
Abstract
The objective of this study was to evaluate the effect of adding of C-Type Natriuretic (CNP) to the in vitro culture medium of bovine embryos on cryotolerability through modulation of lipid content and profile, as well as modulation of gene transcripts linked to embryonic metabolism. Initially, a concentration of 400 nM of CNP was used throughout the in vitro culture and blastocysts were collected for lipid content analysis by Sudan Black B. In addition, blastocysts were selected by morphological quality and developmental stage, and the samples collected were analyzed using MRM- profiling. After, blastocysts were vitrified using OPS. Subsequently the warming, hatched blastocysts were collected and evaluated for transcript abundance in a microfluidic platform. Differences of probabilities lower than P < 0.05, and/or fold change ˃1.5 were considered significant. The CNP group presented a reduction in the relative abundance of ions belonging to different lipid subclasses, such as acylcarnitine, sphingomyelin, cholesteryl esters, free fatty acids, and glycerophospholipid. Furthermore, the triacylglycerol lipids TG 52:3 NL 16:1, TG 56:3 NL 18:1, and the glycerophospholipid C22:6, were increased in the CNP group. A modulation of blastocyst transcripts was also observed by increased transcription of ATF4, and a trend statistical significance of BMP15 and GFPT2 transcripts. There was no difference in blastocyst development rates after warming of CNP-treated embryos.
Similar content being viewed by others

Introduction
Reproduction biotechniques make a decisive contribution to the beef and dairy production chain1. Among the available biotechniques, in vitro production (IVP) embryo stands out on the world stage. About 1,876,591 bovine embryos were in vitro produced (IVP) worldwide in 2023, about 15,8% more than in 2022 (1,876,591 vs.1,620,347, respectively)2.
In addition, since the first successful cryopreservation, several procedures have been developed. Cryopreservation methods can basically be classified into two main strategies: cryopreservation with a slow cooling curve and vitrification3. Embryos produced in vitro have a high sensitivity to cryopreservation, which is apparently associated with the higher lipid content present in the cells of these embryos when compared to those produced in vivo4.
Thus, IVP embryos have a high lipid deposit, and the addition of fetal bovine serum (FBS) in the culture of these embryos is one of the possible causes leading to it5. Lipids such as triacylglycerides (TG), the main lipid class found in the cytoplasm of mammalian cells, are stored as lipid droplets3 and, in cryopreservation processes, their presence is a possibly compromising factor due to the cellular damage it causes.
Despite recent advances related to IVP, low efficiency in cryopreservation processes is still an obstacle6,7,8. Some invasive strategies, such as mechanical removal of lipids, were proposed before cryopreservation9. However, important changes occurred in the developmental potential of blastocysts transferred to recipients10. Further, as there was damage of zona pellucida due to micromanipulation, it also led to a higher chance of transmission of pathogens11.
In this way, numerous substances have already been tested with the aim of reducing or modulating the lipid profile12,13,14,15,16. Recently, the inclusion of the C-type natriuretic peptide (CNP) with different concentrations were tested, showing that a concentration of 400 nM of CNP in the in vitro culture (IVC), not previously described in the literature and which was not embryotoxic, modulated some transcripts related to embryonic metabolism. However, in the previous study no aspect regarding interference in cryotolerance or lipid modulation processes was measured17.
Thus, the objective of our study was to evaluate whether the addition of CNP in the in vitro culture of bovine embryos alters its cryotolerability through the modulation of the lipid content and profile, as well as the modulation of transcripts linked to embryonic metabolism.
Materials and methods
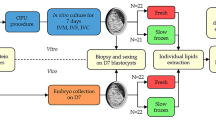
All animal procedures were approved by the Ethics and Animal Handling Committee of the São Paulo State University (UNESP), Botucatu, São Paulo, Brazil, certificate #1180. All medium to produce bovine embryos and solutions used in vitrification and warming were provided by ABS Global Brasil®, Mogi Mirim, São Paulo, Brazil.
In vitro embryo production
Sample collection
Ovaries from a commercial slaughterhouse and from phenotypically resembling Nellore breed females (that is, with the phenotype mostly from Bos taurus indicus animals of the Nellore breed) were collected, packaged and transported to the laboratory in 0.9% saline solution between 30 and 35ºC. Follicles with a diameter of 2–8 mm were aspirated with hypodermic needles (30 × 8; 21G) attached to 10 mL syringes for the recovery of cumulus-oocyte complexes (COCs), with a maximum interval of one hour between the receipt of the ovaries in the laboratory and the completion the aspiration of the follicles present in the bovine ovaries. Only COCs of qualities I and II were used, and the classification was performed conforming International Embryo Technology Society (IETS) oocyte classification. The number and presentation of cumulus cell layers and ooplasm homogeneity are considered. It should be noted that degenerated oocytes were not included with viable oocytes for in vitro maturation (IVM)18,19.
In vitro maturation
Previously to in vitro maturation (IVM), COCs were washed three times in TCM-HEPES 199 supplemented with 10% (v/v) fetal bovine serum, (FBS), 0.20 nM sodium pyruvate and 83.4 mg/mL of gentamicin. The COCs were matured in drops of 100 µL of TCM-199 medium bicarbonate supplemented with 10% (v/v) FBS, 5 µg of luteinizing hormone, 0.5 µg of follicle-stimulating hormone, 1 µg of estradiol, 2.2 µg of pyruvate and 50 µg of gentamycin/mL and incubated for 24 h in an environment with maximum humidity, 38.5 °C and, 5% CO2.
In vitro fertilization
After maturation, the COCs were washed in HEPES-buffered TCM-199 medium and transferred to 100-µL droplets of the fertilization medium that consisted of tris-buffered medium (TBM) supplemented with 8 mg/mL fatty acid-free bovine serum albumin (BSA) and 1 mM glutamine. For fertilization in drops of 100 µL, semen from a single Nellore bull was used (Adamo Fiv Kubera; Código 011NE03127, registry ACF 3522, Alta Genetics). The bull was previously tested in our laboratory for the production of IVF embryos. The cryopreserved semen was heated at 36 °C for 30 s. Sperm selection was performed by Percoll gradient (Percoll 45% in the upper part and 90% in the lower part) by centrifugation (12,100 g, for 2 min), the supernatant (600 µL) was discarded, and the pellet sperm resuspended in 300 µL of fertilization medium and homogenized. The semen was centrifuged again (8,127 g, for 45 s) and, after discarding the supernatant, the sperm concentration was adjusted to obtain a final concentration of 1 × 10⁶ mL⁻¹ live spermatozoa in each drop containing 20 COCs. They were co-incubated for 20 to 22 h in an environment with maximum humidity, 5% CO2, and 38.5° C. The day of insemination was considered day zero (Day 0).
In vitro culture
For in vitro culture, presumptive zygotes were subjected to the removal of cumulus cells by successive pipetting and then incubated in synthetic oviduct fluid (SOF) medium supplemented with 8 mg/mL fatty acid-free BSA under mineral oil at the same temperature and gaseous atmospheric condition used in the previous steps. On the first day of culture (D1) the presumptive zygotes were divided into experimental groups: control (without the addition of CNP) and CNP groups (400 nM of CNP, Sigma–Aldrich/St. Louis, MO, United States). After day three (D3), 50% of the culture media volume was replaced by fresh media (1st feeding). At D5, 50% of the culture media volume was replaced with fresh SOF medium but this time was supplemented with glucose (2nd feeding). During culture, blastocyst (D7) and hatching rates (D8 and D9) were recorded.
Lipid content by Sudan black B
Expanded blastocysts from the control and CNP groups (D7; n = 10/group 6 replicates) were randomly selected during the experimental replicates and had their lipid content measured as described by Sudano et al.. (2012)20. Briefly, the embryos were fixed in a 10% formaldehyde solution (diluted in PBS- pH 7.4) for 2 h at room temperature. After that, they were washed in distilled water with 0.05% polyvinyl alcohol (PVA) and transferred to drops with 50% ethanol (diluted in distilled water). After 2 min, the embryos were transferred to drops with a 1% Sudan Black B solution (w/v) diluted in 70% ethanol (diluted in distilled water) for 1 to 2 min. Subsequently, they were washed in three 50% ethanol baths for 5 min each and then with distilled water containing 0.05% PVA. For mounting the slides, 10 µL of glycerol was used. The evaluation was performed using light microscopy. To estimate lipid content, digital images were captured and processed using Image J 1.41 Software (Wayne Rasband, National Institutes of Health, Bethesda, MD, USA) based on the methodology described by Sudano et al. (2012)20and Costa et al. (2019)15.
Lipid profiling method by the MRM
Blastocysts from the control and CNP groups (D7; n = 10/group, 5 replicates) were selected by morphological quality (excellent or good) and developmental stage (blastocysts to expanded blastocysts) for the analysis using Multiple Reaction Monitoring (MRM) profiling technique21,22. Briefly, the embryos were collected, washed for a few seconds in methanol: water (1:3 v/v) to remove the culture medium and stored at −80° C. Lipid extraction was performed according to Bligh and Dyer (1959)23 and adapted for small volume samples. Briefly, 40 µL of ultrapure water was added to a microtube containing the embryos, and the mixture was to promote cell lysis. Then, 50 µL of chloroform and 90 µL of methanol were added and mixed by pipetting for 15 s (one-phase solution). After, another 50 µL of chloroform and 50 µL of ultrapure H2O were added and the samples were incubated for 5 min at room temperature. Samples were centrifuged to enhance polar from nonpolar phase separation (two-phase solution). The combined organic and water phases were dried in a centrifugal evaporator and samples were vacuum-sealed and stored at −80ºC until MS analysis24.
Vitrification of embryo
On D7 and D8 (n = 50 blastocysts/group, 5 replicates), only expanded blastocysts and IETS grade I blastocysts (excellent or good quality25) were subjected to vitrification using the open pulled straw (OPS) technique developed by Vajta et al. (1997)26. The blastocysts were washed in three drops of H-SOF solution and submitted to a vitrification solution 1 until the beginning of vitrification. Groups of no more than 5 blastocysts at a time were transferred to one well of a four-well plate containing 400 µL of holding solution 1 (TCM-HEPES 199 supplemented with 20% (v/v) FBS), with 7.5% of ethylene glycol (50 µL), and 7.5% of dimethylsulfoxide (DMSO- 50 µL). After 1 min, the structures were transferred to a drop of 10 µL of vitrification solution 2 (TCM-HEPES 199 supplemented with 20% (v/v) FBS and, 0.5 M of sucrose), solution plus 16.5% of ethylene glycol (100 µL) and, 16.5% of DMSO (100 µL). They remained in this drop for 20 s and then were placed at the end of the OPS with as little medium as possible. Immediately after loading, the OPS was immersed in liquid nitrogen, where it was stored for a week, for later warming.
For warming, the OPS was removed from the liquid N2, and one OPS was warmed at a time. The tip of the OPS, where the blastocysts were located, was immersed in a well of a four-well plate containing 800 µL of holding solution (TCM-HEPES 199) and 400 µL of holding solution supplemented with sucrose (HS + S; PBS - pH 7.4 supplemented with 5% (v/v) FBS and, 0.5 M of sucrose). Then, the blastocysts were transferred to the second well containing 400 µL of holding solution and 200 µL of HS + S. The blastocysts spent time between well 1 and 2 did not exceed 5 min. The blastocysts were transferred to well 3 containing 400 µL of holding solution and 100 µL of HS + S for 5 min and finally to well 4 in which there was only holding solution. After that, the blastocysts were washed in three drops of SOF medium and transferred to the culture plate. After 12 h of warming, the rate of re-expansion and hatching was evaluated; and at 24 and 48 h the hatching rate was recorded.
Reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA from blastocysts after vitrification (3 embryos/group in 4 replicates) was extracted using the PicoPure RNA Isolation kit (Life Technologies, Foster City, CA, United States) following the manufacturer’s protocol. Extracted RNA was stored at − 80° C until further analysis by qPCR. RNA concentration was quantified using a spectrophotometer (Nanodrop, Thermo Fisher Scientific, Waltham, MA, United States). For each sample, we used a pool of three blastocysts for reverse transcription. cDNA synthesis was performed using a High-Capacity Reverse Transcription kit (Applied Biosystems, Foster City, CA, United States), following the manufacturer’s instructions. All samples were treated with DNase according to the manufacturer’s instructions before reverse transcription.
Pre-amplification and qPCR
Gene expression analyses of bovine blastocysts were performed independently using Applied BiosystemsTM TaqMan©R Assays specific for B. taurus and based on Fontes et al. (2020)27. We analyzed the abundance of 56 transcripts using a panel of genes formatted to investigate embryonic competence and quality (apoptosis, oxidative stress, proliferation, differentiation), and lipid metabolism in a microfluidic platform (Supplementary Table S2 describing all the genes and their signaling pathways). Prior to qPCR thermal cycling, each sample was subjected to a sequence specific preamplification process as follows: 1.25 mL assay mix (TaqMan©R Assay was pooled to a final concentration of 0.2× for each of the 56 assays), 2.5 mL TaqMan PreAmp Master Mix (Applied Biosystems, #4391128), and 1.25 mL cDNA (5 ng/mL). The reactions were activated at 95° C for 10 min, followed by denaturation at 95° C for 15 s, annealing, and amplification at 60° C for 4 min for 14 cycles. These preamplified products were diluted fivefold (embryos) prior to RT-qPCR analysis. Assays and preamplified samples were transferred to an integrated fluidic circuit plate. For gene expression analysis, the sample solution preparation consisted of 2.25 mL cDNA (preamplified products), 2.5 mL of TaqMan Universal PCR Master Mix (2×, Applied Biosystems), and 0.25 mL of 20× GE Sample Loading Reagent (Fluidigm, South San Francisco, CA, United States); the assay solution included 2.5 mL 20× TaqMan Gene Expression Assay (Applied Biosystems) and 2.5 mL of 2× Assay Loading Reagent (Fluidigm). The 96.96 Dynamic ArrayTM Integrated Fluidic Circuits (Fluidigm) chip was used for data collection. After priming, the chip was loaded with 5 mL each of the assay solution and each sample solution and loaded into an automated controller that prepares the nanoliter-scale reactions. The qPCR thermal cycling was performed in the Biomark HD System (Fluidigm) using the protocol TaqMan GE 56 × 56 Standard, which involved one stage of Thermal Mix (50° C for 2 min, 70° C for 20 min, and 25° C for 10 min) followed by a hot start stage (50° C for 2 min and 95° C for 10 min), 40 cycles of denaturation (95° C for 15 s), primer annealing, and extension (both at 60° C for 60 s).
Statistical analysis
To estimate the lipid content, the results of the analysis were evaluated of data distribution, being non-parametric data, the Kruskal-Wallis test was used followed by the post hoc Student-Newman-Keuls test. The data are present in the form of median, 1sd and 3rd interquartiles, and analyzes were performed with SigmaStat 4.0 software.
To evaluate blastocyst rate and hatching kinetics after warming, data were tested for normality using the Shapiro–Wilk and Bartlett tests. After then, Tukey’s test was applied. Differences with probabilities less than 0.05 were considered significant. The data is presented as mean value and standard error of the mean (SEM) and analyzes were performed with SigmaStat 4.0 software.
To the lipid profile data (MRM profiling), analysis was performed with the first step (discovery) applying a list of MRMs generated by combining the m/z of the analysis in relation to the ions based on the online platform LipidMAPS (http://www.lipidmaps.org/) with expected product ion resulting from precursor scan (Prec) and neutral loss scans (NL; Supplementary Table S1)28. MetaboAnalyst 5.0 (http://www.metaboanalyst.ca) was used for multivariate statistics by Principal Component Analysis (PCA). The two experimental groups, control and CNP group (400 nM of CNP) were evaluated with Student’s t-test. Fold-change values were also calculated and considered significant when fold changes (FC) > ± 1.5 and P < 0.05.
Quantitative PCR data were assessed using the ∆Cq values relative to the geometric mean of the reference genes (genes that maintained stability in their values) among the 56-gene set, i.e., GAPDH, and ACTB. Fold-changes (FC) were calculated using the 2−∆Cq method29. All analyses were performed using SigmaStat 4.0 and MetaboAnalyst 5.0. The evaluation of the transcripts was initially performed with the univariate statistical analysis method, using FC, t-test, and Volcano Plot. In a second step, we analyzed the data by multivariate methods, considering Principal Component Analysis (PCA) and Partial Least Squares- Discriminant Analysis (PLS-DA) and their variations. Weak statistical significance (i.e., a trend that we consider indicative of biological effect) was determined based on 0.06 < P-value ≤ 0.08, while strong significance was considered when p-value ≤ 0.01. Differences with probabilities less than P < 0.05 were considered intermediately significant.
Results
Lipid content by Sudan black B
There was no difference in lipid content analysis between embryos without CNP supplementation (control) compared to embryos supplemented with 400nM CNP (Table 1; Fig. 1).

Illustrative light microscopy images of IVP bovine embryos submitted to culture supplemented with CNP. (A) Bovine embryo without CNP supplementation (control); (B) Bovine embryo submitted to 400 nM CNP supplementation. (1) Images captured by light microscopy; (2) Images converted to grayscale using Image J 1.41 Software. Original magnification x 500.
Lipid profile of embryos cultured with CNP
Embryos cultured with CNP showed a reduction in the relative abundance of ions belonging to different lipid subclasses, such as acylcarnitine, sphingomyelin, cholesteryl esters, free fatty acids, and glycerophospholipid. Furthermore, the triacylglycerol lipids TG 52:3 NL 16:1, TG 56:3 NL 18:1, and the glycerophospholipid C22:6, were increased in the CNP group (Table 2).
Warming, Re-expansion and hatching rates of blastocysts
Regarding the rate of re-expanded and hatched blastocysts, there was no difference between the rates of re-expansion and hatching at 12, 24, and 48 h after warming between the evaluated groups (Tables 3 and 4).
The effect of CNP on the abundance of target-transcripts in IVC embryos post-cryopreservation
There were weak differences between groups on the transcript abundance in ATF4 (P = 0.052), BMP15 (P = 0.063), and GFPT2 (P = 0.063; Fig. 2).

Effect of CNP treatment in IVC on differential gene expression in blastocyst after warming. Data represent the fold change of relative target abundance related to the reference gene. Upregulated transcription ATF4 (P = 0.052), BMP15 (P = 0.063), and GFPT2 (P = 0.063) with the addition of CNP (400 nM) on the D1 of culture after vitrification and warming. Results are represented by least-squares means ± SEM of four replicates/group. Different letters above each bar represent significant differences (P ≤ 0.05) and trend statistical significance was determined based on 0.06 < P-value ≤ 0.08. Control (no treatment) and CNP (400 nM of CNP).
In Partial Least Squares - Discriminant Analysis (PLS-DA) it was possible to observe an overlap in PCA plot, and in PLS-DA a slight separation between the control and CNP-treated groups (Fig. 3A). The Heatmap shows the abundance of transcriptional profiles of bovine embryos produced in vitro with 400 nM CNP and the control group, which obtained fold change less than 1.5. There was a slight clustering between the samples (Fig. 3B). All the others target transcripts analyzed in blastocysts did not differ in a statistically significant way (Supplementary Table S2).

Plots derived from multivariate analysis of the abundance of transcripts derived from untreated (control) and CNP-treated blastocyst after cryopreservation. (A) PLS-DA shows the 2D score plot between groups. (B) Heatmap showing transcriptional profiles abundance of in vitro–produced bovine embryos with 400 nM CNP and control group, which obtained fold change ˃1.5. Control group: n = 4; CNP group: n = 5.
Discussion
Our study is the first to present the use of CNP in in vitro culture of bovine embryos to modulate the lipid profile and content, aiming to make it more cryo-tolerable. Changes in the analysis of transcripts - related to lipid metabolism, embryonic development, and oxidative stress - were observed in blastocysts treated with CNP. In addition, when analyzing the embryonic lipid by the MRM profiling, it was possible to observe changes in ions belonging to different lipid subclasses, such as acylcarnitine, sphingomyelin, cholesterol esters, free fatty acids, and glycerophospholipid. However, no changes in embryonic lipid content were observed in the Sudan Black B analysis.
The use of exogenous CNP in pre-IVM and IVM has been used for years, with different concentrations and in different species14,30,31,32,33. However, the relationship between CNP and the embryo has few reports in the literature14,16,34. It is known that CNP binds to its receptor NPR2 and modulates intracellular cAMP concentrations in oocytes in the in vitro maturation phase30. In this way, the presence of the NPR2 receptor was already detected in bovine17 and murine embryos32. Moreover, it is suspected that CNP can stimulate cGMP, which exerts an action similar to cAMP, stimulating lipolysis and reducing the amount of lipids16.
Unlike the results with the semiquantitative analysis by Sudan Black B, we demonstrated that the culture of embryos with CNP altered the lipid profile. Costa et al.. (2020)16in a similar way to our study, previously also did not observe changes in the Sudan Black B profile after the use of CNP (100 nM). In the present study, a reduction in the relative concentrations of C16:0 (palmitic acid) and C18:0 (stearic acid) was observed. Changes in biological functions associated with metabolic disease, pro-inflammatory response, and reduction in glucose uptake were demonstrated by RNA sequencing in a study performed on ovarian cortex culture and oocyte maturation in a medium containing palmitic, oleic, and stearic acid (NEFA- non-esterified fatty acids)34. In this context, Shibahara et al.. (2020)35 showed that the oocytes developed in the presence of palmitic acid had low developmental ability and differential histone modifications. Also, the exposure of COCs to palmitic acid during IVM reduced mitochondrial membrane potential, and increased oxidative stress in the resulting bovine embryos36.
In our study, embryos of the CNP group (with addition of 400 nM of CNP) have C22:6 (docosahexaenoic acid-DHA) increased. The DHA supplementation (physiological dose of 1 µM) during IVM was able to increase the blastocyst rate of bovine oocytes after IVF37. DHA addition in IVM medium enhances the homeostasis of energy metabolism by improving mitochondrial function and lipid metabolism. Also, DHA improved the quality of matured oocytes and enhanced the embryonic developmental potential of IVP porcine embryos38. Some triacylglycerols (TG) decreased in embryos CNP treatment. TG are found in intracytoplasmic droplets in mammalian cells and represent an energy reserve for the cell3. In this context, our group previously reported that the embryos derived from high-AFC (antral follicles count) cows presented a higher concentration of TG than the embryos from low-AFC females39.
Cholesterol esters are present in all cell membranes and play an important role during cell division after the fertilization stage40. Sphingomyelin (SM), is implicated in proliferation, migration, inflammation, and cell survival41. Glycerophospholipids play an important role in maintaining the function and integrity of cellular and subcellular structures and are the major constituents of cellular membranes42. Similarly, CAR 5:1 is an acylcarnitine, which refers to the compounds that play a crucial role in the metabolism of long-chain fatty acids and are utilized to transport acyl-CoAs out of the mitochondria43. Despite the decrease of some cholesteryl esters, sphingomyelin, and acylcarnitine (Table 2) in the embryos CNP group, we did not observe interferences in the hatching kinetics and rate of re-expansion and hatching after warming blastocysts cultured with CNP. However, the results of in vivo embryo transfer are still needed, whether cryopreserved or freshly transferred, so that there is greater clarity about the possible effects of CNP on the IVC of these embryos.
The transcript of bone morphogenetic protein 15 (BMP15) was upregulated in blastocysts that received CNP in the IVC and evaluated after cryopreservation. BMP15, in mammals, is related to oocyte maturation and cholesterol biosynthesis, with the aim of improving oocyte competence and consequently the development of the bovine embryo44,45. It has already been observed that mutation in the BMP15 gene affects glycolysis and cholesterol biosynthesis in cumulus cells46. A possible inference for the finding in the present study would be that CNP could be intensifying the expression of BMP15 by modulating cholesterol biosynthesis through a potential increase in cGMP, in addition to the stress that the cryopreservation process can cause in cells.
In the blastocysts evaluated after vitrification and warming, it was possible to observe a greater abundance of the ATF4 gene transcript. Activating transcription factor 4 (ATF4) is a transcription factor that upregulates genes involved in amino acid import, glutathione biosynthesis, and antioxidant stress response47,48. In addition, ATF4 is related to the prevention of endoplasmic reticulum stress14,19. Despite the ATF4 action related to the antioxidant stress response, no oxidative stress tests were performed in the present article to support this statement.
In summary, this study showed changes in embryonic metabolism based on the abundance of transcripts and analysis of the lipid profile. Furthermore, maintenance of production rates was observed after vitrification of embryos cultured with CNP, in addition to no change in embryonic lipid content. Since the differences in gene expression were not statistically significant, more studies are needed to establish whether those observed changes - in gene expression and lipid metabolism - will impact embryo implantation and pregnancy development. So far, our work has shown no harmful effects with CNP treatment on the studied endpoints.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Lopes, B. C. et al. Genética Bovina brasileira: Mercado internacional e Mapeamento Das competências e tecnologias Mineiras. O Embriao 51, 111 (2012).
IETS. Statistics of embryo production and transfer in domestic farm animals. Embryo Technology Newsletter, v. 42, n.4, 2024. (2023). Available at: https://www.iets.org/Portals/0/Documents/Public/Committees/DRC/IETS_Data_Retrieval_Report_2023.pdf
Sudano, M. J. et al. Lipidome signatures in early bovine embryo development. Theriogenology 86, 472–484e1 (2016).
Camargo, L. S. A. et al. Osmotic challenge and expression of Aquaporin 3 and na/k ATPase genes in bovine embryos produced in vitro. Cryobiology 63, 256–262 (2011).
Paschoal, D. M. et al. Cell apoptosis and lipid content of in vitro–produced, vitrified bovine embryos treated with forskolin. Theriogenology 87, 108–114 (2017).
Marsico, T. V., de Camargo, J., Valente, R. S. & Sudano, M. J. Embryo competence and cryosurvival: molecular and cellular features. Anim. Reprod. 16, 423–439 (2019).
Ferré, L. B., Kjelland, M. E., Taiyeb, A. M., Campos-Chillon, F. & Ross, P. J. Recent progress in bovine in vitro‐derived embryo cryotolerance: impact of in vitro culture systems, advances in cryopreservation and future considerations. Reprod. Domest. Anim. 55, 659–676 (2020).
Ferré, L. B. et al. Review: recent advances in bovine in vitro embryo production: reproductive biotechnology history and methods. Animal 14, 991–1004 (2020).
Nagashima, H., Kashiwazaki, N., Ashman, R. J., Grupen, C. G. & Nottle, M. B. Cryopreservation of Porcine embryos. Nature 374, 416–416 (1995).
Diez, C. et al. Delipidating in vitro-produced bovine zygotes: effect on further development and consequences for freezability. Theriogenology 55, 923–936 (2001).
Stringfellow, D. A. No Title. in Manual of the International Embryo Transfer Society. (ed. D.A. Stringfellow, S. M. S.) 79–84 (Savoy, 1998).
Sanches, B. V. et al. Cryosurvival and pregnancy rates after exposure of IVF-derived Bos indicus embryos to forskolin before vitrification. Theriogenology 80, 372–377 (2013).
Paschoal, D. M. et al. In vitro embryos production after oocytes treatment with forskolin. Zygote 24, 161–171 (2016).
Botigelli, R. C. et al. Supplementing in vitro embryo production media by NPPC and sildenafil affect the cytoplasmic lipid content and gene expression of bovine cumulus-oocyte complexes and embryos. Reprod. Biol. 18, 66–75 (2018).
Costa, C. B. et al. Influence of forskolin supplementation on embryos produced in vitro. Livest. Sci. 221, 15–18 (2019).
Costa, C. B. et al. Influence of cAMP modulator supplementation of in vitro culture medium on Bos taurus indicus embryos. Theriogenology 141, 134–141 (2020).
Costa, C. B. et al. Developmental and molecular effects of C-Type natriuretic peptide supplementation in in vitro culture of bovine embryos. Int. J. Mol. Sci. 25, 10938 (2024).
Pioltine, E. M. et al. Treatment of in vitro-Matured bovine oocytes with Tauroursodeoxycholic acid modulates the oxidative stress signaling pathway. Front Cell. Dev. Biol 9, 1-12 (2021).
Pioltine, E. M., Costa, C. B., Franchi, F. F., dos Santos, P. H. & Nogueira, M. F. G. Tauroursodeoxycholic acid supplementation in in vitro culture of indicine bovine embryos: molecular and cellular effects on the in vitro cryotolerance. Int. J. Mol. Sci. 24, 14060 (2023).
Sudano, M. J. et al. Phosphatidylcholine and sphingomyelin profiles vary in Bos taurus indicus and Bos taurus taurus in Vitro- and in Vivo-Produced Blastocysts1. Biol Reprod 87,1–11 (2012).
de Lima, C. B. et al. Effect of lipid extraction and room temperature transportation of bovine oocytes determined by MRM profiling. Res. Sq. Dec 23:rs.3.rs-3788683 (2023).
de Lima, C. B. et al. Impact of extraction methods and transportation conditions on lipid profiles of bovine oocytes. Reprod. Sci. 31, 1948–1957 (2024).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
de Lima, C. B. et al. Comprehensive lipid profiling of early stage oocytes and embryos by MRM profiling. J. Mass. Spectrom. 53, 1247–1252 (2018).
Bó, G. A. & Mapletoft, R. J. Evaluation and classification of bovine embryos. Anim. Reprod. 10, 344–348 (2013).
Vajta, G., Holm, P., Greve, T. & Callesen, H. Vitrification of Porcine embryos using the open pulled straw (OPS) method. Acta Vet. Scand. 38, 349–352 (1997).
Fontes, P. K., Castilho, A. C. S., Razza, E. M. & Nogueira, M. F. G. Bona Fide gene expression analysis of samples from the bovine reproductive system by microfluidic platform. Anal. Biochem. 596, 113641 (2020).
Reis, L. G., Casey, T. M., Sobreira, T. J. P., Cooper, B. R. & Ferreira, C. R. Step-by-Step Approach to Build Multiple Reaction Monitoring (MRM) Profiling Instrument Acquisition Methods for Class-based Lipid Exploratory Analysis by Mass Spectrometry. J. Biomol. Tech. 34, 3fc1f5fe.c438 (2023). (1972).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2 – ∆∆CT method. Methods 25, 402–408 (2001).
Franciosi, F. et al. Natriuretic peptide precursor C delays meiotic resumption and sustains gap Junction-Mediated communication in bovine Cumulus-Enclosed Oocytes1. Biol Reprod 91, 1-9 (2014).
Soares, A. C. S. et al. Steroid hormones interact with natriuretic peptide C to delay nuclear maturation, to maintain oocyte–cumulus communication and to improve the quality of in vitro-produced embryos in cattle. Reprod. Fertil. Dev. 29, 2217 (2017).
Xi, H. et al. Expression and localization of Npr2 in mouse oocytes and pre-implantation embryos. Biotech. Histochem. 94, 320–324 (2019).
Ang, L. et al. Supplementation of c-type natriuretic peptide during in vitro growth period benefits the development of murine preantral follicles. Zygote 29, 150–154 (2021).
Pedroza, G. H. et al. Exposure to non-esterified fatty acids in vitro results in changes in the ovarian and follicular environment in cattle. Anim. Reprod. Sci. 238, 106937 (2022).
Shibahara, H. et al. Mechanism of palmitic acid-induced deterioration of in vitro development of Porcine oocytes and granulosa cells. Theriogenology 141, 54–61 (2020).
Marei, W. F. A. et al. Mitochondria-targeted therapy rescues development and quality of embryos derived from oocytes matured under oxidative stress conditions: a bovine in vitro model. Hum. Reprod. 34, 1984–1998 (2019).
Oseikria, M., Elis, S., Maillard, V., Corbin, E. & Uzbekova, S. N-3 polyunsaturated fatty acid DHA during IVM affected oocyte developmental competence in cattle. Theriogenology 85, 1625–1634e2 (2016).
Lee, Y. et al. Docosahexaenoic acid supplementation during Porcine oocyte in vitro maturation improves oocyte quality and embryonic development by enhancing the homeostasis of energy metabolism. Theriogenology 227, 49–59 (2024).
Rosa, C. O. et al. Lipid profile of in vitro embryos produced from Bos indicus cows with low and high antral follicle counts. Livest. Sci. 250, 104586 (2021).
González-Serrano, A. F. et al. Desorption electrospray ionization mass spectrometry reveals lipid metabolism of individual oocytes and embryos. PLoS One. 8, e74981 (2013).
Taniguchi, M. & Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration—from cell and animal models to human disorders. Biochim. Biophys. Acta - Mol. Cell. Biol. Lipids. 1841, 692–703 (2014).
Sarkar, C. & Lipinski, M. M. Glycerophospholipid dysregulation after traumatic brain injury. Neurochem Int. 175, 105701 (2024).
Placidi, M. et al. Carnitines as mitochondrial modulators of oocyte and embryo bioenergetics. Antioxidants 11, 745 (2022).
Caixeta, E. S. et al. Bone morphogenetic protein 15 and fibroblast growth factor 10 enhance cumulus expansion, glucose uptake, and expression of genes in the ovulatory cascade during in vitro maturation of bovine cumulus–oocyte complexes. Reproduction 146, 27–35 (2013).
Cajas, Y. N. et al. Antioxidant nobiletin enhances oocyte maturation and subsequent embryo development and quality. Int. J. Mol. Sci. 21, 5340 (2020).
Su, Y. Q. et al. Oocyte regulation of metabolic cooperativity between mouse cumulus cells and oocytes: BMP15 and GDF9 control cholesterol biosynthesis in cumulus cells. Development 135, 111–121 (2008).
Harding, H. P. et al. Regulated translation initiation controls Stress-Induced gene expression in mammalian cells. Mol. Cell. 6, 1099–1108 (2000).
Harding, H. P. et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 11, 619–633 (2003).
Acknowledgements
The authors would like to thank ABS Global Brasil®, Mogi Mirim, São Paulo, Brazil for availability to use its media in in vitro produced bovine embryos. We would like to thank Rio da Prata slaughterhouse, Bela Vista do Paraíso, Paraná, Brazil, for supplying us with bovine ovaries, and Multiuser Laboratory FitoFarmaTec (managed by Prof. Luiz Claudio Di Stasi), UNESP, Botucatu, São Paulo, for full access to Biomark HD platform.
Funding
This work received financial support by grants #2019/10732-5 and #2021/11747-6, São Paulo Research Foundation (FAPESP). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001 and INCT-LEITE 465725/2014.
Author information
Authors and Affiliations
Contributions
CBC contributed to the conception and design of the study, collected and analyzed data, and wrote the manuscript. NCS and TKS performed most of the experiments. EFC performed RNA extraction and reverse transcription for cDNA of samples subjected to gene expression analysis. CRF contributed to the MRM profiling analyses, as well as the interpretation of the results. AFZ contributed to revision and wrote the manuscript. MMS and FVM provided technical support. MFGN contributed to the conceptualization and design of the entire study and supervised and contributed to critical revision and intellectual input to the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Costa, C.B., Silva, N.C., Costa, É.F. et al. Modulation of lipid composition and gene expression by CNP supplementation in in vitro cultured bovine embryos. Sci Rep 15, 22972 (2025). https://doi.org/10.1038/s41598-025-07453-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07453-0
