
Abstract
Dengue virus (DENV) has emerged as a formidable global health challenge, with a surging incidence rate across the world. Despite numerous research initiatives aimed at developing effective antiviral therapy, no clinically proven drug or vaccine has been identified to combat all four genetically diverse serotypes of DENV. Therefore, comparative analysis of repurposed drugs and phytocompounds against all DENV serotypes is critical in the search for an effective long-term solution to this menacing disease. 93 phytocompounds and 15 drugs were shortlisted from the literature and screened using DataWarrior 5.5.0, from which 10 phytocompounds and 10 drugs were selected for further analysis. Molecular docking was performed by using AutoDockVina tool. Toxicity and drug likeness activity of standard drugs and phytoconstituents was done by using online servers. The current study showed that among all the selected phytoconstituents, lupiwighteone showed the best binding energy, favorable pharmacokinetics and no toxicity with all the selected serotypes of DENV. The MD simulation result supported the stability of lupiwighteone in complexes with NS3, NS5 and E-Protein. This study identifies lupiwighteone as a promising antiviral candidate with favorable drug-like properties against DENV-2, DENV-3, and DENV-4 serotypes. Furthermore, in vitro and in vivo study is required for the validation of antiviral activity of Lupiwighteone against dengue virus.
Similar content being viewed by others
Introduction
Dengue Virus (DENV), belonging to the Flaviviridae family, is one of the rapidly growing arthropod-borne viral diseases which has become a primary concern of public health globally1,2,3. WHO reported an 8-fold increase in dengue cases between 2000 and 20192. This rise, which is most noticeable in many tropical and subtropical countries, is the result of inadequate mosquito control, climate change, resistant mosquito emergence, unplanned urbanization, and a significant increase in population. DENV is transmitted by mosquitoes of the Aedes genus, essentially Aedes aegypti3,4. Clinical features of dengue virus infection can range from being asymptomatic to mild febrile illness and to having severe dengue haemorrhagic fever (DHF)5. But the treatment has been difficult due to the lack of effective antiviral therapy, as well as the ordeal of accurately diagnosing the disease due to its diverse presentation. Moreover, the use of these therapies in the presence of non-neutralizing antibodies may actually increase the risk of antibody-dependent enhancement (ADE), worsening the symptoms of dengue fever1,6. As a result, several initiatives in the domains of computational biology and drug docking have been undertaken to use medicinal phytocompounds and repurposed pharmaceuticals against DENV serotypes.
Understanding the genomics and proteomics of dengue at the serotype level is essential to effectively combat this virus. In 2019, our study delved into exploring conserved epitopes across all four serotypes and revealed 100% conservation in the Envelope protein, NS3, NS4A, NS4B, and NS5 proteins1. Subsequently, in 2022, we investigated the conservancy in genomic regions of evolutionarily significant sequences, focusing on CpG islands spanning all serotypes. Our findings demonstrated that three DENV proteins (NS5, NS3, and E) exhibit highly conserved motifs across all four serotypes7. Thus, based on the previous studies, present investigation focused on NS5, NS3, and E proteins due to their consistency across serotypes. NS5 is an integral and highly conserved protein, with RNA-dependent RNA polymerase (RdRp) and methyltransferase activities, making NS5 a promising target for several anti-DENV medications8. Similarly, NS3, a multifunctional protein; and E surface envelope protein are also additional potent drug targets9,10,11. Due to high mutation rates in RNA viruses and high genetic diversity, targeting conserved proteins of DENV may result in consistent binding of phytochemicals and drugs with all serotypes12. Thus, these three proteins become the most attractive targets for vaccines and antiviral drugs. In this study, a comparative analysis of all serotypes of DENV is done against some of the repurposed drugs and phytochemicals, thus aiding in the selection of new therapeutics effective for all serotypes.
Various studies reported that the phytocompounds extracted from medicinal plants like Thymus serpyllum, Cymbopogancitratus, Foeniculumvulgare, Bacopamonnieri, Rheum emodihave demonstrated a broad spectrum of pharmacological properties against various diseases13,14,15,16,17,18,19,20,21,22. On the other hand, drug repurposing is a viable approach for developing new therapeutic applications while minimising adverse side effects and cytotoxicity. Existing pharmacological compounds are carefully examined to uncover new therapeutic uses. The use of phytocompounds also offers a compelling array of benefits, emphasised by their perfect safety profile, which includes low toxicity potential and a remarkably low level of detrimental effects. As a result, phytocompounds indicate they are potential candidates for the development of therapeutic interventions that are both efficient and well-tolerated. Phytocompounds like Naringin, Maslinic acid, Steroic acid, and Rhodiolin have been successfully docked with DENV’s NS2B/NS3 protease and NS5 methyltransferase in previous studies23,24. However, a comparative analysis of phytocompounds across all serotypes requires further attention. Repurposed drugs like chloroquine, prednisone and lovastatin, to name a few, have shown limited efficacy in reducing viremia during clinical evaluation25, necessitating the identification of more robust therapeutic alternatives. Several inhibitors targeting key DENV proteins, such as NS3 protease, NS5 and the envelope (E) protein, have been reported in literature including natural phytocompounds, synthetic small molecules, and repurposed drugs.Inhibitors like (1S,5R)-3-methyl-2-thioxo-1,2,3,4,5,6-hexahydro-8H-1,5-methanopyrido[1,2-a][1,5]diazocin-8-one (E-protein inhibitor); (2S,4S)-4-(2-Hydroxyphenyl)-2-(methylthio)-1,2,3,4-tetrahydropyrrolo[1,2-a]quinolin-3-one (NS3 inhibitor); 2-Oxo-1,3-benzoxathiol-5-yl acetate (NS5 inhibitor) of DENV have shown promising in silico or in vitro activity26,27,28. However, many of these studies are limited to individual serotypes or lack validation through dynamic simulations, highlighting the need for a more comprehensive and comparative analysis across all four DENV serotypes. In this study, a comparative analysis of all serotypes of DENV is done against some of the repurposed drugs and phytocompounds, thus aiding in the selection of new therapeutics effective for all serotypes.
Methods
Ligand preparation
The investigation began with a comprehensive review of the literature to resulted in identifying 93 phytoconstituents and 15 standard drugs with purported antiviral properties. Employing the DataWarrior 5.5.0 tool (https://openmolecules.org/datawarrior), a meticulous screening process was initiated to discern the most promising compounds. Following an exhaustive analysis of the generated data, the top 10 phytocompounds and 10 drugs were curated for subsequent evaluation (Fig. 1).

Schematic flowchart illustrating the comprehensive methodology.
To facilitate molecular docking simulations, the three-dimensional structures of the selected phytoconstituents and standard drugs were procured from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and DrugBank (https://go.drugbank.com/), in .sdf format. These structures were then subjected to rigorous energy minimization using Chem3D 15.0 to refine their conformations and enhance stability. Following this optimization step, the ligands were converted to PDB format to ensure compatibility with subsequent docking procedures.
The prepared ligands underwent further refinement using AutoDock Tools 1.5.6 to ensure proper configuration and compatibility with the docking software. Subsequently, the ligand structures were saved in pdbqt format, a prerequisite for conducting accurate and reliable docking simulations. This meticulous preparation of the ligands laid a solid foundation for robust and insightful molecular docking studies.
Preparation of target proteins
Based on the availability of 3D crystal structures, the protein structures for NS3 of DENV-4 (PDB ID: 2VBC)29 and DENV-2 (PDB ID: 2FOM)30; NS5 of DENV-3 (PDB ID: 4V0Q)31 and DENV-2 (PDB ID: 5ZQK)32; and E protein of DENV-2 (PDB ID: 1OKE)33 were retrieved from the RCSB-PDB database (https://www.rcsb.org/). These structures were saved in PDB format for further analysis.
To prepare the protein structures for docking simulations, AutoDock Tools 1.5.6 was employed. This involved the removal of water molecules and the addition of missing hydrogen atoms and charges to ensure the accuracy of the protein models. The prepared protein structures were then saved in pdbqt format, rendering them compatible with subsequent docking procedures.
The grid box for each protein was set over the entire protein (blind docking) using the default center coordinates and extending the box size until the entire protein is covered in most cases: for NS5 of DENV-2 (x = 33.572, y=-11.688, z = 25.66), NS5 of DENV-3 (x = 24.892, y = 162.151, z = 24.605), NS3 of DENV-2 (x = 0.455, y=-16.942, z = 13.968), NS3 of DENV-4 (x=-6.068, y=-2.850, z = 20.867), and for E of DENV-2 (x=-13.129, y = 69.049, z = 24.451).
Virtual screening and molecular Docking
Molecular docking analysis was conducted using the AutoDockVina tool34 to investigate the interactions between the selected ligands and the active sites of the target proteins with the exhaustiveness set to “8” in the configuration file. This approach aimed to elucidate the nature and causes of the interactions between the sketched compounds and the protein targets.
The docking study encompassed all prepared phytocompounds and drugs against the three target proteins of DENV. Subsequently, the most favorable conformations, characterized by the highest binding energies, were selected. The 10 compounds with the highest affinity for target protein for each docking run was selected Protein-ligand complexes were then analyzed using Discovery Studio Visualizer to examine the array of interactions between the proteins and ligands.
Furthermore, the compounds demonstrating the most promising interactions with respective protein were chosen for further analysis for their Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties. This comprehensive approach provided insights into the potential therapeutic efficacy and safety profiles of the selected compounds.
In silico toxicity prediction of phytocompounds and drugs
ADMET screening was conducted to evaluate the absorption, toxicity, and drug-likeness properties of the ligands. The analysis utilized several tools, including SwissADME (http://www.swissadme.ch), MolInspirationCheminformatics (https://www.molinspiration.com), and PROTOX 3.0 (https://tox-new.charite.de/protox_II/).
SwissADME was employed to identify predictive models for the physicochemical properties, drug-likeness, medicinal chemistry, and pharmacokinetics of the compounds. This facilitated the selection of compounds with optimal characteristics for inhibiting the action of DENV proteins. To further assess the safety profile of the compounds, their structures were uploaded to the Protox 3.0 to predict toxicity endpoints such as cytotoxicity, hepatotoxicity, carcinogenicity, and mutagenicity. LD50 values were also considered for quantitative analysis.
Additionally, the MolInspiration server was utilized to predict bioactivity scores and assess molecular descriptors and drug likeliness properties. This analysis was based on Lipinski’s Rule of Five21,35, which stipulates that for a compound to be orally active, it should not violate more than one of the five criteria.
Molecular dynamics (MD) simulation
The MD simulation of the protein-ligand complex was done using GROMACS 2021.536 software which was installed in ubuntu 18.04 LTS on a GPU enabled PC having the nvidiaGeforce RTX 2070 maxQ graphics card. Simulations were conducted for complexes involving lupiwighteone with all three proteins,(1S,5R)-3-methyl-2-thioxo-1,2,3,4,5,6-hexahydro-8H-1,5-methanopyrido[1,2-a][1,5]diazocin-8-one (E-protein inhibitor); (2S,4S)-4-(2-Hydroxyphenyl)-2-(methylthio)-1,2,3,4-tetrahydropyrrolo[1,2-a]quinolin-3-one (NS3 inhibitor); 2-Oxo-1,3-benzoxathiol-5-yl acetate (NS5 inhibitor) to evaluate their structural stability and dynamic interactions. Docked structures of the protein-ligand complexes were used as starting points in the simulation study. Proteins were processed and the topology files were prepared by using pdb2gmx and charmm36 force field, while the ligands topology files were prepared by using the online server, SwissParam. The solvent (using the TIP3P water model) addition was done in a cubic box by using a box distance 1.0 nm from closest atom in the protein. The addition of Cl− ions was used to neutralize the system. Energy minimization was done by using steepest descent algorithm taking 50,000 steps, 5 kJ/mol maximum force and Verlet cut-off scheme taking Particle-Mesh Ewald (PME) columbic interactions. Position restraints were applied in the equilibration step. After that, NVT equilibration was done in 300 K and 100 ps in 50,000 steps and NPT equilibration taking Parrinello-Rahman (pressure coupling), 1 bar reference pressure and 100 ps in 50,000 steps. LINCS algorithm was applied to constraint all the bond lengths. For long-range electrostatics, Particle-mesh Ewald (PME) algorithm was used. The snapshots of poses from the last frame of the trajectory are presented in Fig. 3. After successful completion of molecular dynamic simulation for 100 ns, Root mean square deviation (RMSD) of backbone residues, number of Hydrogen bonds, Root mean square fluctuations (RMSF), Radius of gyration (RG) and solvent accessible surface area (SASA)were calculated from the trajectory files from each complex.
The free energy of binding calculations was done using the standalone program, gmx_MMPBSA37 based on the MM-PBSA (From AMBER) functionalities, a well-known and excellent tool for performing end-point binding free energy calculations and to predict the stability of the complex. In this study, MM-PBSA method was employed to calculate the interaction free energy between the proteins and ligands complex under investigation.
Results
The screening process was meticulously conducted following an extensive literature search to identify 93 phytocompounds and 15 drugs with documented potential for antiviral activity. These compounds were subjected to rigorous screening using DataWarrior based on predefined criteria encompassing drug likeness, total molecular weight, and molecular flexibility to gauge their suitability as antiviral agents. The top ten drugs and phytocompounds were identified based on their optimal compliance with the screening filters (Fig. 2). These compounds emerged as promising candidates warranting further investigation in subsequent studies. The outcomes gleaned from the screening process offered valuable insights into their potential for antiviral activity, underscoring their suitability for advanced development as potential antiviral agents.

3D plot showing the drug likeness, total molecular weight, and molecular flexibility of the top 10 (A) Phytocompounds (B) Drugs. The x-axis represents the drug likeness score. The y-axis shows the total molecular weight of the drugs in Daltons (Da), and the z-axis shows the molecular flexibility score. The plot was generated using Data Warrior software.
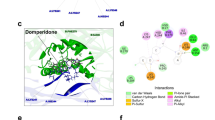
Subsequently, docking simulations were performed utilizing AutoDockVina to assess the interactions of the top 10 phytocompounds and drugs with NS3, NS5, and E proteins of DENV. The selected phytocompounds included Aloe-emodin, Avicularin, Baicalein, Catechin, Epicatechin, Flavone, Kaempferol, Kushenol W, lupiwighteone, and Quercitrin. Remarkably, Lupiwighteone emerged as the most promising phytocompound, exhibiting average binding affinity (-8.46 kcal/mol) across all target proteins, particularly with NS3 and NS5, suggesting its potential efficacy against dengue virus infection (Table 1). The NS3 protein exhibited notable binding affinities with lupiwighteone, registering values of -8.5 kcal/mol and − 9.5 kcal/mol for DENV-2 and DENV- 4 serotypes, respectively. Following NS3, the NS5 protein of DENV- 2 and DENV- 3, as well as the E protein of DENV- 2, also displayed considerable binding interactions with lupiwighteone. Among the 10 phytocompounds examined, lupiwighteone consistently demonstrated the highest binding affinity with all target proteins. Notably, in the case of NS3, lupiwighteone established crucial hydrogen bonding with ASN152, alongside hydrophobic interactions involving residues LEU85, GLY148, LEU76, LEU149, MET49, TRP69, TRP83, ILE123, ILE165, LYS74, ASN167, ALA164, VAL154, and ALA166 as shown Fig. 3. Similarly, in interactions with NS5, lupiwighteone was observed to form hydrogen bonds with SER661, ASN610, and ASP663, while engaging in hydrophobic interactions with SER796, GLN603, ASP539, ILE474, TRP475, ALA473, THR606, TYR607, GLY662, ASP664, ILE797, HIS798, and CYS709 (refer to Fig. 4). Furthermore, in its interaction with the Envelope (E) protein, lupiwighteone established hydrogen bonds with THR239 and exhibited hydrophobic interactions with LEU253, LEU218, and PRO217 (refer to Fig. 5). The comprehensive summary of lupiwighteone’s interactions with the target proteins is presented in Table 3. Following lupiwighteone, Baicalein exhibited notable binding affinities with NS5 of DENV-2 and DENV-3, with binding energies of -8.2 kcal/mol and − 9.8 kcal/mol, respectively. Additionally, Baicalein demonstrated binding affinities with NS3 of DENV-2 and DENV-3, as well as with the E protein of DENV-2, with binding energies of -8.3 kcal/mol, -7.6 kcal/mol, and − 8.7 kcal/mol, respectively. This trend was consistent with other phytocompounds, which also displayed binding affinities with all protein sequences considered in the study. However, the variations in binding scores across different serotypes raised concerns regarding their efficacy against all serotypes. Comparative analyses of docking scores were performed to contextualize the findings. Notably, lupiwighteone demonstrated higher binding affinities compared to other compounds, corroborating its potential as a lead candidate for further investigation. Moreover, the docking results were benchmarked against established standards or previous studies to validate the efficacy of the selected compounds38.
The study identified a selection of top 10 drugs, comprising Asunaprevir, Boceprevir, Bromocriptine, CoPP, Daclatasvir, Doravirine, Grazoprevir, Ledipasvir, Raltegravir, and Sofosbuvir. Comprehensive data on the binding affinities of each compound with all serotypes can be found in Table 2 for detailed reference (Table 3).

Interactions of lupiwighteone with NS3 (A, B), NS5 (C, D) and envelop (E, F) target proteins: (A, C, E) 2D interactions and (B, D, F) 3 D interactions.
ADME prediction of drugs and phytoconstituents
To evaluate the drug-likeness of the inhibitors, all candidate molecules underwent analysis using the Protox II server, as detailed in Tables 4 and 5. As stated above, Protox 3.0 provided insights into the compounds’ LD50 values, toxicity classes, and potential hepatotoxic, carcinogenic, mutagenic, immunotoxic, and cytotoxic activities. Additionally, Molinspiration and SwissADME39, both ADMET-based drug scan tools, were utilized for further assessment, as summarized in Supplementary Tables S1–S4.
Among these compounds, lupiwighteone (C20H18O5), chemically known as 5,7-dihydroxy-3-(4-hydroxyphenyl)-8-(3-methylbut-2-enyl) chromen-4-one, exhibited notable properties. With a molecular weight of 338.36 g/mol, a miLogP value of 4.52, and a solubility of 1.008 mg/L @ 25 °C (estimated), lupiwighteone emerged as a promising phytocompound warranting further investigation.
The toxicity data obtained from the Protox 3.0 online server revealed that all the phytocompounds were categorized under Class 4–6. Specifically, Catechin (LD50 = 10000 mg/kg), Epicatechin (LD50 = 10000 mg/kg), Kaempferol (LD50 = 3919 mg/kg), and lupiwighteone (LD50 = 2500 mg/kg) were among the phytocompounds with no active toxicity. Furthermore, among the drugs, Bromocriptine and Daclatasvir were classified as Class 3 drugs, while Ledipasvir was categorized as Class 4. These findings provide valuable insights into the toxicity profiles of the tested compounds, guiding further investigations into their safety and efficacy for potential therapeutic applications.
The MolInspiration online server was employed to estimate the drug likeness of both the active phytocompounds and drugs. Notably, all the phytocompounds, with the exception of Quercitrin and Avicularin, were predicted to have zero violations of drug likeness rules. Among the drugs, Doravirine exhibited zero violations, indicating its potential suitability for drug development. Additionally, Raltegravir, CoPP, Bromocriptine, and Boceprevir were found to violate only one of the rules, suggesting that they are promising candidates warranting further analysis. These results highlight the drug likeliness of the tested compounds and provide valuable guidance for future investigations into their pharmacological properties and therapeutic potential.
Based on our docking results, Baicalein, KushenolW, and lupiwighteone exhibited higher binding affinities for all the target proteins considered in our study. However, upon conducting toxicity analysis, it was revealed that Baicaleindemonstrated activity for carcinogenicity and mutagenicity, while KushenolW showed activity for immunotoxicity. Consequently, these findings disqualified Baicalein and KushenolW as potential candidates for further investigation.
Notably, lupiwighteone displayed promising ADME characteristics, further supporting its potential for MD simulation studies. Its higher binding affinity with all three target proteins of different serotypes reaffirms its potential as an effective phytocompound for combating dengue. The consistent high binding scores observed for lupiwighteone underscore its promising candidacy for therapeutic development against dengue virus infections.
Likewise, among repurposed drug candidates, although Ledipesvir showed average highest docking scores with all the serotypes, but it was found to be immunotoxic. Another drug, Bromocriptine had an average binding score of -9.7 with all proteins along all serotypes, but it was also found to be immunotoxic as well as cytotoxic. Daclatasvir was found to bind to all the proteins across serotypes efficiently with zero toxicity thus making it the best candidate for further studies.
MD simulation of protein ligand complexes
MD simulation was done to study the stability of protein ligand complexes. Molecular docking and toxicity data revealed that lupiwighteone has the best pharmacokinetic and toxicity profile as well as excellent binding affinity for all target proteins. The complexes of the three target proteins with Lupiwighteone were subjected to 100ns Molecular Dynamics simulation in a box of water to determine what the trajectory would look like and then estimate a profile of stability for the complexes and every MD simulation was done relative to a reference for each protein. Recently reported potent inhibitors were docked with the target proteins at the sites described in literature where they are thought to bind to in order to elicit their protein inhibition and the complexes obtained were subjected to MD simulation under the same conditions as the lupiwighteone complexes of the three studied proteins. The Free Energy of Binding of the complexes were also estimated using the gMMPBSA method. The Free energy of binding of the complexes showed that lupiwighteone has a very high binding energy when complexed with E-protein and NS3 protein relative to the inhibitors (Table 6). The binding energy of lupiwighteone when bound to E-Protein is almost 10x that of the inhibitor and that of lupiwighteone when bound to NS3 protein is more than double that of the inhibitor used as a reference compound. However, the free energy of binding of lupiwighteone with NS5 protein is lower compared to the reference compound which shows that lupiwighteone may not be very potent against the NS5 protein compared the E-Protein and NS3protein. Though the free energy of binding of lupiwighteone with NS5 is significant, it may still be considered as an inhibitor of NS5 and this means that if lupiwighteone is administered orally as a Dengue virus therapeutic agent, it has a very tendency of being effective at reducing the viral load because of its high potential for inhibiting three different proteins that are critical for the virus metabolism.
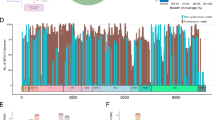
The RMSD plots (Fig. 4) of the complex of lupiwighteone and the study proteins also show that the complexes are stable within the 100ns time frame for which the complexes were studied in the molecular dynamics simulation. The complexes of lupiwighteone achieved equilibrium shortly after the commencement of the simulation relative to the complexes of the inhibitors for the three study proteins. The RMSF plots (Fig. 5) of the three studied proteins bound with lupiwighteone and corresponding inhibitor shows that the fluctuations of amino acid residues in the site to which the ligands bound are similar in all cases for individual proteins except for E-Protein where residues in the range of 330–340 fluctuated more intensely when bound to lupiwighteone compared to when it was bound to the E-protein inhibitor. The plots of radius of gyration as well as solvent accessible surface area (Figs. 6 and 7) also showed that the proteins were not overly perturbed by the lupiwighteone during the course of the simulation (very little conformational changes) which suggests that the protein maintained a stable confirmation while bound to the lupiwighteone. Essentially, the plots from the trajectory obtained from the MD simulation showed that the complexes obtained for lupiwighteone bound to the target proteins were stable and the estimated Free Energy of Binding (FEB) suggests that lupiwighteone is worth considering for a follow up biological study involving the target proteins directly or an animal model (Fig. 8).

RMSD (A) E-protein inhibitor and Lupiwighteone bound to E-protein, (B) NS3 inhibitor and Lupiwighteone bound to NS3, (C) NS5 inhibitor and Lupiwighteone bound to NS5 protein.

RMSF (A) E-protein inhibitor and Lupiwighteone bound to E-protein, (B) NS3 inhibitor and Lupiwighteone bound to NS3, (C) NS5 inhibitor and Lupiwighteone bound to NS5 proteins.

Radius of gyration (A) E-protein inhibitor and Lupiwighteone bound to E-protein, (B) NS3 inhibitor and Lupiwighteone bound to NS3, (C) NS5 inhibitor and Lupiwighteone bound to NS5 proteins.

Solvent Accessible Surface Area (SASA) (A) E-protein inhibitor and Lupiwighteone bound to E-protein, (B) NS3 inhibitor and Lupiwighteone bound to NS3, (C) NS5 inhibitor and Lupiwighteone bound to NS5 proteins.

Estimated Binding free energy Breakdown (gMMPBSA): (A) E-protein, (B) NS3 and (C) NS5 proteins in complex with lupiwighteone and corresponding inhibitors.
Discussion
The evolution of dengue virus and the emergence of its four distinct serotypes (DENV1-4) have been the subject of much research and interest in the scientific community. Despite the earliest recorded dengue epidemics occurring in the 18th and 19th centuries40, the virus is thought to have originated even earlier, with evidence suggesting it may have circulated in primates for thousands of years. The diversification of the virus into multiple serotypes is attributed to genetic drift and to some extent recombination events that occurred during its evolutionary journey, as it spread to new regions and encountered different host populations41. The circulation of these serotypes presents a significant challenge for controlling the spread of the disease and developing effective vaccines and drugs42.
The main challenge in vaccine development is to create a tetravalent formulation such that it has equally effective immunization for all the four DENV serotypes43. Even though vaccine development for dengue started in the 1920s, till date around 7 vaccines have gone through various clinical trials44. The first commercialized and FDA approved vaccine, Dengvaxia® (CYD-TDV) is currently available and licensed in 20 countries but it has raised some safety issues which even lead to retraction of Dengvaxia’s license in some countries including Philippines8. Despite these setbacks, two other vaccine candidates, LAV-TDV from NIAID/Butantan and TAK-003 from Takeda, have shown promising results in phase III clinical trials. However, concerns about vaccine efficacy and safety persist, particularly in light of historical immunogenicity data and recent disclosures of variable efficacy against all DENV serotypes. Previous study underscores the importance of prioritizing vaccine formulations that encompass a comprehensive array of epitopes essential for inducing protective immunity, spanning both cellular and humoral responses. This imperative remains relevant despite comparable population coverage percentages achieved by LAV-TDV and TAK-003 vaccine formulations45,46.
In light of these challenges, immunoinformatics has emerged as a pivotal tool in designing multi-epitope vaccines capable of targeting all DENV serotypes. Recent research highlights the potential of immunoinformatics to identify conserved epitopes from structural and non-structural proteins, essential for robust immune responses. Kaushik et al. designed a candidate vaccine targeting DENV-1 to DENV-4 through conserved epitopes, demonstrating its immunogenicity and antibody production in vivo. These findings underscore the importance of immunoinformatics in developing next-generation vaccines capable of providing comprehensive protection against dengue virus47.
The drug discovery for dengue had commenced decades ago but is not an easy process. The lack of a potent drug covering all serotypes signifies that more resources need to be explored. Firstly, an anti-DENV drug should be inhibitory to all the serotypes and even combat the antibody dependent enhancement which is seen during secondary dengue infection48. Secondly, drug development in general is a long and tedious process making it tough to develop a de novo drug. Hence, repurposing of drugs comes into picture. This strategy reduces the time and expense of drug development because the majority of the pharmaceuticals that are being repurposed are already FDA-authorized thereby expediting the process49.Therefore, we conducted a comparative study to find a common repurposed drug which will be effective against the three most common and threatening serotypes of dengue including DENV-2, DENV-3 and DENV-450.
Based on comparative analysis of the binding affinities, our data indicates that three drugs including Daclatasvir, Ledipasvir and Bromocriptine can serve as potential candidates for inhibiting the selected target proteins of Dengue virus (NS3, NS5 and E protein). Interestingly, Daclatasvir and Ledipasvir were reported to belong to the class of FDA approved direct-acting antiviral drugs (DAA) which were previously used in the treatment of Hepatitis C (phylogenetically related to DENV)51,52. The role of Bromocriptine against DENV has also been shown in one of the previous studies53, and our comparative analysis justifies its use against other DENV serotypes as well.
A previous study also reported Daclatasvir to be an effective agent against the E protein of DENV-254. Our analysis supports their finding and also indicates the effectiveness of this drug against NS3 of DENV-2 and DENV-4 (binding affinity of -10.2 kcal/mol each) and NS5 protein of DENV-2 and DENV-3 (binding affinities of -8.8 kcal/mol each). Ledipasvir was found to have consistently high binding affinities with all three conserved proteins (NS5, NS3 and E proteins). Ledipasvir is FDA-approved direct-acting antiviral drug (DAA) recommended for viral infections like HCV53,55 and COVID-1956,57,58. It has shown little to no side effects on patients. Our analysis indicates the effectiveness of this drug against NS3 of DENV-2 and DENV-4 (binding affinity of -10.6 kcal/mol each) and NS5 protein of DENV-2 and DENV-3 (binding affinities of -11.2 kcal/mol and − 9.6 kcal/mol respectively). Apart from the two drugs discussed above, Bromocriptine also showed consistent high binding affinities with NS3 protein of DENV-2 and DENV- 4 (binding affinities of -11.7 and − 10.2 kcal/mol) and NS5 for DENV- 2 and DENV- 3. This is also supported by an in vitro study where bromocriptine has been shown to have potent anti-dengue virus (DENV) activity and low cytotoxicity53. Thus, we suggest in vitro as well as in vivo analysis of Daclatasvir, Ledipasvir and Bromocriptine as drug candidates for treating Dengue infection and preventing Antibody (Ab)-dependent enhancement of infection.
While the enlisted synthetic compounds showed encouraging results against the targeted proteins from the insilico study, because they are known drugs being used clinically already, there is plenty of documentation about their side effects and actual toxicity profile. Declatasvir, though shows successful all-oral regimens of antiviral therapy in patients with chronic hepatitis C and cirrhosis is occasionally complicated by hepatic decompensation (chronic liver injury) and may cause reactivation of hepatitis B in susceptible patients coinfected with the hepatitis B virus (HBV). Ledipasvir has also been linked to an adverse effect that causes lung diseases59 just has bromocriptine has been found to have potential for liver injury (especially when overdosed which happens accidentally in children). Our pharmacokinetics and toxicity profile predictions also show that the synthetic compounds may be toxic when administered especially at high doses.
On the other hand, the phytochemical, lupiwighteone (8-prenylgenistein), a naturally occurring polyphenol-based phytocompound caught our attention. This pyrano-isoflavone compound derived from Erythrina variegate (the Indian Coral TrADEee)60 has exhibited remarkable binding affinities with all the target proteins, indicating its potential role as an inhibitor. Lupiwighteone demonstrates extraordinary organic properties which makes it the finest prospective compound for therapeutical purposes in our list. It belongs to toxicity class 5 and was observed to be negative for all toxicity profiles on ProTox 3.0 analysis with LD50 as high as 2500 mg/kg. Lupiwighteone has high affinity with NS5 of DENV-2 and DENV-3; NS3 of DENV-2 and DENV-4 and E-protein of DENV-2. Additionally, it follows Lipinski’s rule of five with no violations, indicating the highest drug likeliness. The MD Simulation results underscore the potential of lupiwighteone as a lead candidate for antiviral drug development against dengue virus.
The findings of this study not only unravel the molecular mechanisms governing the interaction between lupiwighteone and target proteins but also offer valuable insights into its pharmacological profile. These results set the stage for subsequent experimental validation and clinical translation, addressing the critical need for effective antiviral therapies against dengue virus infections. Therefore, based on this in silico comparative analysis, we propose lupiwighteone as a prospective phytocompound for the treatment of dengue infection, potentially exhibiting inhibitory effects against DENV-2, DENV-3, and DENV-4 serotypes. Leveraging computational methodologies and advanced molecular modeling techniques, this research contributes to ongoing endeavors in drug discovery and development to combat dengue virus infections. By conducting a comparative analysis of promising phytocompounds and drugs against different serotypes, the present study offers a rational approach to identifying broad-spectrum therapeutics.
Conclusion
Amidst the alarming statistics provided by the World Health Organization (WHO), which indicate that roughly half of the global population is now vulnerable to Dengue virus (DENV) infection, with an estimated 100–400 million cases reported annually, the urgency to combat this pervasive threat has reached unprecedented levels. Despite the significant global burden of Dengue, there remains a lack of anti-DENV antivirals, prophylactics, or therapeutics availability. Addressing this pressing global health challenge, our study pioneers a coherent approach to identify potential therapeutics against DENV. Through meticulous screening, integrating molecular docking studies, ADMET analysis, and drug likeness profiling, we have uncovered promising repurposed drugs and phytocompounds exhibiting consistent binding affinity across DENV serotypes. In summary, our study underscores the promising antiviral activity of phytocompoundlupiwighteone and also highlights the candidature of daclatasvir as repurposed drug which calls for further exploration of its therapeutic potential. These novel findings not only offer a ray of hope in the battle against DENV but also set the stage for further experimental validation and clinical translation, promising a brighter future in the fight against Dengue infections.
Data availability
All data generated or analysed during this study are included in this published article. Additional data will be made available on a reasonable request.
Abbreviations
- ADMET:
-
Absorption, distribution, metabolism, excretion, and toxicity
- LD50:
-
Lethal dose 50
- MD Simulation:
-
Molecular dynamics simulation
- MMBPSA:
-
Molecular mechanics Poisson Boltzmann surface area
- NCBI:
-
National Center of Biotechnology Information
- PDB:
-
Protein data bank
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuations
- ROG:
-
Radius of gyration
- SASA:
-
Solvent accessible surface area
References
Verma, M. et al. Highly conserved epitopes of DENV structural and non-structural proteins: Candidates for universal vaccine targets. Gene 695, 18–25. https://doi.org/10.1016/j.gene.2019.02.001 (2019).
Murugesan, A. & Manoharan, M. Dengue virus. In Emerging Reemerging Viral Pathogens 281–359. https://doi.org/10.1016/B978-0-12-819400-3.00016-8 (2020).
Gubler, D. J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11, 480–496 (1998).
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507. https://doi.org/10.1038/nature12060 (2013).
Guzman, M. G. et al. Dengue: A continuing global threat. Nat. Rev. Microbiol. 8, S7–S16. https://doi.org/10.1038/nrmicro2460 (2010).
Verma, M., Phartyal, R. & Bhatt, A. Introduction to flaviviruses and their global prevalence. In Human Viruses: Diseases, Treatments and Vaccines: The New Insights (ed Ahmad, S. I.) 411–439 (Springer International Publishing, 2021). https://doi.org/10.1007/978-3-030-71165-8_19.
Jaglan, A., Satija, S., Singh, D., Phartyal, R. & Verma, M. Intra-genomic heterogeneity in CpG dinucleotide composition in dengue virus. Acta Trop. 232, 106501. https://doi.org/10.1016/j.actatropica.2022.106501 (2022).
Thomas, S. J. & Yoon, I-K. A review of Dengvaxia®: Development to deployment. Hum. Vaccines Immunother. 15, 2295–2314. https://doi.org/10.1080/21645515.2019.1658503 (2019).
Lim, S. P., Noble, C. G. & Shi, P-Y. The dengue virus NS5 protein as a target for drug discovery. Antiviral Res. 119, 57–67. https://doi.org/10.1016/j.antiviral.2015.04.010 (2015).
Pujar, S. A. H., Sethu, G. V., Bhagyalalitha, A. K. & Singh, M. Dengue structural proteins as antiviral drug targets: Current status in the drug discovery & development. Eur. J. Med. Chem. 221, 113527. https://doi.org/10.1016/j.ejmech.2021.113527 (2021).
Sukhralia, S. et al. From dengue to zika: The wide spread of mosquito-borne arboviruses. Eur. J. Clin. Microbiol. Infect. Dis. 38, 3–14. https://doi.org/10.1007/s10096-018-3375-7 (2019).
Sharma, A., Krishna, S. & Sowdhamini, R. Bioinformatics analysis of mutations sheds light on the evolution of dengue NS1 protein with implications in the identification of potential functional and druggable sites. Mol. Biol. Evol. 40, msad033. https://doi.org/10.1093/molbev/msad033 (2023).
Salaria, D. et al. In vitro and in Silico antioxidant and anti-inflammatory potential Of essential oil of Cymbopogon citratus (DC.) stapf. Of North-Western Himalaya. J. Biomol. Struct. Dyn. 40, 14131–14145. https://doi.org/10.1080/07391102.2021.2001371 (2022).
Salaria, D. et al. Phytoconstituents of traditional Himalayan herbs as potential inhibitors of human papillomavirus (HPV-18) for cervical cancer treatment: An in silico approach. PLoS One. 17, e0265420. https://doi.org/10.1371/journal.pone.0265420 (2022).
Salaria, D. et al. In vitro and in silico analysis of Thymus serpyllum essential oil as bioactivity enhancer of antibacterial and antifungal agents. J. Biomol. Struct. Dyn. 40, 10383–10402. https://doi.org/10.1080/07391102.2021.1943530 (2022).
Rolta, R. et al. Phytocompounds of Rheum emodi, Thymus serpyllum, and Artemisia annua inhibit spike protein of SARS-CoV-2 binding to ACE2 receptor: in silico approach. Curr. Pharmacol. Rep. 7, 135–149. https://doi.org/10.1007/s40495-021-00259-4 (2021).
Rolta, R. et al. Molecular Docking studies of phytocompounds of Rheum emodi wall with proteins responsible for antibiotic resistance in bacterial and fungal pathogens: in Silico approach to enhance the bio-availability of antibiotics. J. Biomol. Struct. Dyn. 40, 3789–3803. https://doi.org/10.1080/07391102.2020.1850364 (2022).
Rolta, R. et al. Identification of novel inhibitor phytoconstituents for influenza A H3N2: An in silico approach. J. Biomol. Struct. Dyn. 43, 4287–4296. https://doi.org/10.1080/07391102.2024.2305313 (2025).
Mehta, J. et al. Antibacterial potential of Bacopa monnieri (L.) wettst. and its bioactive molecules against Uropathogens—An in Silico study to identify potential lead Molecule(s) for the development of new drugs to treat urinary tract infections. Molecules 27, 4971. https://doi.org/10.3390/molecules27154971 (2022).
Mehta, J. et al. Phytocompounds from Himalayan medicinal plants as potential drugs to treat multidrug-resistant Salmonella typhimurium: An in silico approach. Biomedicines 9, 1402. https://doi.org/10.3390/biomedicines9101402 (2021).
Chugh, A. et al. Comparative Docking studies of drugs and phytocompounds for emerging variants of SARS-CoV-2. 3 Biotech 13, 36. https://doi.org/10.1007/s13205-022-03450-6 (2023).
Kaur, B. et al. An in Silico investigation to explore Anti-Cancer potential of Foeniculum vulgare mill. Phytoconstituents for the management of human breast Cancer. Molecules 27, 4077. https://doi.org/10.3390/molecules27134077 (2022).
Purohit, P., Dash, J. J. & Meher, B. Exploration of phytocompounds as potential anti-dengue agents against Denv Ns2b-Ns3 protease: Structure-based virtual screening and Md simulation studies (2020).
Hasan, M., Mia, M. M., Munna, S. U., Talha, M. M. H. & Das, K. Seawater fungi-derived compound screening to identify novel small molecules against dengue virus NS5 methyltransferase and NS2B/NS3 protease. Inf. Med. Unlocked 30, 100932. https://doi.org/10.1016/j.imu.2022.100932 (2022).
Anasir, M. I., Ramanathan, B. & Poh, C. L. Structure-Based design of antivirals against envelope glycoprotein of dengue virus. Viruses 12, 367. https://doi.org/10.3390/v12040367 (2020).
Lin, C-S. et al. Inhibition of dengue viruses by N-methylcytisine Thio derivatives through targeting viral envelope protein and NS2B-NS3 protease. Bioorg. Med. Chem. Lett. 99, 129623. https://doi.org/10.1016/j.bmcl.2024.129623 (2024).
Shimizu, H. et al. Discovery of a small molecule inhibitor targeting dengue virus NS5 RNA-dependent RNA polymerase. PLoS Negl. Trop. Dis. 13, e0007894. https://doi.org/10.1371/journal.pntd.0007894 (2019).
Starvaggi, J., Previti, S., Zappalà, M. & Ettari, R. The Inhibition of NS2B/NS3 protease: A new therapeutic opportunity to treat dengue and Zika virus infection. Int. J. Mol. Sci. 25, 4376. https://doi.org/10.3390/ijms25084376 (2024).
Luo, D. et al. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 82, 173–183. https://doi.org/10.1128/JVI.01788-07 (2008).
Erbel, P. et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West nile virus. Nat. Struct. Mol. Biol. 13, 372–373. https://doi.org/10.1038/nsmb1073 (2006).
Zhao, Y. et al. A crystal structure of the dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog. 11, e1004682. https://doi.org/10.1371/journal.ppat.1004682 (2015).
El Sahili, A. et al. NS5 from dengue virus serotype 2 can adopt a conformation analogous to that of its Zika virus and Japanese encephalitis virus homologues. J. Virol. 94, e01294–e01219. https://doi.org/10.1128/JVI.01294-19 (2019).
Modis, Y., Ogata, S., Clements, D. & Harrison, S. C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100, 6986–6991. https://doi.org/10.1073/pnas.0832193100 (2003).
Trott, O. & Olson, A. J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2010).
Frey, J. G. & Bird, C. L. Cheminformatics and the semantic web: Adding value with linked data and enhanced provenance. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 465–481. https://doi.org/10.1002/wcms.1127 (2013).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291. https://doi.org/10.1021/acs.jctc.1c00645 (2021).
Purohit, P., Sahoo, S., Panda, M., Sahoo, P. S. & Meher, B. R. Targeting the DENV NS2B-NS3 protease with active antiviral phytocompounds: Structure-based virtual screening, molecular Docking and molecular dynamics simulation studies. J. Mol. Model. 28, 365. https://doi.org/10.1007/s00894-022-05355-w (2022).
Magdaleno, J. S. L. et al. Toward α-1,3/4 fucosyltransferases targeted drug discovery: In Silico Uncovering of promising natural inhibitors of fucosyltransferase 6. J. Cell. Biochem. 124, 1173–1185. https://doi.org/10.1002/jcb.30440 (2023).
Whitehorn, J. & Farrar, J. Dengue Br. Med. Bull. 95, 161–173. https://doi.org/10.1093/bmb/ldq019 (2010).
Weaver, S. C. & Vasilakis, N. Molecular evolution of dengue viruses: Contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 9, 523–540. https://doi.org/10.1016/j.meegid.2009.02.003 (2009).
Pollett, S. et al. Understanding dengue virus evolution to support epidemic surveillance and counter-measure development. Infect. Genet. Evol. 62, 279–295. https://doi.org/10.1016/j.meegid.2018.04.032 (2018).
Izmirly, A. M., Alturki, S. O., Alturki, S. O., Connors, J. & Haddad, E. K. Challenges in dengue vaccines development: Pre-existing infections and cross-reactivity. Front. Immunol. 11 (2020).
Torres-Flores, J. M., Reyes-Sandoval, A. & Salazar, M. I. Dengue vaccines: An update. BioDrugs 36, 325–336. https://doi.org/10.1007/s40259-022-00531-z (2022).
Thomas, S. J. Is new dengue vaccine efficacy data a relief or cause for concern? npj Vaccines 8, 1–6. https://doi.org/10.1038/s41541-023-00658-2 (2023).
Pinheiro, J. R. et al. Comparison of neutralizing dengue virus B cell epitopes and protective T cell epitopes with those in three main dengue virus vaccines. Front. Immunol.. https://doi.org/10.3389/fimmu.2021.715136 (2021).
Kaushik, V., Gupta, G. S. K., Kalra, L. R., Shaikh, U. & Cavallo, A. R. Immunoinformatics aided design and in-vivo validation of a cross-reactive peptide based multi-epitope vaccine targeting multiple serotypes of dengue virus. Front. Immunol. 13, 865180. https://doi.org/10.3389/fimmu.2022.865180 (2022).
de Oliveira, A. S. et al. NS3 and NS5 proteins: Important targets for anti-dengue drug design. J. Braz. Chem. Soc. 25, 1759–1769. https://doi.org/10.5935/0103-5053.20140057 (2014).
Tian, Y-S., Zhou, Y., Takagi, T., Kameoka, M. & Kawashita, N. Dengue virus and its inhibitors: A brief review. Chem. Pharm. Bull. 66, 191–206. https://doi.org/10.1248/cpb.c17-00794 (2018).
Law, G. L., Tisoncik-Go, J., Korth, M. J. & Katze, M. G. Drug repurposing: A better approach for infectious disease drug discovery? Curr. Opin. Immunol. 25, 588–592. https://doi.org/10.1016/j.coi.2013.08.004 (2013).
Sabry, N., Kamel, A. M., Cordie, A. & Esmat, G. Daclatasvir as a hepatitis C infection treatment option: An up-to-date evaluation. Expert Opin. Pharmacother. 24, 159–170. https://doi.org/10.1080/14656566.2022.2145883 (2023).
Balatow, P., Sandlin, A. & Cory, T. J. An evaluation of ledipasvir + sofosbuvir for the treatment of chronic hepatitis C infection. Expert Opin. Pharmacother. 22, 1839–1846. https://doi.org/10.1080/14656566.2021.1943359 (2021).
Kato, F. et al. Novel antiviral activity of Bromocriptine against dengue virus replication. Antiviral Res. 131, 141–147. https://doi.org/10.1016/j.antiviral.2016.04.014 (2016).
Montgomery, M., Ho, N., Chung, E. & Marzella, N. Daclatasvir (Daklinza): A treatment option for chronic hepatitis C infection. Pharm. Ther. 41, 751–755 (2016).
Afdhal, N. et al. Ledipasvir and Sofosbuvir for untreated HCV genotype 1 infection. N. Engl. J. Med. 370, 1889–1898. https://doi.org/10.1056/NEJMoa1402454 (2014).
Medhat, M. A. et al. Sofosbuvir/ledipasvir in combination or Nitazoxanide alone are safe and efficient treatments for COVID-19 infection: A randomized controlled trial for repurposing antivirals. Arab. J. Gastroenterol. 23, 165–171. https://doi.org/10.1016/j.ajg.2022.04.005 (2022).
Nourian, A. et al. Efficacy and safety of sofosbuvir/ledipasvir in treatment of patients with COVID-19; A randomized clinical trial. Acta Biomed. 91, e2020102. https://doi.org/10.23750/abm.v91i4.10877 (2020).
Pirzada, R. H., Haseeb, M., Batool, M., Kim, M. & Choi, S. Remdesivir and Ledipasvir among the FDA-Approved antiviral drugs have potential to inhibit SARS-CoV-2 replication. Cells 10, 1052. https://doi.org/10.3390/cells10051052 (2021).
Omotani, S. et al. Drug-induced lung disease adverse effect with Ledipasvir acetonate/sofosbuvir. J. Pharm. Health Care Sci. 6, 6. https://doi.org/10.1186/s40780-020-00162-y (2020).
Naresh, P. et al. Targeting a conserved pocket (n-octyl-β-D-glucoside) on the dengue virus envelope protein by small bioactive molecule inhibitors. J. Biomol. Struct. Dyn. 40, 4866–4878. https://doi.org/10.1080/07391102.2020.1862707 (2022).
Acknowledgements
MV acknowledges Principal, Hansraj College, University of Delhi for her constant support. IS, AV, MV acknowledge SRI VIPRA internship program, Sri Venkateswara College for providing research opportunity.
Author information
Authors and Affiliations
Contributions
Conceptualization, M.V.; methodology, M.V., P.V., R.R., D.S.; software, R.R., O.A.F., P.V., I.S.; validation, M.V., P.V., R.R.; formal analysis, P.V., I.S., R.R.; O.A.F. Performed MD Simulation; data curation, R.R., D.S., P.V,. I.S.; writing—P.V., R.R., D.S., I.S., O.A.F,. M.V.; editing, M.V., P.V,. R.R., A.V.; supervision, M.V., O.A.F. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Human and animals rights
This research article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vats, P., Rolta, R., Salaria, D. et al. Comparative analysis of phytocompounds and repurposed drugs against dengue virus serotypes employing an in silico study. Sci Rep 15, 27878 (2025). https://doi.org/10.1038/s41598-025-06974-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06974-y
