDM), and Texel Tamlet × Moghani (TTM). We hypothesize that genomic adaptations in these crossbred lambs will reveal insights into balancing productivity and environmental resilience, contributing to optimized breeding strategies and the long-term sustainability of sheep farming in Iran.
Materials and methods
Ethics statement
The Animal Care and Use Committee at the Agricultural Biotechnology Research Institute of Iran (ABRII), Karaj, Alborz, approved this research. All experiments and procedures involving animals were performed in strict accordance with relevant guidelines and regulations, including veterinary best practices, as outlined in the approved protocol. Samples were obtained from the Jovain Agricultural & Industrial Corporation breeding facility in Razavi Khorasan, Iran. No invasive procedures were conducted during the study. Furthermore, all methods are reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org) to ensure transparency and reproducibility.
Genetic material
The animals in this study are part of a breeding program in Iran aimed at enhancing Moghani sheep productivity through crossbreeding, with Booroola selected for prolificacy and Texel for muscularity. The F1 generation was produced by inseminating 380 Moghani ewes with frozen sperm from two Texel sires (Tamlet and Dalzell) and two Booroola sires (Romney and Merino). The resulting F1 rams were then backcrossed to purebred Moghani ewes to produce the first paternal backcrossing (PBC1) generation of crossbred lambs. The PBC1 groups include Booroola Merino × Moghani (BMM), Booroola Romney × Moghani (BRM), Texel Dalzell × Moghani (TDM), and Texel Tamlet × Moghani (TTM)10,11,12.
DNA extraction and sequencing
A GBS project was conducted with 65 male sheep from five breeds/strains, as shown in Fig. 1: Moghani pure sheep (M, n = 9), Booroola Merino × Moghani (BMM, n = 13), Booroola Romney × Moghani (BRM, n = 19), Texel Dalzell × Moghani (TDM, n = 11), and Texel Tamlet × Moghani (TTM, n = 13). Blood samples (1 ml per lamb) were collected from the jugular vein into EDTA tubes, and genomic DNA was extracted as described by Talebi et al.49. GBS libraries were constructed following the protocol by Elshire et al.50, with minor modifications, and were genotyped by BGI Genomics, China. Sequencing was performed using paired-end Illumina HiSeq sequencing, generating 150 bp reads with an average sequencing depth of approximately 8 × per sample. This sequencing strategy ensured sufficient variant discovery while maintaining cost efficiency for population genomic analyses.

Crossbreeding of five sheep breeds/strains. Texel sire strains (Texel Dalzell [TD] and Texel Tamlet [TT]) and Booroola sire strains (Booroola Merino [BM] and Booroola Romney [BR]) were initially crossed with Moghani pure ewes (M), producing F1 crossbred rams. These F1 rams were then backcrossed with purebred Moghani ewes, resulting in the paternal backcross generation (PBC1): Booroola Merino × Moghani (BMM), Booroola Romney × Moghani (BRM), Texel Dalzell × Moghani (TDM), and Texel Tamlet × Moghani (TTM).
Raw data processing
The raw GBS data were demultiplexed using Illumina Experiment Manager v1.16.0. Quality checks were performed with FastQC v.0.11.5, and low-quality reads (quality < 20), nucleotides, minimal read length 36 to discards any reads shorter than 36 bases, and adapters were trimmed using Trimmomatic v0.3951. Paired FASTQ files were aligned to the Ovis aries reference genome (OAR_v.3.1) with BWA software v0.7.12 using bwa-mem algorithm52. Duplicate reads were marked with Picard MarkDuplicates v1.115. SAM files were compressed and sorted into BAM files using SAMtools v.1.6, and Picard added read group information. Reference files (fai and dict) were generated thereafter. The statistics of the BAM files including total read counts, mapped reads, average mapping quality and average coverage were calculated using SAMtools v1.6 (Supplementary Table 1).
Variant calling
Variants were called using the Genome Analysis Toolkit (GATK)53. After recalibrating base quality scores, BAM files were processed with GATK-HaplotypeCaller to call SNPs and INDELs via local de-novo haplotype assembly. Sixty-five VCF files were generated for individual samples and then combined using the GenotypeGVCFs module. The raw VCFs from five sheep populations including M (n = 9), BMM (n = 13), BRM (n = 19), TDM (n = 11), and TTM (n = 13) were merged using bcftools.
Data preparation and filtration
Raw VCF files were filtered to retain variants with at least 5 reads of coverage using VCFtools v0.1.1354. This initial filtration resulted in a total of 8,769,289 variants, including both SNPs and indels, across five sheep populations (Table 1). Subsequent filtering was performed using PLINK v1.955, applying criteria including a missing genotype rate (–geno 0.5), a minor allele frequency (–maf 0.01), significant deviations from the Hardy–Weinberg equilibrium (PHWE < 10e−5), and clustering parameters (Cluster Size = 4, Cluster Window Size = 10).
Annotation of genetic variants
Variants were annotated using SnpEff v3.456 based on functional categories, including regions 5 kb upstream and downstream of genes, intergenic regions, missense and synonymous mutations, introns, UTRs, and stop gain/loss mutations. For simplicity, variants in upstream/downstream regions and UTRs were grouped into single categories.
Population structure and relatedness analysis
Two approaches were used to study population structure:
-
(1)
PCA: Principal component analysis (PCA) was performed using PLINK v1.9 (–pca)55. Eigenvectors and eigenvalues from the covariance matrix were used to create a 3D PCA plot in R using the ggplot2 and scatterplot3d packages57. PCA reduces dimensionality by transforming variables into uncorrelated principal components (PCs) that explain the maximum variance. PC1 typically reflects broad genetic differences, while PC2 and PC3 capture finer-scale variation46,58,59.
-
(2)
ADMIXTURE: ADMIXTURE60 was employed to infer individual ancestry proportions under a maximum likelihood framework. K values from 2 to 6 were tested, with the best model selected based on cross-validation errors. CLUMPP v1.1.161 was used to align and average cluster memberships across multiple ADMIXTURE runs, ensuring consistent ancestry estimates. This method is similar to STRUCTURE 2.3.462 for detecting genetic substructure and shared ancestry.
Diversity metrics and fixation index
Diversity metrics, including observed heterozygosity (HO) and expected heterozygosity (HE), were calculated using the PLINK v1.9 software package55. The polymorphic marker ratio (PN), representing the fraction of polymorphic loci within the population, was determined by averaging the proportion of non-missing SNPs per individual across each population.
The fixation index (FST), a measure of population differentiation, was computed for each SNP to assess genetic variation among breeds. Each breed was compared to the combined samples from the other breeds using the PLINK –fst and –within functions55.
Runs of homozygosity (ROH) analysis
ROH were analyzed using PLINK v1.955 with the –homozyg module, which scans SNPs using a sliding window approach63. ROH were defined with the following criteria: 30-SNP sliding window, 0.05 overlap, minimum of 30 consecutive SNPs, 100 kb minimum length, 1000 kb max gap, 1 SNP per 50 kb density, and allowance for up to ten missing and zero heterozygous SNPs. Allowance of ten missing data was utilized because of the GBS data structure which tends to possess a large portion of missing data than microarray datasets. ROHs were then classified into four length classes: 0.1–0.3, 0.3–1, 1–2, and 2–4 Mb using an in-house R script.
Inbreeding coefficients
Two inbreeding coefficients were calculated for each population using PLINK v1.955. Wright’s inbreeding coefficient (FIS) was calculated according to Wright64 as:
where HO is the observed heterozygosity and HE is the expected heterozygosity. Genomic inbreeding based on ROH (FROH) was estimated according to McQuillan et al.21 as:
where LROH is the total length of ROH, and Ltotal (~ 2452 Mb) is the autosomal chromosome length covered by SNPs (Oar_v3.1). The correlation between FROH and FIS was calculated for all homozygous stretches across the five sheep populations. Plots were generated using the ‘ggplot2’ package in R.
Detection of ROH islands and enrichment analysis
To identify genomic regions associated with ROH, we calculated the SNP frequency in ROH across individuals. ROH islands were defined as genomic regions containing the top 1% of SNPs with the highest ROH frequencies across all individuals. Using a custom AWK script, these regions were compared with the sheep QTL database release 53 (https://www.animalgenome.org/cgi-bin/QTLdb/OA/index). KOBAS-i was used for all gene ontology (GO) analysis based on KEGG database65.
Results and discussion
Building on our prior research, we conducted a crossbreeding program involving Moghani sheep and selected Texel (Tamlet and Dalzell) and Booroola (Merino and Romney) sires to enhance muscularity and prolificacy traits10,11,12. Although the PBC1 crossbred lambs exhibited improvements in these productive traits due to hybrid vigor effects66, they also demonstrated reduced adaptability and survivability compared to purebred Moghani sheep12. This phenomenon raises significant concerns regarding the long-term viability of such crossbreeding strategies, emphasizing the need for a nuanced understanding of the genetic interplay at work. This trade-off highlights concerns about the long-term sustainability of such crossbreeding strategies, emphasizing the need for a deeper understanding of the genetic mechanisms underlying these phenotypic outcomes. The observed challenges reinforce the importance of balancing productivity gains with maintaining adaptive resilience, particularly under local environmental conditions.
Overview of genotyping-by-sequencing (GBS) outcomes
The total and mapped read counts per sample showed variability across the 65 samples, with some having significantly higher read numbers (Fig. 2A and Supplementary Table 1). Despite this, the percentage of reads successfully mapped was consistently high, averaging around 99%, indicating robust alignment quality. The average mapping quality was stable across samples, with a negligible correlation between sample number and mapping quality (R2 = 0.008, Fig. 2B), suggesting consistent alignment and reliable variant calling. Average coverage also varied among samples, with a slight increase as sample numbers rose (R2 = 0.024, Fig. 2C). This variability in coverage could influence variant detection, particularly in lower-coverage samples. To assess the impact of missing data, we examined the distribution of SNP-level missingness across the genome. The results indicated that missing sites were randomly distributed across autosomes without regional clustering (Supplementary Fig. S1), consistent with the stochastic nature of GBS.

Genotyping-by-sequencing (GBS) data summary. (A) Total raw reads generated and the percentage mapped across 65 samples. (B) Average mapping quality across 65 samples. (C) Average GBS coverage across 65 samples.
Table 1 summarizes the GBS data processing results across five sheep populations. The Moghani sheep (M) exhibited the highest average total reads, with 3,803,688.22 reads, significantly exceeding the overall average of 3,131,446 ± 1,394,648 reads (Table 1 and Supplementary Table 1). Despite this, the Booroola Romney × Moghani (BRM) population demonstrated the greatest genetic diversity based on the highest counts of SNPs (1,953,625), indels (181,542), and multiallelic sites (34,278) compared to the other populations. In contrast, the Moghani population had the lowest counts of SNPs (1,437,615) and indels (133,553), reflecting a lower level of genetic diversity. These findings suggest that, at least in terms of SNP, indel, and multiallelic site counts, crossbred populations, particularly BRM, possess greater genetic complexity compared to the purebred Moghani sheep. However, we acknowledge that GBS data primarily captures variation at the SNP and indel levels and may not fully encompass all aspects of genetic diversity.
Distribution and annotation of genetic variants
Following quality control, 579,027 genetic variants were identified, distributed uniformly across the 26 autosomes and the X chromosome (Fig. 3A). Of these, 567,493 were mapped to chromosomes 1–26, with 99.7% representing previously reported variants, and 0.3% classified as novel. These variants were associated with 24,377 genes and 55,560 transcripts. Intronic variants were the most abundant (62%), followed by intergenic (18%) and upstream gene variants (6%). In coding regions, missense variants were the most common (60%), while synonymous (27%) and frameshift variants (6%) were also present (Fig. 3B). This variant distribution reflects the predominance of non-coding variants, which are likely to play significant roles in gene regulation and expression, consistent with studies on sheep genomes that emphasize the importance of regulatory variants67. The high prevalence of missense variants in coding regions suggests potential functional impacts on protein structure, particularly relevant in crossbred populations where adaptive introgression can introduce advantageous alleles68. Novel variants, although less frequent, may offer crucial insights into unique genetic adaptations that contribute to the sustainability and resilience of sheep breeds, especially in traits related to local adaptation and productivity. These findings align with earlier research that has identified the importance of both coding and non-coding regions in shaping the complex traits of sheep, such as disease resistance, reproductive success, and environmental69,70,71. Investigating the role of these novel variants in future breeding programs will be critical for improving traits such as resilience to climate change and enhancing productivity in sheep populations72.

Distribution of variants on sheep chromosomes. (A) SNP density across each sheep chromosome, displayed with SNP counts in 1 Mb window sizes. (B) Proportion of variant consequences across autosomes, with a majority being intronic (62%) and missense (60%).
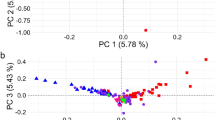
Principal component analysis (PCA) of genetic relationships
The PCA of genotypic data (Fig. 4) shows distinct genetic differentiation among the five sheep populations. The first three principal components (PC1: 35%, PC2: 25%, PC3: 20%) collectively explain a significant proportion of the genetic variance. The M population is clearly separated along PC1, while the crossbred populations (TDM, TTM, BRM, and BMM) form well-defined clusters with minimal overlap, suggesting structured genetic variation. Specifically, BMM shows strong differentiation along PC2 and PC3, highlighting unique genetic influences within this population. Studies in livestock genetics have demonstrated that PCA effectively identifies population stratification, with PC1 typically capturing major genetic differences, while additional components reveal finer-scale structure46,58,59. These findings highlight the utility of PCA in unraveling genetic structure and informing breeding strategies in crossbred populations.

3D PCA plot of genomic variation across five sheep populations. The plot visualizes the first three principal components (PC1, PC2, and PC3) capturing 35%, 25%, and 20% of the variation, respectively. Moghani pure sheep (M), Texel Dalzell × Moghani (TDM), Texel Tamlet × Moghani (TTM), Booroola Romney × Moghani (BRM), Booroola Merino × Moghani (BMM).
Population admixture
Based on the cross-validation (CV) error values shown in Fig. 5A, the model with K = 4 (CV = 0.44) provided the best fit for our data. The Structure analysis in Fig. 5B displays the genetic ancestry of the five sheep populations (BMM, BRM, M, TDM, and TTM) across different numbers of assumed ancestral populations (K = 2 to K = 4). As K increases from 2 to 4, the populations begin to differentiate, with BMM and BRM showing mixed ancestry from two distinct genetic clusters. This differentiation is likely related to historical crossbreeding events aimed at enhancing desirable traits like prolificacy and muscularity73. The increasing complexity of substructure observed at 4, particularly in BMM and BRM, suggests multiple ancestral sources and reflects the impact of admixture events over time74. We acknowledge that the populations are not homogeneous, as observed in the Structure analysis, especially within BMM, BRM, and M populations. These populations exhibit mixed ancestry, suggesting admixture rather than homogeneous genetic backgrounds. The relatively homogeneous structure within TDM and TTM even at higher K values suggests recent divergence or limited gene flow between these populations. This could result from breeding practices designed to preserve specific traits, leading to genetic isolation75. The M population mixed ancestry at K = 4 highlights its intermediate position, likely reflecting genetic contributions to multiple populations. Similar patterns in other sheep studies suggest shared ancestry resulting from recent gene flow18,67,76. Cross-validation (CV) error values for each K were calculated to determine the best K value. The lowest CV error was observed at K = 4, which suggests that K = 4 represents the most stable and informative population structure for our data.

Genetic ancestry inference and model selection. (A) Cross-validation (CV) error as a function of the number of clusters (K). The CV error decreases initially, reaching a minimum at K = 4 (CV = 0.44), before increasing at higher values. Blue circles represent data points, and the red line connects them to highlight the trend. (B) Genetic ancestry analysis of five populations (BMM, BRM, M, TDM, and TTM) for different assumed ancestral populations (K = 2 to K = 4). Each vertical bar represents an individual, with colors indicating estimated ancestry proportions.
These findings are consistent with the PCA results (Fig. 4), reinforcing the genetic distinctiveness of BMM and BRM, which show complex ancestry patterns likely due to historical admixture. The relatively homogeneous structure within TDM and TTM suggests recent divergence or limited gene flow between them. The M population’s mixed ancestry at higher K values further underscores its role as a genetic bridge, highlighting the intricate genetic relationships and historical admixture among the studied populations.
Genetic diversity and fixation index
The genetic diversity analysis of the five sheep populations, as presented in Table 2, shows that the Moghani pure sheep (M) had the highest observed heterozygosity (HO = 0.521 ± 0.10) and the lowest Wright’s inbreeding coefficient (FIS = − 0.474), indicating greater genetic diversity compared to the crossbred lambs. Conversely, the crossbred lambs, such as BRM and TTM, exhibited lower heterozygosity (HO = 0.410 ± 0.09 and 0.431 ± 0.10, respectively) but higher polymorphic marker ratios (PN), with BRM at 84% and TTM at 82%. To further assess differences in heterozygosity levels among the breeds, we performed pairwise Wilcoxon rank-sum tests. The results revealed that Moghani sheep had significantly higher HO compared to the BMM and BRM crossbred populations (p = 0.016 and p = 0.011, respectively), while no statistically significant differences were observed among the remaining group comparisons (p > 0.05). These results are presented in Supplementary Fig. S2. Notably, the observed heterozygosity in our five sheep populations surpasses the values reported by Eydivandi et al. for Moghani sheep from Ardabil province, Iran (HO = 0.373 ± 0.010)77. Despite these differences, all populations exhibited negative FIS values, with BRM at − 0.385 and TTM at − 0.383, similar to the Lori-Bakhtiari sheep breed reported by Abdoli et al.19, indicating an excess of heterozygotes and low levels of inbreeding. Moreover, the FST results in Table 3 support these findings. The Moghani pure sheep (M) shows the highest genetic differentiation from TDM (FST = 0.01812), consistent with its superior genetic diversity. In contrast, lower FST values between BRM and TTM (e.g., 0.00714) align with their lower heterozygosity and higher polymorphic marker ratios (%PN). These results suggest that while crossbreeding has increased genetic variation, the pure Moghani population retains higher genetic diversity, potentially offering greater resilience against inbreeding depression.
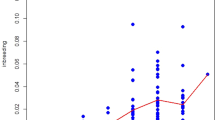
Genomic inbreeding coefficient estimated from ROH (FROH)
The analysis of ROH across the five sheep populations provides important insights into their genetic architecture and inbreeding history. Across all populations, the majority of ROH segments fall within the shortest length category (0.1–0.3 Mb), accounting for 42.70% to 70.76% of the total ROH (Table 4). However, it is important to note the limitations of calculating ROH from GBS data. Long ROHs tend to break into shorter tracts due to the presence of frequent heterozygous gaps in sequencing resolution22, whereas microarrays would likely miss them, given the uneven distribution of markers across the genome. Ceballos et al.24 demonstrated that low-coverage whole-genome sequencing (WGS), including GBS, tends to produce a higher rate of erroneous heterozygous SNP calls compared to array-based genotyping. This can compromise the accuracy of ROH detection by fragmenting longer homozygous segments. These limitations highlight the need for flexible ROH detection criteria, such as allowing a small number of heterozygous SNPs within segments, to improve sensitivity in low-coverage datasets. Although WGS provides higher resolution than SNP arrays, we recognize that the choice of ROH parameters, particularly the minimum segment length, can significantly influence the results. This is especially important when working with datasets that have substantial missing data or when identifying shorter ROH segments. Notably, the Moghani population (M) has the highest proportion of its ROH in this shortest category (70.76%), suggesting a history of distant inbreeding or effective genetic management that has prevented the accumulation of longer homozygous segments (Table 4 and Supplementary Table 2). This is in contrast to the TDM and TTM populations, which have higher proportions of ROH in the 0.3–1 Mb category (54.15% and 64.39%, respectively). These crossbred populations also exhibit the longest average ROH lengths (425.29 ± 71.44 Mb and 416.80 ± 166.42 Mb, respectively), reflecting more recent inbreeding events or reduced effective population sizes (Table 4 and Fig. 6).

A schematic representation of ROH profile in sheep genome. The profile is given by the average genomic inbreeding coefficient (FROH) and the average ROH lengths based on Kb (KBAVG).
The total ROH length (Sum L.) varies significantly among the populations, with the crossbred lambs, particularly BRM (6885.41 Mb) and BMM (4563.57 Mb), displaying notably higher total ROH lengths compared to the Moghani sheep (2733.12 Mb). This disparity underscores the differences in genetic architecture between the purebred and crossbred groups, with crossbreeding likely contributing to the increased ROH coverage, potentially due to genetic bottlenecks or recent inbreeding. While evaluating ROH, we also considered the influence of sequencing depth, finding a moderate Spearman correlation between average coverage and ROH counts (ρ = 0.496, p = 2.62e-05), suggesting that coverage may slightly impact ROH detection across individuals without affecting population-level trends (Supplementary Fig. S3). Further highlighting the extent of inbreeding, the maximum ROH lengths (Max. L.) observed in the TTM (603.86 Mb) and TDM (571.43 Mb) populations indicate the presence of notably long homozygous segments. These values are substantially higher than those found in the Moghani population (425.45 Mb) (Table 4 and Supplementary Table 2), suggesting that the crossbred populations have undergone more intense inbreeding or possess smaller effective population sizes that exacerbate the formation of extended ROH. The mean ROH length (Mean L.) also underscores these differences, with TDM (425.29 ± 71.44 Mb) and TTM (416.80 ± 166.42 Mb) populations having higher mean ROH lengths compared to Moghani sheep (303.68 ± 111.61 Mb). This pattern is consistent with their higher FROH values, where TDM and TTM have the highest FROH values (0.173 ± 0.029 and 0.170 ± 0.068, respectively), indicating that a substantial proportion of their genomes is covered by ROH (Table 4, Fig. 6 and Supplementary Table 2). In contrast, the Moghani population shows a lower FROH value (0.124 ± 0.046, Table 4), reflecting its superior genetic diversity and a history of managed inbreeding, as evidenced by its highest observed heterozygosity (HO = 0.521 ± 0.10) and the lowest FIS (− 0.474) (Table 2). Moreover, the disparity in minimum ROH lengths (Min. L.) across populations, with TDM (323.18 Mb) and TTM (101.39 Mb) showing considerably higher minimum ROH lengths than the Moghani sheep (63.48 Mb), provides further evidence of genetic bottlenecks or recent inbreeding in the crossbred populations (Table 4 and Supplementary Table 2). Comparatively, the FROH values reported for other Iranian sheep breeds are considerably lower than those observed in our study. Eydivandi et al.77 found FROH values of 0.0475 ± 0.0048 for Afshari, 0.0443 ± 0.0026 for Moghani, 0.0450 ± 0.0043 for Qezel, 0.0417 ± 0.0025 for Zel, and 0.0428 ± 0.0030 for Lori-Bakhtiari. Abdoli et al. (2023) reported an FROH of 0.031 ± 0.003 for Lori-Bakhtiari sheep19, while Barani et al. (2023) found FROH values of 0.0410 for Baluchi, 0.0090 for Zel, and 0.0153 for Lori-Bakhtiari78. In contrast, the FROH values for the five sheep populations in our study are notably high, similar to those observed in Holstein dairy cattle (FROH = 0.15,44). This increased inbreeding likely results from the intensive selective breeding practices at the Jovain Agricultural & Industrial Corporation breeding facility, which resemble those used in dairy cattle. However, it’s important to note that FROH values in Holsteins were probably calculated using microarray data, which offers higher marker density and a more even distribution across the genome compared to the GBS method used in our study. This methodological difference might affect the comparability of FROH values, as microarrays tend to capture longer ROH segments more accurately79. Despite this limitation, the elevated FROH values in our study suggest increased inbreeding in the crossbred populations, unlike the lower FROH values and greater genetic diversity reported for other Iranian sheep breeds. Additionally, Ceballos et al.24 emphasized the importance of considering minimum ROH length thresholds when calculating ROH, as shorter segments are more susceptible to bias.
These findings underscore the distinct inbreeding dynamics among the populations, as reflected in the FROH ranking of TDM > TTM > BRM > BMM > M (Table 4 and Fig. 6). The higher FROH values and longer ROH segments in the crossbred populations, particularly in TDM and TTM, suggest that while crossbreeding has introduced greater genetic variation, it has also led to substantial recent inbreeding or genetic bottlenecks. This is evident in the higher average ROH lengths and the greater proportion of the genome covered by ROH in these populations compared to the Moghani pure sheep (Fig. 6, Table 4, and Supplementary Table 2). Furthermore, Fig. 7 presents a schematic visualization of the Pearson correlation between two measures of molecular inbreeding (FIS and FROH). In Moghani sheep, which exhibit the highest genetic diversity, there is a strong negative correlation between FIS and FROH (r = − 0.936, p = 0). This supports the conclusion that higher genetic diversity in Moghani sheep is associated with fewer extended homozygous regions, indicating a history of distant inbreeding and effective preservation of genetic diversity. This negative correlation indicates that higher genetic diversity, as evidenced by lower FIS values, is associated with fewer extended homozygous regions, reflected in a lower FROH (Table 4). In contrast, crossbred populations such as BRM and TTM show more complex relationships between FIS and FROH. BRM demonstrates a significant negative correlation similar to Moghani, suggesting a close relationship between inbreeding measures. TTM also shows a significant negative correlation, but with a different strength and pattern, reflecting its unique genetic dynamics. Despite crossbreeding introducing more genetic variation, the extent of homozygosity (as captured by FROH) still strongly influences inbreeding measures. The higher inbreeding levels observed in crossbred lambs are largely due to the limited number of exotic founder sires (two Texel sires: Dalzell and Tamlet, and two Booroola sires: Romney and Merino) used to generate the crossbred lamb populations. This process restricted the inheritance of exotic germplasm, enhancing productivity but reducing adaptability and survivability due to limited genetic diversity. Forutan et al.44 conducted a simulation study using Holstein dairy cattle data, demonstrating that inbreeding coefficients derived from runs of homozygosity were more accurate than those based on pedigree (FPED) or genomic relationship matrices (GRM). In the absence of pedigree data, several studies have underscored the utility of ROH segments for evaluating an individual’s inbreeding level in sheep populations18,28,69,77,80.

Inbreeding within sheep populations. The Pearson correlation is presented between two measures of molecular inbreeding metrics. Populations are coded as M = Moghani pure sheep, BMM = Booroola Merino × Moghani, BRM = Booroola Romney × Moghani, TDM = Texel Dalzell × Moghani, TTM = Texel Tamlet × Moghani.
Overall, these findings underscore the intricate interplay between genetic diversity and inbreeding dynamics. The Moghani population maintains shorter ROH lengths and moderate FROH values, indicating effective management of genetic diversity and less recent inbreeding. In contrast, the BRM and BMM populations reflect a blend of ancient and recent inbreeding, while the TTM and TDM populations, with their longer ROH segments and higher FROH values, are indicative of more recent inbreeding or smaller effective population sizes. These results highlight how different populations manage their genetic diversity and how inbreeding dynamics vary across different genetic backgrounds and breeding histories.
Genomic regions within ROH islands and their functional annotation
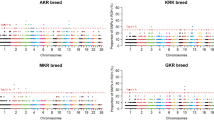
ROH islands could indicate genomic regions that have undergone natural or artificial selection. To explore this, we aimed to identify the most homozygous variants within ROH islands as potential candidates for recent adaptation. Our focus was on the outlying SNPs in the top of ROHs for each population. Due to varying levels of polymorphism, effective population sizes, and historical selection pressures, the homozygosity threshold for defining an ROH island was specific to each population. Breeds with higher genetic diversity, such as Moghani (M), required a lower threshold (67%) to identify ROH islands, while crossbred populations with more extensive homozygosity, such as BRM (78%) and TDM (73%), necessitated higher thresholds. These thresholds, representing the top 1% of SNPs with the highest ROH frequencies, reflect breed-specific differences in linkage disequilibrium, selection history, and genetic diversity. Consequently, we established SNP homozygosity thresholds of ≥ 67%, 76%, 78%, 73%, and 69% for M, BMM, BRM, TDM, and TTM, respectively (Fig. 8). The average length of ROH islands across all breeds was 250.52 kb ± 20.84, with an average of 50.31 ± 0.60 consecutive SNPs per region (details in Supplementary Table 3). As a result, ROH islands often broke into shorter segments due to the presence of short heterozygous gaps within the ROH sequences43. Summary statistics are provided in detail in Supplementary Table 3 and are illustrated schematically in Fig. 8.

Genome wide distribution of runs of homozygosity (ROH) hotspots. The x-axis represents the SNP genomic coordinate chromosome-wise, and the y-axis shows the proportion of overlapping ROH shared among individuals based upon number in population. Populations are coded as M = Moghani pure sheep, BMM = Booroola Merino × Moghani, BRM = Booroola Romney × Moghani, TDM = Texel Dalzell × Moghani, TTM = Texel Tamlet × Moghani.
As detailed in Table 5, the identification of ROH islands co-located with QTLs across the five sheep populations (M, BMM, BRM, TDM, and TTM) underscores critical genetic regions linked to key production traits. This analysis not only enhances our understanding of the genetic architecture within these populations but also provides valuable insights for breeding programs focused on improving economically important traits, including wool quality, exterior characteristics, reproduction, and health81. In the Moghani pure breed, the focus on QTL regions linked to wool quality (e.g., QTL_IDs 263609, 263599) and exterior traits (e.g., QTL_ID 263629) aligns with previous studies that have identified similar regions in other sheep breeds known for their wool production and adaptability80,82,83. The Moghani breed’s adaptability and resilience, coupled with these QTL findings (see Table 5), suggest that these genetic regions could be critical for maintaining these traits under various environmental conditions, reinforcing the importance of these loci in breeding programs aimed at improving wool quality. The overlaps in QTLs related to wool quality, reproduction (QTL_IDs 263641, 263643), and meat traits (QTL_IDs 95880, 95881) observed in the BMM and BRM crossbreeds reflect the dual-purpose breeding objectives of these populations (Table 5). Similar findings have been reported in other prolific sheep breeds, such as the Texel28, Finnsheep81, Romanov69, Balouchi84, where QTLs linked to reproductive traits and meat production have been identified, underscoring the genetic complexity underlying these economically important traits. The identification of these overlapping QTLs in BMM and BRM crossbreeds suggests that these populations share a common genetic basis for prolificacy and meat production, which could be exploited in breeding programs to optimize both traits. The TDM and TTM crossbreeds, known for their muscularity and meat production, showed significant QTL regions associated with wool quality, reproduction (e.g., QTL_ID 263648), and particularly meat and carcass traits (QTL_IDs 95878, 95879) (Table 5). These findings are consistent with studies on other terminal sire breeds, such as Texel and Suffolk85,86,87, where QTLs related to muscle development and carcass quality have been well documented (Table 5). The presence of these QTLs in TDM and TTM crossbreeds reinforces their potential as terminal sires in breeding programs aimed at improving carcass traits and meat yield. The identification of common QTL regions across chromosomes 1, 4, 7, 9, and 22, particularly the prominent region on chromosome 1 (POS: 111294404–122825051) associated with wool quality, suggests the existence of conserved genetic loci influencing wool traits across different sheep populations. This finding is in line with previous reports that have identified similar QTL hotspots on chromosome 1 in other wool-producing breeds88, indicating that these regions may play a crucial role in the genetic regulation of wool quality across diverse genetic backgrounds.
Further analysis of these ROH islands, as illustrated in the Venn diagram (Fig. 9) and detailed in Supplementary Table 4, reveals both shared and unique variants (Fig. 9A) and genes (Fig. 9B) across the five sheep populations. The genes identified in each population, as listed in Supplementary Table 4, include: M [CYB5, GGTA1P, GGTA2P, Calpha], BMM [RXRG, PSMD4, SLC25A33, U6, LHB, APIP, SLC25A20, LIN28A, PAG4, AVPR1A, PDGFB, ASNA1, CD14, DGAT1], BRM [IGFBP4, mTOR, ARFRP1, SLC25A20, MRCL3], TDM [ARFRP1, F2, TLR9, DYA, MRCL3, GCK], and TTM [UROD, PSMD4, ARFRP1, F2, DYA, VEGFA, ANXA2].

Venn diagrams illustrating the overlap and uniqueness of variants and genes within ROH islands across five sheep populations (M, BMM, BRM, TDM, and TTM). (A) Venn diagram showing shared and unique variants within ROH islands among the populations. (B) Venn diagram highlighting shared and unique genes within ROH islands across the populations.
Figure 10 highlights the pathways enriched by KOBAS analysis for genes located within ROH islands across four crossbred lamb populations: BMM, BRM, TDM, and TTM. In the BMM (Fig. 10A) and BRM (Fig. 10B) populations, which are noted for their prolificacy due to the introgression of the FecB/Booroola gene, key pathways related to reproductive and health traits were identified. For instance, in the BMM population, the "Prolactin signaling pathway" and “Ovarian steroidogenesis” are linked to reproductive efficiency through the LHB gene (Fig. 10A), findings that align with previous studies showing the role of prolactin and steroidogenesis in enhancing reproductive traits in prolific sheep breeds89. In the BRM population, the "Central carbon metabolism in cancer" pathway, associated with the mTOR gene, underscores the importance of this pathway in overall health maintenance, potentially influencing both growth and reproductive traits90,91. Additionally, in the BMM population, pathways such as "Cysteine and methionine metabolism" and "Glycerolipid metabolism," associated with the genes APIP and DGAT1 respectively, suggest enhanced metabolic adaptability. These metabolic pathways have been previously linked to energy efficiency and fat deposition, critical traits in sheep bred for specific production purposes92,93,94,95,96. Conversely, the TDM and TTM populations, bred for enhanced muscularity through the introduction of the myostatin hyper-muscular gene, showed pathways related to muscle growth, metabolic efficiency, and immune response. In the TDM population (Fig. 10C), the "Insulin signaling pathway" and "Starch and sucrose metabolism," both associated with the GCK gene, are critical for energy utilization and muscle development97. These pathways are well-known for their roles in optimizing muscle growth and feed efficiency in livestock.

KOBAS-enriched pathways associated with genes in ROH islands across four crossbred lamb populations. (A) BMM: Pathways related to reproductive traits and metabolic adaptability. (B) BRM: Pathways linked to overall health maintenance. (C) TDM: Pathways supporting muscle development and energy utilization. (D) TTM: Pathways involved in muscle growth and immune response.
The TTM population (Fig. 10D) exhibited key pathways such as the "VEGF signaling pathway" and the "PI3K-Akt signaling pathway," associated with the VEGFA gene, which are vital for muscle growth, as reviewed by Talebi et al.98. These pathways, along with the three major signaling pathways: (1) insulin/insulin-like growth factor 1 (IGF1)–Akt–rapamycin (mTOR); (2) transforming growth factor (TGF)β/myostatin/activin/BMP; and (3) β-adrenergic signaling, are known to control skeletal muscle growth, as evidenced by studies in hyper-muscular sheep breeds99,100,101. Immune-related pathways, such as the "Intestinal immune network for IgA production" and the "Complement and coagulation cascades" (Fig. 10D), linked to the DYA and F2 genes respectively, underscore the importance of a robust immune system for the health and productivity of these meat-type lambs. This is consistent with the growing understanding that immune competence is a critical factor in the productivity of livestock, particularly in intensive breeding systems102,103.
The findings of the present study reflect the targeted breeding strategies employed in these sheep populations, with reproductive and metabolic enhancements observed in BMM and BRM, and improved muscle growth and metabolic efficiency in TDM and TTM. The shared genetic pathways suggest common underlying mechanisms contributing to overall productivity and health, despite distinct breeding objectives. This enhanced understanding of the genetic architecture underlying important traits provides valuable insights for future breeding programs aimed at improving productivity and resilience. However, it is important to acknowledge that GBS, while a valuable tool for variant discovery, has limitations such as missing data and heterozygosity gaps, which may impact estimates of genetic diversity and population structure. The limited genotype representation calls for caution when interpreting the results. Future studies should aim to incorporate broader genotype representation to overcome these limitations, which would strengthen the findings. Continued exploration of ROH islands and their associated pathways will be essential for advancing genomic selection strategies and optimizing the genetic potential of livestock populations.
Conclusions
This investigation presents the initial systematic comparison of the genomic structure of Moghani sheep and their crossbred counterparts with Texel and Booroola sheep, providing valuable insights into the genetic diversity, admixture, and adaptation of these populations. Besides, it makes a valuable contribution to the comprehension of the genetic diversity and selection signatures of pure and crossbred sheep populations. Our data indicated that crossbred populations displayed longer ROH segments and higher FROH values, which suggested recent inbreeding or genetic bottlenecks, as revealed by GBS data analysis. In contrast, the Moghani strain exhibited shorter ROH segments, which was indicative of its more distant inbreeding history and its greater genetic diversity. The four identified candidate genes comprising of Prolactin signaling (LHB) central carbon metabolism in cancer (mTOR) are associated with reproductive and metabolic pathways in the Booroola populations while VEGF signaling and Insulin linked to muscle development and immune responses in the Texel populations. These gene may provide valuable insights into the biological processes that underlie fundamental production traits and may be used as breeding program targets to enhance flesh production, reproductive efficiency, and wool quality. Nonetheless, recommending genetic improvement strategies is unfeasible without addressing the limited sample size and the limitations of GBS, including heterozygosity gaps and absent data, which require careful evaluation. Thus, increasing the sample size, utilizing more comprehensive genotyping methodologies, and addressing data gaps are all areas that require further investigation in future study.
Data availability
The datasets generated and/or analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) repository under BioProject PRJNA1166956. The raw Illumina sequencing reads can be accessed at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1166956.
References
Alberto, F. J. et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 813 (2018).
World Livestock 2011—Livestock in food security. http://www.fao.org/3/i2373e/i2373e00.htm.
Talebi, R. et al. Detection of single nucleotide polymorphisms at major prolificacy genes in the Mehraban sheep and association with litter size. Ann. Anim. Sci. 18, 685 (2018).
Talebi, R. et al. Association of ovine follistatin gene polymorphisms with body measurements, fat-tail traits and morphometric of head in Iranian Mehraban sheep. Small Rumin. Res. 225, 107020 (2023).
Majd, S. A. et al. Polymorphism identification in ovine KISS1R/GPR54 gene among pure and crossbreeds of Iranian sheep. Small Rumin. Res. 173, 23–29 (2019).
Ahmadi, A., Mohammadizade, M., Zamani, P., Farah-avar, A. & Talebi, R. Identification of polymorphism in HSP70A1A gene in Mehraban and Romanov sheep breeds using PCR-SSCP technique. Res. Anim. Prod. 12, 169–179 (2021).
Esfandyari, H., Aslaminejad, A. A. & Rafat, S. A. Wool characteristics in the third generation of Arkharmerino × Ghezel and Arkharmerino × Moghani crossbreed sheep. Trop. Anim. Health Prod. 43, 1337–1343 (2011).
Mokhber Yousefabad, M., Shodja, J., Alijani, S. & Behruzlak, M. Evaluation of fleece characteristics of first and second generations of ArkharMerinos × Ghezel and ArkharMerinos × Moghani crossbred sheep. J. Agric. Sci. Nat. Resour. 15, 149–156 (2008).
Shodja, J., Farahvash, T. & Rafat, S. A. The evaluation of fleece characteristics in Arkharmerino Moghani F1. In Proceedings of the 11”AAAP Animal Science Congress 624–626 (2004).
Talebi, R., Ghaffari, M. R., Fabre, S. & Qanbari, S. Comparison of the growth performance traits in F1 crossbred lambs between two strains Booroola Merino × Moghani and Booroola Romney × Moghani. In 12th National and 4th International Biotechnology Congress of Islamic Republic of Iran 1–11 (2021).
Talebi, R., Ghaffari, M. R., Fabre, S., Mardi, M. & Kazemi Alamouti, M. Comparison of the growth performance between pure Moghani sheep and crosses with Texel or Booroola sheep carrying major genes contributing to muscularity and prolificacy. Anim. Biotechnol. 34, 3495–3506 (2023).
Talebi, R. et al. Assessing the performance of Moghani crossbred lambs derived from different mating systems with Texel and Booroola sheep. PLoS ONE 19, 1–20 (2024).
Kumar, H. et al. Genome-wide detection of copy number variations in Tharparkar cattle. Anim. Biotechnol. 34, 448–455 (2023).
Nayak, S. S. et al. Comprehensive selection signature analyses in dairy cattle exploiting purebred and crossbred genomic data. Mamm. Genome 34, 615–631 (2023).
Arefnejad, B. et al. Unveiling the population genetic structure of Iranian horses breeds by whole-genome resequencing analysis. Mamm. Genome 35, 201–227 (2024).
Bruford, M. W. et al. Prospects and challenges for the conservation of farm animal genomic resources, 2015–2025. Front. Genet. 6, 1–11 (2015).
Domrazek, K. & Jurka, P. Application of next-generation sequencing (NGS) techniques for selected companion animals. Animals 14, 1578 (2024).
Ghoreishifar, S. M., Rochus, C. M., Moghaddaszadeh-ahrabi, S. & Davoudi, P. Shared ancestry and signatures of recent selection in Gotland sheep. Genes https://doi.org/10.3390/genes12030433 (2021).
Abdoli, R. et al. Runs of homozygosity and cross-generational inbreeding of Iranian fat-tailed sheep. Heredity 130, 358–367 (2023).
Larsson, M. N. A. et al. Ancient sheep genomes reveal four Millennia of North European short-tailed sheep in the Baltic Sea region. Genome Biol. Evol. 16, 1–16 (2024).
McQuillan, R. et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372 (2008).
Talebi, R., Szmatoła, T., Mészáros, G. & Qanbari, S. Runs of homozygosity in modern chicken revealed by sequence data. G3 Genes Genomes Genet. 10, 4615–4623 (2020).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234 (2018).
Ceballos, F. C., Hazelhurst, S. & Ramsay, M. Assessing runs of Homozygosity: A comparison of SNP Array and whole genome sequence low coverage data. BMC Genomics 19, 1–12. https://doi.org/10.1186/s12864-018-4489-0 (2018).
Zhong, T. et al. Comparative whole-genome resequencing to uncover selection signatures linked to litter size in Hu Sheep and five other breeds. BMC Genomics 25, 1–14 (2024).
Lukic, B. et al. Genomic signatures of selection, local adaptation and production type characterisation of East Adriatic sheep breeds. J. Anim. Sci. Biotechnol. https://doi.org/10.1186/s40104-023-00936-y (2023).
Tsartsianidou, V. et al. A comprehensive genome—wide scan detects genomic regions related to local adaptation and climate resilience in Mediterranean domestic sheep. Genet. Sel. Evol. 53, 1–17 (2021).
Purfield, D. C., McParland, S., Wall, E. & Berry, D. P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 12, 1–23 (2017).
Argun Karsli, B. et al. Genome-wide discovery of selection signatures in four Anatolian sheep breeds revealed by ddRADseq. Sci. Rep. 14, 1–10 (2024).
Kusza, S., Badaoui, B. & Wanjala, G. Insights into the genomic homogeneity of Moroccan indigenous sheep breeds though the lens of runs of homozygosity. Sci. Rep. 14, 1–11 (2024).
Rajawat, D. et al. Revealing genomic footprints of selection for fiber and production traits in three Indian sheep breeds. J. Nat. Fibers 19, 14963–14974 (2022).
Bertolini, F. et al. Signatures of selection and environmental adaptation across the goat genome post-domestication 06 Biological Sciences 0604 Genetics. Genet. Sel. Evol. 50, 1–24 (2018).
Colli, L. et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 50, 1–20 (2018).
Onzima, R. B. et al. Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds. Front. Genet. 9, 318 (2018).
Sun, X. et al. Genetic diversity and selection signatures in Jianchang black goats revealed by whole-genome sequencing Data. Animals 12, 1–15 (2022).
Jiang, Y. et al. Genome-wide detection of genetic structure and runs of homozygosity analysis in Anhui indigenous and Western commercial pig breeds using PorcineSNP80k data. BMC Genomics 23, 1–14 (2022).
Di Gregorio, P., Perna, A., Di Trana, A. & Rando, A. Identification of ROH Islands conserved through generations in pigs belonging to the Nero Lucano Breed. Genes 14, 1503 (2023).
Schiavo, G. et al. Runs of homozygosity islands in Italian cosmopolitan and autochthonous pig breeds identify selection signatures in the porcine genome. Livest. Sci. 240, 104219 (2020).
Colpitts, J., McLoughlin, P. D. & Poissant, J. Runs of homozygosity in Sable Island feral horses reveal the genomic consequences of inbreeding and divergence from domestic breeds. BMC Genomics 23, 1–18 (2022).
Kasarda, R. et al. Level of inbreeding in Norik of muran horse: Pedigree vs. Genomic data. Acta Univ. Agric. Silvic. Mendelianae Brun. 67, 1457–1463 (2019).
Ablondi, M., Viklund, Å., Lindgren, G., Eriksson, S. & Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genomics 20, 1–12 (2019).
Santos, W. B. et al. Genome-wide scans for signatures of selection in Mangalarga Marchador horses using high-throughput SNP genotyping. BMC Genomics 22, 1–17 (2021).
Nandolo, W. et al. Misidentification of runs of homozygosity islands in cattle caused by interference with copy number variation or large intermarker distances. Genet. Sel. Evol. 50, 1–13 (2018).
Forutan, M. et al. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genomics 19, 1–12 (2018).
Toro Ospina, A. M. et al. Genome-wide identification of runs of homozygosity islands in the Gyr breed (Bos indicus). Reprod. Domest. Anim. 55, 333–342 (2020).
Rajawat, D. et al. Identification of important genomic footprints using eight different selection signature statistics in domestic cattle breeds. Gene 816, 146165 (2022).
Yuan, J. et al. Genome-wide run of homozygosity analysis reveals candidate genomic regions associated with environmental adaptations of Tibetan native chickens. BMC Genomics 23, 1–16 (2022).
Strillacci, M. G. et al. Looking at genetic structure and selection signatures of the Mexican chicken population using single nucleotide polymorphism markers. Poult. Sci. 97, 791–802 (2018).
Talebi, R., Seighalani, R. & Qanbari, S. A handmade DNA extraction kit using laundry powder; insights on simplicity, cost-efficiency, rapidity, safety and the quality of purified DNA. Anim. Biotechnol. 32, 388–394 (2021).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6, e19379 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1330 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Chhotaray, S. et al. Genomics Ancestry informative markers derived from discriminant analysis of principal components provide important insights into the composition of crossbred cattle. Genomics 112, 1726–1733 (2020).
Pal, D. et al. Unraveling genetic admixture in the Indian crossbred cattle by different approaches using Bovine 50K BeadChip. Trop. Anim. Health Prod. 54, 135 (2022).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Howrigan, D. P., Simonson, M. A. & Keller, M. C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genomics 12, 1–15 (2011).
Wright, S. Evolution in Mendelian populations. Genetics 16, 97–159 (1931).
Bu, D. et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325 (2021).
Leymaster, K. A. Fundamental aspects of crossbreeding of sheep: Use of breed diversity to improve efficiency of meat production. Sheep Goat Res. J. 17, 50–59 (2002).
Kijas, J. W. et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10, e1001258 (2012).
Lv, F. H. et al. Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 31, 3324–3343 (2014).
Nosrati, M., Asadollahpour Nanaei, H., Amiri Ghanatsaman, Z. & Esmailizadeh, A. Whole genome sequence analysis to detect signatures of positive selection for high fecundity in sheep. Reprod. Domest. Anim. 54, 358–364 (2019).
Zhang, C. L. et al. Analysis on the desert adaptability of indigenous sheep in the southern edge of Taklimakan Desert. Sci. Rep. 12, 1–16 (2022).
Bedhiaf-Romdhani, S. et al. Efficiency of genotyping by sequencing in inferring genomic relatedness and molecular insights into fat tail selection in Tunisian sheep. Anim. Genet. 54, 389–397 (2023).
Yang, J. et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biol. 25, 1–46 (2024).
Nicoloso, L. et al. Genetic diversity of Italian goat breeds assessed with a medium-density SNP chip. Genet. Sel. Evol. 47, 1–10 (2015).
Deniskova, T. E. et al. Population structure and genetic diversity of 25 Russian sheep breeds based on whole-genome genotyping. Genet. Sel. Evol. 50, 1–16 (2018).
Ciani, E. et al. Merino and Merino-derived sheep breeds: A genome-wide intercontinental study. Genet. Sel. Evol. 47, 1–12 (2015).
García-Gámez, E. et al. GWA analysis for milk production traits in dairy sheep and genetic support for a QTN influencing milk protein percentage in the LALBA gene. PLoS ONE 7, 1–9 (2012).
Eydivandi, S., Sahana, G., Momen, M., Moradi, M. H. & Schönherz, A. A. Genetic diversity in Iranian indigenous sheep vis-à-vis selected exogenous sheep breeds and wild mouflon. Anim. Genet. 51, 772–787 (2020).
Barani, S. et al. Genome-wide study of linkage disequilibrium, population structure, and inbreeding in Iranian indigenous sheep breeds. PLoS ONE 18, 1–19 (2023).
Szmatoła, T., Gurgul, A., Jasielczuk, I., Fu, W. & Ropka-Molik, K. A detailed characteristics of bias associated with long runs of homozygosity identification based on medium density SNP microarrays. J. Genomics 8, 43 (2020).
Patiabadi, Z. et al. Whole-genome scan for selection signature associated with temperature adaptation in Iranian sheep breeds. PLoS ONE 19, e0309023 (2024).
Li, X. et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 11, 2815 (2020).
Gutiérrez-Gil, B. et al. High-resolution analysis of selection sweeps identified between fine-wool Merino and coarse-wool Churra sheep breeds. Genet. Sel. Evol. 49, 1–24 (2017).
Zhao, B. et al. Integration of a single-step genome-wide association study with a multi-tissue transcriptome analysis provides novel insights into the genetic basis of wool and weight traits in sheep. Genet. Sel. Evol. 53, 1–14 (2021).
Gholizadeh, M., Rahimi-Mianji, G., Nejati-Javaremi, A., De Koning, D. J. & Jonas, E. Genomewide association study to detect QTL for twinning rate in Baluchi sheep. J. Genet. 93, 489–493 (2014).
Hadjipavlou, G., Matika, O., Clop, A. & Bishop, S. C. Two single nucleotide polymorphisms in the myostatin (GDF8) gene have significant association with muscle depth of commercial Charollais sheep. Anim. Genet. 39, 346–353 (2008).
Macfarlane, J. M. et al. Effect and mode of action of the Texel muscling QTL (TM-QTL) on carcass traits in purebred Texel lambs. Animal 8, 1053–1061 (2014).
Walling, G. A. et al. Mapping of quantitative trait loci for growth and carcass traits in commercial sheep populations. J. Anim. Sci. 82, 2234–2245 (2004).
McRae, A. F. et al. Linkage disequilibrium in domestic sheep. Genetics 160, 1113–1122 (2002).
Davis, G. H. et al. DNA tests in prolific sheep from eight countries provide new evidence on origin of the Booroola (FecB) mutation. Biol. Reprod. 66, 1869–1874 (2002).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Guo, Z. & Yu, Q. Role of mTOR signaling in female reproduction. Front. Endocrinol. 10, 1–13 (2019).
Suárez-Vega, A., Gutiérrez-Gil, B., Klopp, C., Tosser-Klopp, G. & Arranz, J. J. Variant discovery in the sheep milk transcriptome using RNA sequencing. BMC Genomics 18, 1–14 (2017).
Taghizadeh, S. et al. Genome-wide identification of copy number variation and association with fat deposition in thin and fat-tailed sheep breeds. Sci. Rep. 12, 1–13 (2022).
Gebreselassie, G., Berihulay, H., Jiang, L. & Ma, Y. Review on genomic regions and candidate genes associated with economically important production and reproduction traits in sheep (Ovies aries). Animals 10, 33 (2020).
Tuersuntuoheti, M. et al. Exploring the growth trait molecular markers in two sheep breeds based on Genome-wide association analys. PLoS ONE 18, 1–18 (2023).
Li, Y. et al. Uncovering the candidate genes related to sheep body weight using multi-trait genome-wide association analysis. Front. Vet. Sci. 10, 1–11 (2023).
Irwin, D. M. & Tan, H. Evolution of glucose utilization: Glucokinase and glucokinase regulator protein. Mol. Phylogenet. Evol. 70, 195–203 (2014).
Talebi, R., Ghaffari, M. R., Zeinalabedini, M., Abdoli, R. & Mardi, M. Genetic basis of muscle-related traits in sheep: A review. Anim. Genet. 53, 723–739 (2022).
Sartori, R., Romanello, V. & Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 12, 1–12 (2021).
Sartori, R. et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 296, 1248–1257 (2009).
Clop, A. et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 38, 813–818 (2006).
Garrick, D. J., Taylor, J. F. & Fernando, R. L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 41, 1–8 (2009).
Staelens, L., Langenaeken, T., Rega, F. & Meuris, B. Difference in coagulation systems of large animal species used in cardiovascular research: A systematic review. J. Artif. Organs https://doi.org/10.1007/s10047-024-01446-y (2024).
Acknowledgements
The authors express their heartfelt appreciation to Jovain Agricultural & Industrial Company, Khorasan Razavi, Jovain, for supplying valuable data. Gratitude is extended to Mr. Hamid Jafar-Abadi, Mr. Mohammad Reza Ghale-Noei, Mr. Valiollah Annabestani, and the diligent animal husbandry team for their collaborative contributions to data collection and blood sampling.
Funding
This research was financially supported by the Agricultural Biotechnology Research Institute of Iran (ABRII), Karaj, Iran (96036-020-05-05-01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.T. and M.R.G.; Data curation: R.T.; Formal analysis: R.T. and T.S.; Funding acquisition: R.T., M.M., M.Z., and M.R.G.; Investigation: R.T.; Methodology: R.T., T.S., M.K.A., and SAJT; Project administration: R.T. and M.R.G.; Resources: R.T., M.Z., and M.R.G.; Software: R.T. and T.S.; Supervision: R.T. and M.R.G.; Validation: R.T. and T.S.; Visualization: R.T. and T.S.; Writing – original draft: R.T.; Writing – review & editing: R.T., T.S., and M.R.G.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Talebi, R., Mardi, M., Zeinalabedini, M. et al. Genomic architecture of purebred and crossbred Moghani lambs with Texel and Booroola sheep. Sci Rep 15, 22833 (2025). https://doi.org/10.1038/s41598-025-06152-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06152-0
