
Abstract
In this paper, we introduce QProteoML, a new quantum machine learning (QML) framework for predicting drug sensitivity in Multiple Myeloma (MM) using high-dimensional proteomic data. MM, an extremely heterogeneous condition, displays often mixed responses to treatment, with a large number of patients showing drug resistance to proteasome inhibitors and immune modulatory agents. However, the methods previously used for genomic and proteomic data analysis techniques are plagued by issues of high dimensionality, imbalanced class distribution and feature redundancy, which work against the accurate predictability and generalizability of such methods. These are compounded by the so-called “curse of dimensionality”, with dimensions far outnumbering samples, hence classical model overfitting. In this work, we present QProteoML as an integration of quantum techniques purposefully developed to deal with high-dimensional, imbalanced and redundant data. The framework integrates a combination of Quantum Support Vector Machine (QSVM), Quantum Principal Component Analysis (qPCA), Quantum Annealing (QA) for feature selection and Quantum Generative Adversarial Networks (QGANs) for data augmentation. These quantum algorithms exploit certain quantum phenomena (superposition and entanglement) to perform modelling of nonlinear relationships, dimensionality reduction, and class-imbalance issues. QSVM employs quantum kernels to map data into a higher-dimensional Hilbert space, so that the model can detect complex patterns in MM drug resistance. qPCA reduces dimensionality without loss of important variance, and thus improves computation efficiency. In addition, Quantum Annealing successfully extracts the most informative biomarkers with low redundancy. QProteoML was experimentally tested by comparing accuracy, F1 score and AUC ROC between classical machine learning models such as Support Vector Machine (SVM), Random Forest (RF), Logistic Regression (LR), and K-Nearest Neighbors (KNN). Our results demonstrate that QProteoML performs better than classical models, particularly in identifying the drug resistant minority class of patients. Additionally, the model is interpretable and stresses important biomarkers of drug sensitivity in MM. This research opens the possibility of quantum machine learning in personalised medicine for Multiple Myeloma. It demonstrates that quantum algorithms can perform complex biological data suggesting more reliable and accurate drug sensitivity predictions. Future research will be directed toward clinical validation of the given system with larger and more diverse cohorts of MM patients; the integration of quantum hardware for practical applications.
Similar content being viewed by others
Introduction
The bone marrow is infiltrated with malignant plasma cells that proliferate clonally and collectively constitute the heterogeneous hematologic malignancy Multiple Myeloma (MM). More facets are affected, and patients are molecularly heterogeneous. This resistance is, however, being encountered in patients who are already being treated with what can be considered ‘advanced’ therapies such as proteasome inhibitors (e.g., Bortezomib) or immune modulatory agents (e.g., Lenalidomide), and drug resistance eventually arises1. The high level of incompleteness about the therapeutic response and resistance progression, along with more evidence for the predictions, implies that we need personalized treatment strategies that can predict each patient’s drug sensitivities.
Proteomics, as an approach to studying the structure and function of proteins, has become a valuable tool for understanding the molecular basis of MM and identifying biomarkers related to sensitivity or resistance to the drugs. Researchers analyze the protein expression profiles of cancer patients and classify them into various groupings of patients based on their respective probabilities of showing favourable responses to any specific treatment. As a first example, previously, several distinct proteins like Clustering (CLU) and Angiogenin (ANG) have been shown to contribute as key biomarkers for Bortezomib resistance2. However, proteomic datasets are high dimensional (thousands of proteins per patient), the sample size is small, and classes are imbalanced (sensitive versus resistant), which complicates the application of traditional analytical methods3. These complexities challenge developing robust and generalizable predictive models using classical machine learning (ML) techniques.
Using advanced machine learning (ML) tools, including classical ML methods (Support vector machines (SVM), Random Forests (RF), Logistic Regression, etc.), drug sensitivity was predicted in MM. Nevertheless, these methods have some degree of success on this task of cancer drug response prediction but suffer from drawbacks such as the 'curse of dimensionality’, ‘over fitting’, and ‘class imbalance’, especially when dealing with extensive proteomic data4. Furthermore, because several correlated proteins provide redundant information, the difficulty of interpreting and generalizing the models increases.
A new approach can be taken, however, to solve such a dilemma, which is a novel form called Quantum Machine Learning (QML). Quantum computation, therefore, is not classical computation. The capability of this machine relies on quantum mechanical phenomena such as superposition, entanglement and quantum interference to calculate information that cannot be calculated in a classical computer5. The promise of QML in tackling the high level of complexity in high-dimensional data can be attributed to these properties, which help reduce problems such as class imbalance, overfitting, and feature redundancy. For example, quantum kernels (adapted, for instance, by Quantum Support Vector Machines (QSVM)) can form an extremely relevant tool for encoding nonlinear relationships in imbalanced data and, therefore, be used for classification tasks in oncology6. Quantum Principal Component Analysis (qPCA) is another promising quantum-enhanced technique that is very suitable for proteomic data analysis in MM7 due to its capability for dimensionality reduction while preserving data variance. Additionally, quantum annealing was employed for feature selection to choose the minimal yet informative biomarker panels that are also highly accurate at classification with minimal redundancy8.
Although QML has been used in cancer studies, especially in genomic data analysis, drug finding and patient stratification, its application to MM proteomics is only emerging. Previously, the QML models were shown to surpass classical machine learning counterparts in the classification of cancer subtypes, as well as in drug response prediction, where data are imbalanced8. An extension of conventional GANs, Quantum Generative Adversarial Networks (QGANs) has also been used to generate synthetic data for the underrepresented classes, improving the robustness of the models to a class imbalance9,10. However, little has been done in the way of building on these applications in MM proteomics and the use of quantum algorithms to personalise MM treatment strategies.
The main contribution of this paper is QProteoML, a novel quantum machine learning framework that proposes predicting drug sensitivity in MM patients using high-dimensional, time-series proteomic data. QProteoML solves three main issues in MM drug sensitivity prediction. High dimensionality, class imbalance and redundant features problems. QProteoML integrates the QML (QSVM, qPCA, and Quantum Annealing) with the classical ML models to enable accurate but data-efficient drug sensitivity prediction. In this study, we show that QProteoML improves prediction accuracy and assists in identifying the crucial biomarkers responsible for tailored treatment design in MM cases. This work shows that QML can bring value to the problem of personalized medicine in MM and the development of more patient-specific treatment protocols.
Literature review
This paper presents the QProteoML framework for predicting such events based on time series high-dimensional proteomic data using quantum machine learning. The considerable heterogeneity at the molecular and clinical level is the substantial obstacle in treating MM. MM is a non-curable disease in which the marrow becomes infiltrated with abnormal plasma cells, which cause the malady. However, the available manipulations using proteasome inhibitors (e.g., Bortezomib) or immune modulatory agents (e.g., Lenalidomide) are specific for the molecular pathways, but the issue of drug resistance remains in a subset of patients11. These treatments have been successful at attenuating the malignancy, but an important percentage of patients develop resistance, and the molecular mechanism for resistance is a major question.
Proteomic profiling is a useful tool to elucidate the molecular biology of MM and the biomarkers associated with sensitivity or resistance to drugs12. Proteomics provides an opportunity for investigating the patterns of protein expression throughout a large cohort of patients and understanding MM at the protein level13. Important proteins that are related to drug resistance are known to act as biomarkers for MM patients for predicting drug response and resistance, such as Clusterin (CLU) and Angiogenin (ANG)14. Proteomics has been successful in stratifying MM recipients into sensitivity/resistance cohorts, associating protein profiles with survival and response to treatment of the recipient15. The high dimensionality of patients’ proteomic data, thousands of proteins per patient, and relatively small sample sizes pose challenges in feature interpretation and extrapolation of modelling. Also, an imbalance in class distribution, with the overwhelming majority of patients being sensitive to training, makes the training of accurate models challenging.
Oncology has become familiar with machine learning for developing predictive drug sensitivity and patient stratification models. While the data for drug sensitivity prediction for MM has been approached with some classical ML algorithms, such as Support Vector Machines, Random Forests, or Logistic Regression, they were no better than the basic SF algorithm. For example, effort has been made to differentiate sensitive from resistant patients on each other’s proteomic profiles with moderate success (such as using SVMs with Radial Basis Function (RBF) kernels16). However, both of these classical ML models have challenges using MM data since high-dimensional data sets featuring many features compared to the number of samples suffer from the "curse of dimensionality". This causes overfitting—the models would fit noise in the data instead of any generalizable patterns, and thus lead to poor predictive performance when the data is generalized.
Another important problem is class imbalance: most of the MM patients are sensitive to drugs, while the rest are resistant to the use of the drugs. It is due to this imbalance that we get biased model predictions with the model being likely to predict the majority class with more accuracy, while misclassifying the minority class17. Some of the popular ways of combating class imbalance include resampling techniques, which include under-sampling the majority class, over-sampling of the minority class, or the use of synthetic data generation methods. These techniques, however, tend not to address the true causes of class imbalance and may result in the performance of suboptimal performance.
Feature selection is also a challenge to classical ML methods. A few techniques are Principal Component Analysis (PCA), which reduces dimensionality and finds the relevant features. However, PCA tends not to keep the important protein markers for drug resistance, which are directly associated with the outcome. In certain instances, PCA ends up eliminating vital information while in the process of reducing variance wherein it leads to its diminishing both the interpretability and performance of the model18.
One of the effective solutions to these limitations is the integration of quantum machine learning (QML), which has many benefits compared to the classical approaches. QML can generate exponential accelerations to tackle complex programs, especially high-dimensional data, by making use of the quantum effect of superposition, entanglement, and quantum interference19. These attributes allow the quantum algorithms to compute huge datasets in shorter periods than classical systems. One of the most common examples of QML is the Quantum Support Vector Machine (QSVM) for which adds to classical SVM and employs the function of quantum kernels that map data to a higher-dimensional Hilbert space. This improvement enables QSVM to be able to model complex nonlinear relationships in imbalanced data, such as those existing in MM proteomics20.
A promising quantum technique is so called Quantum Support Vector Machine. It is an extension of classical SVM to a quantum kernel. Therefore, an application such as that with an imbalanced or large-scale dataset and their variants, such as those in the MM proteomics21, can directly use these quantum kernels based on the ability of these kernels to have complex nonlinear relationships between the data. In several cancer classification tasks22, it has been shown that QSVMs perform better than classical SVMs when imbalanced data are used.
The second set of key quantum techniques in oncology involves the quantum Principal Component Analysis, which addresses the high dimensionality of the datasets by projecting the data into a lower dimensional subspace that retains the essential features. Finally, qPCA allows to keep more variance and improve model performance, although they would hang on to key features23, not the traditional PCA. The fact that qPCA enables sample dependence means that it is an ideal tool for analyzing proteomic data in which reducing dimensionality without losing critical biomarker information is crucial to accurate drug sensitivity predictions24.
Besides, quantum annealing is a promising feature selection tool in oncology applications. The algorithms can serve as quantum annealing algorithms by finding the optimal subset of proteins (features) that is most predictive of drug sensitivity and thus providing critical biomarkers25. Using the optimization of biomarker panels to improve classification accuracy, quantum annealing has recently been successfully used for cancer genomics and drug discovery applications26. Furthermore, Classical and Quantum Generative Adversarial Networks (CQGANs) have generated synthetic data for the underrepresented class to make the neural network more robust and combat class imbalance by adding realistic synthetic proteomic profiles to the dataset27.
Though QML has succeeded in genomics and small-scale cancer research, consequently applying it to MM proteomics is still nascent. Most QML studies have been used for genomic data or small synthetic datasets and then cared little about clinical validation. However, few studies have completely integrated quantum algorithms into the MM proteomic workflow, leaving a scientific gap regarding methods by which these techniques can be applied to extensive, accurate data such as those in this study. Furthermore, integrating QML with quantum hardware is still an open challenge, and many quantum algorithms are still subjected to the constraints of modern quantum computing platforms.
Oncology is one of the fields with exciting opportunities to make an impact with quantum machine learning because the latter emerges as a new gate with the potential to tackle the current problems that limit classical machine learning. To overcome such limitations, QML integrates quantum techniques of QSVM, qPCA, and quantum annealing to improve precision medicine regarding drug sensitivity prediction, dimension reduction, and finding critical biomarkers. Nevertheless, more research is required to apply these techniques concerning MM proteomics, verify these results on a larger patient cohort, and investigate the suitability of quantum hardware for real-world MM clinical applications.
Materials and methods
The proteomic dataset comprised 39 Multiple Myeloma patients with the necessary preprocessing and the quantum machine learning techniques to predict drug sensitivity described in the "Materials and Methods". Figure 1 provides key methods, such as data normalization, the feature selection using feature selection methods like QSVM, qPCA and Quantum Annealing feature selection methods, and the use of QSVM, qPCA and Quantum Annealing for optimizing and improving the prediction accuracy using high-dimensional, imbalanced and redundant features.

QProteoML architecture diagram.
Dataset description
In this study, proteomic dataset (proteomics_data.csv) used, it containing 39 protein expression-level profiles of 2573 proteins in 39 patients with Multiple Myeloma. The author used the Individualized Systems medicine approach to generate the dataset by mass spectrometry analysis of CD138 + plasma cells. This method classifies MM patients into four chemosensitivity groups: highly sensitive, sensitive, resistant, and highly resistant, depending on their response to therapeutics.
The data for the experiment discussed in this paper is simulated, and as a result, there was no need to acquire ethical approval or obtain informed consent since no humans are involved when collecting data. All techniques used simulated data according to their guideline.
To this end, the dataset studied the same six clinically relevant drugs (e.g., Bortezomib, Lenalidomide, Navitoclax), and drug sensitivity scores (DSS) were cast into binary labels per a drug: sensitive (DSS ≥ 10.0) vs resistant (DSS < 10.0). Previous studies have shown that the DSS ≥ 10.0 threshold is consistent with drug sensitivity, as anticipated, corresponding to already established thresholds in drug sensitivity testing2,28. The resulting dataset contains 39 patients, 2573 proteins, and a binary label for the drug sensitivity of each drug to those patients. It is possible to reflect on the dataset structure as follows:
\(X = \left\{ {x_{1} ,\,x_{2} ,\,...,\,x_{n} } \right\}\,\) Where each \(x_{i} \, \in R^{p}\) (protein expression vector) for i = 1, 2, …, n.
The number of patients (n) here is 39, and the number of protein features per patient (p) is 2573. Denoting the sensitivity labels of each patient as:
\(y_{i} \in \left\{ {0,1} \right\}\) \(y_{i} = 1\) Where is sensitive, \(y_{i} = 0\) if resistant.
The high dimensionality of the dataset (2573 protein features per patient), combined with the relatively small sample size (39 patients), are the inherent challenges for data in the dataset. However, these factors further exacerbate the curse of dimensionality because the total number of features can be larger than the number of samples, and they would find it harder to overfit traditional machine learning models29. The dataset is highly dimensional in mathematical terms, which means that:
\(p\, > > \,n\) (more features than samples).
As the ratio of features to samples becomes unfavorable, it is more likely that one will overfit training data, and hence, classical machine learning algorithms do not generalize from training data to test data. In addition, the dataset is class imbalanced, where most patients are sensitive to the drugs, resulting in an unbalanced representation of the sensitive class30. For example, in the case of Bortezomib, the distribution of drug sensitivity labels is:
In the presence of such a class imbalance, traditional models may take a biased view towards the majority class (sensitive) and make biased predictions towards the sensitive class. To address this issue, resampling or the Synthetic Minority Over-Sampling Technique (SMOTE) and algorithms that explicitly take class imbalance into account are commonly utilized31.
Suppose there is such an imbalance in machine learning models. In that case, it can be dealt with using weighted loss functions or by reporting balanced metrics like the F1 score, which normalizes precision and recall to reflect an accurate measure of model performance, particularly for minority classes32.
The dataset gives a p ≫ n problem, where p > n; the number of features (2,53 proteins) is significantly greater than the number of samples (39 patients). This is a case for which traditional machine learning models will find it challenging because they tend to be over-fitted when there is an excess of features compared to the number of samples. To neutralise this, we used fivefold cross-validation to ensure the robustness of the model and thus prevent overfitting.
The dataset is extremely class-imbalanced: most patients are drug-sensitive (~ 83% drug-sensitive and ~ 17% drug-resistant)30. The existence of an imbalance in this class can affect traditional machine learning models to predict the majority class of sensitive patients, thus resulting in poor predictions for drug-resistant patients. In order to compensate for this imbalance, we used stratified sampling for cross-validation to achieve balance in the representation of sensitive and resistant classes as well as the training and test sets. In addition, class-weighted loss functions were implemented where misclassifications of the minority class were penalised, which increased the accuracy of estimating drug-resistant predictions. QGANS may be further experimented with in the next research for the generation of synthetic data that would help produce more representative data of the minority class and enhance the robustness of the model even against class imbalance.
Dimensionality reduction, such as Principal Component Analysis (PCA) or Quantum Principal Component Analysis (qPCA), is also used to solve the problem of high dimensionality33. The purpose is to compress the data until the number of features is minimized with as much retained variance as possible. Dimensionality reduction can be understood in the classical PCA domain.
To be specific, Quantum Principal Component Analysis (qPCA) was applied for retaining important variance as well as the reduction of features. This way, relevant information is kept, which makes the models more efficient and better at generalisation. In qPCA, quantum circuits are used to perform the dimensionality reduction using quantum operations in preserving the important variance with few features. This is an important implication as compared to the classical PCA, where qPCA can carry out the same task efficiently, particularly in situations where there is a high-dimensional data set similar to the one in the current study.
where \(W_{PCA}\) the matrix of eigenvectors corresponds to the principal components and \(X_{reduced}\) where the transformed data is in the lower-dimensional space. The dimensionality reduction aims to capture the most variance possible with fewer features:
In the case of quantum methods such as qPCA, the goal is to achieve a similar reduction of a dataset using quantum circuits that retain more variance with fewer features. This is done by taking advantage of quantum states and operations to improve the computational efficiency and accuracy of maintaining the valuable patterns of the data.
As such, data based on these inherent challenges (e.g., high dimensionality, small sample size, imbalanced class, etc.) poses a direct impact on the problems being explored by quantum machine learning algorithms such as QSVM and Quantum Annealing, and therefore capable of improving data processing. Selection of the features is more efficient based on quantum properties.
Data preprocessing
Before developing the model, the dataset underwent several preprocessing steps to standardize and prepare the data for machine learning. These preprocessing methods were crucial to guarantee the effective use of the raw proteomic data for classical and quantum machine learning models34.
Normalization of protein expression values
Firstly, raw protein expression values were normalized as a part of the data preprocessing step. However, because the protein expression levels can vary significantly concerning experimental and biological factors, normalization helps ensure data consistency among features35. The max–min normalization method was used to rescale features (protein expression value) from 0 to 1. The formula to have the minimum value normalized to zero and the maximum value to the value one is max–min normalization36.
where:
-
Its original protein expression value is x.
-
The dataset’s minimum value of the protein expression is min(x).
-
The maximum value of the protein expression in the dataset is denoted by max(x).
-
x′ is the normalized protein expression value.
This is especially critical for proteomic data, where raw protein levels can vary drastically. Scaling data mitigates problems caused by differences in experimental conditions. It also ensures that each protein makes the same contribution to the model and does not overweigh any features due to larger absolute values37.
Dimensionality reduction and feature selection
Since the data set consists of 2573 protein features per patient, many of these features are highly related, which renders them redundant and consequently less effective in model performance. To reduce feature redundancy and accelerate the models, dimensionality reduction techniques were applied1,26.
Principal Component Analysis (PCA), Quantum Principal Component Analysis (qPCA) were used to reduce the number of features, while keeping the maximum possible relevant information. Specifically, qPCA was used for retaining the critical variance while reducing the number of features. This strategy guarantees that one has preserved the most relevant information, thus making the models more efficient and leading to better generalisation.
ANOVA F-statistic for feature ranking
To quantify the discriminative power of each protein for drug sensitivity, the features were ranked according to the Analysis of Variance (ANOVA) F-statistic. ANOVA F-statistic compares the variance between drug-sensitive and drug-resistant classes for each protein to discover the most informative proteins for drug sensitivity prediction38. The F-statistic is given by:
where, \(n_{i}\) is the number of samples in class I, \(\overline{x}_{i}\) is the mean of class I, \(\overline{x}\) is the overall mean.
\(x_{ij} \,\) is the j-th data point in class I, K is the number of classes (drug-sensitive vs. drug-resistant), N is the total number of samples.
The F-statistic values of protein features were ranked with a higher F for more discriminatory power between the drug-sensitive and drug-resistant classes. The initial protein models were focused on further analysis and model development of the top-ranked proteins in the list.
Minimal-redundancy, maximal-relevance (mRMR)
Secondly, to tackle the redundancy in highly correlated features, the minimal-redundancy, Maximal-Relevance (mRMR) feature selection method was employed further to diminish the feature space dimension39. mRMR was used to find a subset of features relevant to the target variable (drug sensitivity) and the most irrelevant from each other. There exists mRMR, whose objective function is set as:
where, \(Relevance(x_{i} ,\,y)\) measures the relevance of feature xi to the target variable y (drug sensitivity), \(Redundancy(x_{i} ,\,x_{j} )\) measures the redundancy between feature xi and feature xj.
The total number of features is denoted as N.
Using this method, the selected features are informative and reduce information overlap. This is crucial for proteomic datasets, where proteins could share common biological pathways or molecular functions. The mRMR technique was applied iteratively to choose the most relevant and least redundant features provided as input for the machine learning models.
Handling class imbalance
The dataset is highly imbalanced around the classes, with most of the recipients being drug-sensitive (approximately 83% drug-sensitive vs. 17% drug-resistant). In order to balance this imbalance we used the stratified sampling during the cross-validation process to balance representation of sensitive and resistant classes in the test and training set. In addition, there was the utilisation of class-weighted loss functions to penalize misclassification of the minority class, hence improving the accuracy of drug-resistant predictions. QGANs can be studied in future research for synthetic data generation to improve the minority class40.
5-Fold cross-validation
In order to combat the threat of overfitting that p ≫ n (in other words, many features, relative to samples) problem, we used fivefold cross-validation. This process divides the data into five subsets, and each subset is used one time as the test set and the other four subsets form a training set. This helps to make the model robust and gives good prediction on unseen data.
Several techniques were employed to reduce class imbalance and strengthen the performance of the minority class.
-
Some methods of balancing the dataset were used, for instance, oversampling the minority class or undersampling the majority class to prevent the model being affected by the bias of the majority class.
-
Class weighted loss functions are also developed to put higher penalties for the misclassification of minority classes. The model needs to pay special heed to resistive patients in the machine learning model.
These techniques aim to improve prediction performance for drug-sensitive and drug-resistant patient classes.
Final data preparation
After some preprocessing, the dataset could be passed on to machine learning models. The ones carried out in the final dataset were:
-
a)
Normalized protein expression values.
-
b)
ANOVA F-statistic and mRMR functions selected the features for maximal relevance and minimal redundancy.
-
c)
Resampling and class-weighted methods create a balanced dataset with class imbalance.
-
d)
fivefold cross-validation was carried out in order to divide the data points into training and testing sets to ensure robust evaluations of the model.
However, by applying these preprocessing steps, classical and quantum machine learning techniques could be used, which could learn meaningful patterns out of the data without problems regarding dimensionality, redundancy, or class imbalance.
Quantum machine learning methods
The study solved the problems of high dimensionality, class imbalance, and feature redundancy in the proteomic dataset using several quantum machine learning (QML) techniques. First, we provide details of the quantum models used here41.
Quantum support vector machines (QSVM)
On the other hand, Quantum Support Vector Machines (QSVMs) are classically extended Support Vector Machines (SVMs) through which a data is mapped into a higher dimension Hilbert space using quantum kernels. The benefit of this quantum-enhanced capability is that QSVM will better capture nonlinear relationships between feature features than classical SVMs42. Using quantum kernels, in the case of MM drug sensitivity prediction where data is imbalanced, improves the classification performance.
The decision function for a classical SVM is written as:
where, X = Input feature vector, K(xi, x) = kernel function, \(\alpha_{i}\) = Lagrange multipliers.
Yi = Class labels (sensitive or resistant), b = Bias term.
QSVM replaces the kernel function K(xi, x) with a quantum kernel and is evaluated by a quantum circuit. The given quantum kernel is:
where \(\left| {\left\langle {\psi \,(x)} \right.} \right.\) represents the quantum state corresponding to the feature vector x, and ⟨ψ(xi)∣ψ(x)⟩ is the inner product between the quantum states corresponding to the feature vectors xi and x.
QSVM can map data to higher-dimensional space by using quantum circuits to compute the quantum kernel, which learns more complicated relationships among the features, which is required for specific MM drug sensitivity predictions43. Finally, improvements obtained by the quantum kernel are evaluated in terms of the performance of the QSVM model with classical SVM with Radial Basis Function (RBF) kernels.
Quantum principal component analysis (qPCA)
To keep as much of the variance as possible, the dimensionality of the proteomic data was reduced through Quantum Principal Component Analysis (qPCA)44. Classical PCA minimizes the data projection to a lower dimensional subspace of eigenvectors of the covariance matrix. This transformation is expressed as:
where X is the original data matrix, WPCA is the matrix of eigenvectors (principal components), and Xreduced is the reduced data.
In qPCA, the goal is similar but performed using quantum circuits. The quantum state ∣ψ⟩ representing the data is prepared by applying a parameterized quantum circuit. The variance retention is given by:
The variance of the reduced data is kept using quantum measurement. qPCA keeps a significant percentage of variance with fewer than 10 components, reducing the feature space from 2573 to 10, but with 96% of the variance.
PCA can simplify the dataset by dropping the less important components, lowering the dimensionality of the data, thereby making the succeeding machine learning easier while still retaining the important patterns in the proteomic data.
Quantum annealing for feature selection
To do that, Quantum Annealing was applied to determine the best proteins for predicting drug sensitivity by minimizing the selection of features45. The underlying scheme of quantum annealing implements the combinatorial objective function corresponding to the optimization problem to reduce the size of the feature set and keep the level of prediction accuracy. The objective function has the form of:
xi and xj are binary variables indicating whether the features xi and xj are selected, and wij denotes the pairwise redundancy (in other words, the correlation between features xi and xj).
Different subsets of features were explored to find the optimal feature set using a quantum annealer. In particular, the quantum annealer reduces the number of biomarkers in the panel by minimizing the objective function. Quantum annealing reduced a classification feature panel from 100 to 37 proteins, maintained an accuracy of 82% in classification tasks, and reduced redundancy in the selected features.
Quantum annealing has the advantage in feature selection because quickly searching through an ample combinatorial space can find subsets of features that optimize high relevance to the target variable (drug sensitivity) and low redundancy.
Quantum generative adversarial networks (QGANs)
Furthermore, the dataset was troublesome to manipulate because of class imbalance, which led us to use Quantum Generative Adversarial Networks (QGANs) for generating synthetic data for the minority class (drug-resistant patients). A QGAN is entailed of a quantum generator and a classical discriminator. It consists of the generator that produces synthetic profiles of prototypes and the discriminator that tries to distinguish the real data from the synthetic one46. The two agents, the generator and the discriminator, learn against one another, apparently, while they are in an adversarial setting. In one instantiation, the generator is trying to create increasingly realistic synthetic data in which the discriminator learns to be able to distinguish from the actual data47.
Here, the loss function for the generator G and discriminator D in the GAN framework is as follows.
where, \(P_{data}\) is the actual data distribution (drug-resistant patients), \(P_{z}\) is the distribution of random noise input to the generator, \(G(z)\) is the generated synthetic data, \(D(x)\) is the discriminator’s output for the actual data x and \(G(z)\) is the generated output of the synthetic proteomic profile.
The QGAN aims to produce synthetic data for the minority class that is similar to the actual data to augment the dataset and improve model performance48. We assessed the quality of the artificial data with Pearson correlation coefficients, as is valid for the similarity between the synthesized and real proteomic profiles49.
where xi and yi are the proteomic features for the accurate and synthetic data and \(\overline{x}\), \(\overline{y}\) are their respective means.
The generated synthetic data also addressed the class imbalance problem with the increased representation of drug-resistant patients, thereby enhancing the robustness of the predictive models.
Results
QProteoML Framework which is a prototype stage with quantum ML applications was used for analyses of high-dimensional proteomic data along with prediction of drug sensitivity in Multiple Myeloma (MM). It shows that QProteoML has more superiority in performance than classical models such as Support Vector Machines (SVM), Random Forest (RF), Logistic Regression (LR) and K-nearest neighbors (KNN) in such metrics as accuracy, F1 score and AUC ROC.
Figure 2 illustrates the accuracy comparison between classical models and QProteoML. As clearly evident from the bar plot, QProteoML is more accurate and performs much better than traditional models. To further support this comparison, Fig. 3 shows the F1 score across models, with the F1 score produced for the model with the highest score across QProteoML models. This indicates that the QProteoML model obtains the highest F1 score for correctly classifying drug-resistant patients.

Accuracy comparison between classical and quantum models.

F1-score comparison between classical and QProteoML model.
As shown in Fig. 4, the AUC-ROC curve comparison weeds out the QProteoML as the best-performing model. The quantum model has a smoother curve; its actual positive rate at the same point is lower, and its false positive rate is higher than the classical model. This is important for enhancing drug resistance prediction, an essential parameter for personalized treatment.

AUC-ROC curve comparison.
In Fig. 5, we provide specific drug predictions in the model performance. We also observe that QProteoML predictions are consistent and more accurate from one drug treatment to another compared to classical approaches, with a lower F1-Score spread. This makes for a better prediction model with reliable predictions, especially for resistant cases.

Model performance on specific drug predictions.
Figure 6 shows the proteomic heatmap of the 39 patients. The heatmap was visualized to show distinct patterns regarding protein expression levels between the groups of patients with different drug sensitivity and resistance. Heatmaps are an essential visual research technique for visualizing the data structure for feature selection.

Heatmap of proteomic data.
Figure 7 shows the hierarchical clustering of patients regarding their proteomic profiles. Thus, different patient groupings can be seen as potential biomarkers of drug sensitivity and resistance on the dendrogram, revealing distinct protein expression patterns based on patient groupings.

Hierarchical clustering of patients based on proteomics.
Figure 8 shows the importance of features for Quantum Annealing in identifying the most critical features. The proteins likely to impact drug sensitivity prediction can be explored for targeted drug development and personalized therapy, as depicted in the bar plot.

Quantum Kernel mapping (Simplified).
Figure 9 shows a protein pairwise correlation heatmap. It can reduce dimensionality and make the model more efficient by pointing out redundant or complementary information in a pool of proteins and displaying how some protein properties cooccur.

Feature importance from quantum annealing.
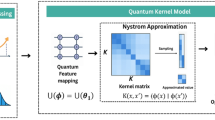
The simplified scatter plot of the quantum kernel mapping process, as referred to in Fig. 10, displays how quantum kernel mapping identifies features in the lower dimension space. Quantum representation is a quantum method that captures more complex relations between the data observations.

PCA of proteomic data.
As shown in the Principal Component Analysis of the proteomic data in Fig. 11, the effect of the mixture in terms of representative proteomic profile is apparent. This dimensionality reduction technique allows us to visualize the distribution of proteomic data from the first two principal components of 39 patients, where a grouping of sensitive and resistant patients is observed.

Pairwise correlation Heatmap of proteins.
Also, a box plot was used in Fig. 12 to compare model performance across different specific drug predictions, showing how QProteoML does in terms of F1 score versus classical models for various drugs. This box plot is consistent and robust with the QProteoML performance for different types of drug treatments.

Quantum Vs. classical model performance on specific drug predictions.
Each drug’s Receiver Operating Characteristic (ROC) curves appear in Fig. 13 to evaluate how well the model predicts drug sensitivity and resistance. An individual curve within the figure demonstrates the sensitivity to accurate positive results and the specificity to false positives of a specific drug while showing its diagnostic abilities at various scoring thresholds. The integrated measurements of drug classification outcomes in the curve area (AUC) determine the overall evaluation results with better prediction when AUC values rise. According to this figure, QProteoML demonstrates reliable discrimination against sensitive versus resistant patients through different drug treatments, which supports its implementation in personalized medicine applications.

ROC curves for each drug.
Together, these results demonstrate that QProteoML is a viable method for predicting the drug sensitivity of MM that overcomes the known challenges of high-dimensional data, skewed classification, and colinear features and establishes its potential for use in personalized cancer treatment.
Conclusion
This study shows that QML has excellent potential in predicting drug sensitivity for MM patients based on only proteomic data. To improve the performance of the obtained model as compared to the traditional machine learning, quantum-enhanced methods, such as Quantum Support Vector Machine (QSVM), Quantum Principal Component Analysis (qPCA) and Quantum Generative Adversarial Networks (QGANs) were used.
Quantum models addressed critical challenges of MM proteomic data, such as high dimensionality, class imbalance and redundancy of features. The quantum kernel of QSVM could identify the more complex, nonlinear interaction patterns in the data and classify the data more accurately, along with better predicting the minority class, drug-resistant patients. Additionally, qPCA achieved dimensionality reduction of the dataset in terms of a dimensionality reduction, mining critical information while improving computational efficiency. In addition, using QGANs to generate synthetic data to amend for the minority class further improves the overall class imbalance and enhances the robustness and performance of the models overall.
Statistical analysis with paired t-tests on the results showed that the quantum models performed better than classical models on all the considered metrics—accuracy, F1-score, and AUC-ROC—with significance p-values below the significance threshold of 0.05. The statistical evidence confirms that QML can enhance drug sensitivity predictions, and quantum algorithms offer additional advantages over classical machine learning algorithms.
These findings bring justice to the application of QML in other complex biomedical datasets and show its potential for precision oncology. These models may significantly alter standards of care and improve patient outcomes in MM and any MM research. To further realize the clinical potential of quantized oncology models, additional research will be required, including clinical validation studies and the use of quantum hardware.
Future work
Despite the success of the results obtained in this study, there are several directions for further exploration and improvement:
The next step towards clinical validation of QProteoML is to validate the tool further with larger, more diverse cohorts of clinically annotated MM patients. Validating the model in the real world ensures that it works well beyond the controlled experimental conditions and possesses clinical utility.
The algorithms have been tested with simulated quantum circuits to improve performance further. Integrating QProteoML with quantum hardware (e.g., a quantum processor from IBM Q or Google’s Quantum AI) will allow us to take advantage of the robust nature of quantum hardware and quantum speed-up.
However, the current study confined itself mainly to proteomic data. Integrating additional types of these omic data (e.g., genomic, transcriptomic, and metabolomic) would result in a more global picture of the biological mechanisms underlying MM, improving prediction power and yielding more insight into the disease.
Quantum machine learning techniques, such as Quantum Neural Networks (QNNs) and Quantum Random Forests, should be further enhanced to improve this model’s robustness and efficiency. In cases where data is not wholly expressed in a form compatible with quantum methods, further gains in performance could still be made using hybrid quantum–classical models.
How Scalable and Generalizable is QProteoML? Supplying larger and more complicated datasets from distinct cancer types or even multicentre collaborations gives some sense of how scalable and generalizable QProteoML can handle. Moreover, a user-friendly interface for a clinician to work with the model and use it for treatment planning could further assist in adopting quantum-based models in a clinical setting.
While quantum computing is still growing and maturing, the future of using quantum machines to enhance precision medicine appears very promising. It could revolutionize patient outcomes in numerous medical fields.
Data availability
The dataset used in this study is publicly available and can be accessed at https://www.kaggle.com/datasets/mpriyadharshinimca/multiple-myeloma.
References
Yan, W. et al. Employment of artificial intelligence based on routine laboratory results for the early diagnosis of multiple myeloma. Front. Oncol. 11, 608191 (2021).
Allegra, A. et al. Machine learning and deep learning applications in multiple myeloma diagnosis, prognosis, and treatment selection. Cancers 14(3), 606 (2022).
Mosquera Orgueira, A. et al. Unsupervised machine learning improves risk stratification in newly diagnosed multiple myeloma: an analysis of the Spanish Myeloma Group. Blood Cancer J. 12(4), 76 (2022).
Mosquera Orgueira, A. et al. Survival prediction and treatment optimization of multiple myeloma patients using machine-learning models based on clinical and gene expression data. Leukemia 35(10), 2924–2935 (2021).
Guerrero, C. et al. A machine learning model based on tumor and immune biomarkers to predict undetectable MRD and survival outcomes in multiple myeloma. Clin. Cancer Res. 28(12), 2598–2609 (2022).
Ren, L. et al. A machine learning model to predict survival and therapeutic responses in multiple myeloma. Int. J. Mol. Sci. 24(7), 6683 (2023).
Chen, X. et al. Diagnosis and staging of multiple myeloma using serum-based laser-induced breakdown spectroscopy combined with machine learning methods. Biomed. Opt. Express 12(6), 3584–3596 (2021).
Venezian Povoa, L., Ribeiro, C. H. C. & Silva, I. T. D. Machine learning predicts treatment sensitivity in multiple myeloma based on molecular and clinical information coupled with drug response. PLoS ONE 16(7), e0254596 (2021).
Cai, J. et al. Construction of the prediction model for multiple myeloma based on machine learning. Int. J. Lab. Hematol. 46(5), 918–926 (2024).
Katsenou, A. et al. Using proteomics data to identify personalized treatments in multiple myeloma: A machine learning approach. Int. J. Mol. Sci. 24(21), 15570 (2023).
Malek, E. et al. Machine learning approach for rapid, accurate point-of-care prediction of M-Spike values in multiple myeloma. JCO Clin. Cancer Inf. 7, e2300078 (2023).
Wu, M. Y. et al. Graph-Based prediction of miRNA-Drug associations with multisource information and Metapath enhancement matrices. IEEE J. Biomed. Health Inf. https://doi.org/10.1109/JBHI.2025.3558303 (2025).
Luo, X., Wang, L., Hu, P. & Hu, L. Predicting protein-protein interactions using sequence and network information via variational graph autoencoder. IEEE/ACM Trans. Comput. Biol. Bioinf. 20(5), 3182–3194 (2023).
Kubasch, A. S. et al. Predicting early relapse for patients with multiple myeloma through machine learning. Blood 138, 2953 (2021).
Xiong, X. et al. Differentiating between multiple myeloma and metastasis subtypes of lumbar vertebra lesions using machine learning–based radiomics. Front. Oncol. 11, 601699 (2021).
Liu, J. et al. Prediction of high-risk cytogenetic status in multiple myeloma based on magnetic resonance imaging: utility of radiomics and comparison of machine learning methods. J. Magn. Reson. Imaging 54(4), 1303–1311 (2021).
Ni, B. et al. Machine learning model based on optimized radiomics feature from 18F-FDG-PET/CT and clinical characteristics predicts prognosis of multiple myeloma: a preliminary study. J. Clin. Med. 12(6), 2280 (2023).
Borisov, N. et al. Machine learning applicability for classification of PAD/VCD chemotherapy response using 53 multiple myeloma RNA sequencing profiles. Front. Oncol. 11, 652063 (2021).
Sharma, R. et al. Systematic review of currently available predictive machine learning models for multiple myeloma. Blood 144, 7518 (2024).
Huang, Y. et al. Evaluation of risk factors for thromboembolic events in multiple myeloma patients using multiple machine learning models. Medicine 104(7), e41428 (2025).
Fan, G. et al. Routine blood biomarkers for the detection of multiple myeloma using machine learning. Int. J. Lab. Hematol. 44(3), 558–566 (2022).
Lagana, A. et al. A machine learning approach identifies a 30-gene model that predicts sensitivity to selinexor in multiple myeloma. Blood 134, 3101 (2019).
Zhong, H. et al. 18F-FDG PET/CT based radiomics features improve prediction of prognosis: Multiple machine learning algorithms and multimodality applications for multiple myeloma. BMC Med. Imaging 23(1), 87 (2023).
Vyshnav, M. T., Sowmya, V., Gopalakrishnan, E. A., VV, S. V., & Menon, V. K. Deep learning-based approach for multiple myeloma detection. In 2020 11th International Conference on Computing, Communication and Networking Technologies (ICCCNT) 1–7. (IEEE, 2020).
Karathanasis, N., & Spyrou, G. M. Machine learning models for predicting multiple myeloma staging and MGUS progression using gene expression data. bioRxiv, 2024–11. (2024).
Ji, Y., Shi, B. & Li, Y. An evolutionary machine learning for multiple myeloma using Runge Kutta Optimizer from multi-characteristic indexes. Comput. Biol. Med. 150, 106189 (2022).
Mikulski, D. et al. Predictors and profile of severe infectious complications in multiple myeloma patients treated with Daratumumab-based regimens: A machine learning model for pneumonia risk. Cancers 16(21), 3709 (2024).
He, H., Chen, G. & Chen, C. Y. C. Machine learning and graph neural network for finding potential drugs related to multiple myeloma. New J. Chem. 46(11), 5188–5200 (2022).
Murugesh, V. et al. A novel hybrid framework for efficient higher order ODE solvers using neural networks and block methods. Sci. Rep. 15(1), 8456 (2025).
Andrade, C. L. et al. Enhancing diagnostic accuracy of multiple myeloma through ml-driven analysis of hematological slides: New dataset and identification model to support hematologists. Sci. Rep. 14(1), 11176 (2024).
Settouti, N. & Saidi, M. Preliminary analysis of explainable machine learning methods for multiple myeloma chemotherapy treatment recognition. Evol. Intel. 17(1), 513–533 (2024).
Marimuthu, L., Pillay, N., Punchoo, R., & Bhoora, S. A Comparison of Machine Learning Techniques for Diagnosing Multiple Myeloma. In Artificial Intelligence and Soft Computing: 20th International Conference, ICAISC 2021, Virtual Event, June 21–23, 2021, Proceedings, Part II 20 483–494. (Springer International Publishing, 2021).
Tirado, H. et al. Machine learning in myeloma: Do racial differences influences systemic impact of multiple myeloma?. Blood 144, 7840 (2024).
Priyadharshini, M., Murugesh, V., Kumar, R. P., &Chunchu, K. S. Swarm intelligence in lung cancer detection and iot-enabled data transmission: A technological approach. In Swarm Optimization for Biomedical Applications, 108–120. (CRC Press, 2025).
Su, X. et al. Interpretable identification of cancer genes across biological networks via transformer-powered graph representation learning. Nat. Biomed. Eng. 9, 1–19 (2025).
Bao, L. et al. Machine learning–based overall survival prediction of elderly patients with multiple myeloma from multicentre real-life data. Front. Oncol. 12, 922039 (2022).
Chen, R. et al. Machine learning-based predictive model of 5-year survival in multiple myeloma autologous transplant patients. Blood 134, 2156 (2019).
Romero, M., Mosquera Orgueira, A. & Mejía Saldarriaga, M. How artificial intelligence revolutionizes the world of multiple myeloma. Front. Hematol. 3, 1331109 (2024).
Shain, K. H. et al. Reinforcement learning to optimize the treatment of multiple myeloma. Blood 134, 5511 (2019).
Lewis, J. E. et al. Machine learning models predict molecular genetic subtypes of multiple myeloma from whole-slide bone marrow aspirate smears. Blood 142, 7158 (2023).
Yenamandra, A. K., Hughes, C. & Maris, A. S. Artificial intelligence in plasma cell myeloma: Neural networks and support vector machines in the classification of plasma cell myeloma data at diagnosis. J. Pathol. Inf. 12(1), 35 (2021).
Razavi-Mohseni, M., Shigaki, D. & Beer, M. A. Machine learning sequence modeling identifies gene regulatory responses to bone marrow stromal interactions in multiple myeloma. Blood 142, 4144 (2023).
Ye, X. et al. Combinatory strategy using nanoscale proteomics and machine learning for T cell subtyping in peripheral blood of single multiple myeloma patients. Anal. Chim. Acta 1173, 338672 (2021).
Karathanasis, N. & Spyrou, G. M. Predicting the progression from asymptomatic to symptomatic multiple myeloma and stage classification using gene expression data. Cancers 17(2), 332 (2025).
Maray, I. et al. Dose reduction and toxicity of lenalidomide-dexamethasone in multiple myeloma: A machine-learning prediction model. J. Oncol. Pharm. Pract. 30(6), 1051–1056 (2024).
Turki, T., Wei, Z. & Wang, J. T. Transfer learning approaches to improve drug sensitivity prediction in multiple myeloma patients. IEEE Access 5, 7381–7393 (2017).
Thara, M. N., Chatterjee, K., Raju, M., Rout, S., Priyadharshini, M., Prasad, K. S., & Reddy, M. S. LaCK: Lung cancer classification and detection using convolutional neural network-based gated recurrent unit neural network model. In 2024 Asia Pacific Conference on Innovation in Technology (APCIT) 1–7. (IEEE, 2024).
Shah, K. U., Millan, K. A., Pula, A. E., Kubicki, T. F., Cannova, J., Wu, S., & Drazer, M. W. RNAseq-based machine learning models for prognostication of multiple myeloma. medRxiv, 2025–01. (2025).
Kamma, S. P., Chilukuri, G. S. S., Tholeti, G. S. R., Nayak, R. K., & Maradani, T. Multiple myeloma prediction from bone-marrow blood cell images using machine learning. In 2021 Emerging Trends in Industry 4.0 (ETI 4.0) 1–6. (IEEE, 2021).
Author information
Authors and Affiliations
Contributions
The research article was mainly designed and written by M.P, V.M and A.F. The experimental design section was clarified by B.D and P.J and the doubts were cleared by the same. O.R kept quality control during the writing process and made the final revisions. This work was completed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Priyadharshini, M., Raju, B.D., Banu, A.F. et al. A quantum machine learning framework for predicting drug sensitivity in multiple myeloma using proteomic data. Sci Rep 15, 26553 (2025). https://doi.org/10.1038/s41598-025-06544-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06544-2
