
Abstract
Glioma is the most common and aggressive malignant tumors in central nervous system, its morbidity and mortality are both high. Lysine acetylation could alter the expression of oncogene and anti-oncogene. Thus, this study explored the potential mechanism and aimed to find new diagnostic and therapeutic methods. The Cancer Genome Atlas Glioblastoma Multiforme, Glioma related dataset were downloaded from UCSC and CGGA database respectively. Lysine acetylation-related genes (LARGs) was acquired from published literature. Differentially expressed genes (DEGs) were analyzed between GBM and control samples. LARGs (DE-LARGs) were obtained by taking the intersection of DEGs and weighted gene co-expression network analysis (WGCNA) module genes. Subsequently, enrichment analysis and protein–protein interaction (PPI) network was processed. Then, prognosis genes were selected, risk model was constructed and verified. After that, independent prognosis factors were used to predict the survival of GBM patients. Corresponding pathways and functions were analyzed between different groups. The difference of immune environment was compared. Finally, the drug prediction and regulatory network construction was performed. Prognosis genes in tumor and normal tissue were identified using immunohistochemistry. Totally 6767 DEGs were screened out. A total of 2890 module genes were identified highly correlated with lysine acetylation score by WGCNA. A total of 313 DE-LARGs were acquired by taking the intersection of DEGs and module genes. PPI network was constructed and 215 genes were obtained. Further, risk model revealed 5 genes (CD79B, STXBP4, DDHD1, FKBP1B and TRAM2) was related with overall survival (OS) of GBM patients. Kaplan–Meier survival and receiver operating characteristic curves were proved to be highly accurate both in training and validation set. Based on nomogram, riskscore was the independent prognosis factor for patients. The immune infiltration level was highly expressed in high risk group. Four drugs (PAC.1, OSI.906, WH.4.023, BMS.536924) were identified as chemotherapeutic drugs in Glioma. The transcription factors (TFs)-mRNA regulatory network was constructed and 76 TFs were obtained using TRRUST. Finally, the expression of prognostic genes in tumor was significantly higher than that in normal tissue. New prognostic genes CD79B, STXBP4, DDHD1, FKBP1B, and TRAM2 were identified for glioma through the new perspective of lysine acetylation, suggesting their importance in the development of the disease and offering potential insights for diagnosis and treatment.
Similar content being viewed by others

Introduction
Glioma is a common type of brain tumor that originates from neuroglial cells and is known for its infiltrative growth, making it challenging to fully remove and likely to come back, putting patients at risk. Glioma symptoms vary depending on tumor size and location, but commonly include headaches, nausea, vomiting, and neurological issues like limb weakness and speech problems. Without prompt treatment, glioma can cause serious neurological damage and be life-threatening1. Glioma research has advanced significantly in understanding its molecular mechanisms and developing novel therapeutic strategies, yet critical challenges persist. At the molecular level, key drivers such as IDH1/2 mutations disrupt cellular metabolism through 2-hydroxyglutarate accumulation2, while EGFR amplification and TP53/ATRX loss drive proliferation and genomic instability3,4. Epigenetic dysregulation, notably MGMT promoter methylation predicting temozolomide sensitivity5, and histone modifications further shape tumor behavior6. The immunosuppressive microenvironment, mediated by glioma-associated microglia and Treg infiltration, alongside hypoxia-induced VEGF-driven angiogenesis, complicates treatment efficacy7. Current therapies rely on surgery, radiotherapy, and chemotherapy, but limitations arise from infiltrative growth and resistance mechanisms. Emerging approaches include IDH inhibitors extending progression-free survival, CAR-T cells targeting IL-13Rα2/EGFRvIII, and tumor-treating fields (TTFields) improving median survival to 20.9 months in newly diagnosed GBM8. Liquid biopsy (ctDNA, exosomal miRNAs) and AI-enhanced imaging (multimodal MRI radiomics) now refine diagnosis and prognosis9,10. However, challenges such as blood–brain barrier limitations, tumor heterogeneity revealed by single-cell sequencing, and GSC-driven recurrence demand innovative solutions like nanoparticle drug delivery, CRISPR-based gene editing, and adaptive clinical trial designs11. Glioma treatment options include surgery, radiotherapy, and chemotherapy. Surgery aims to remove as much tumor tissue as possible, but complete removal is often challenging due to the tumor’s infiltrative growth. Radiotherapy and chemotherapy are used to kill remaining tumor cells, but can have side effects12. Despite multimodal therapeutic interventions, the prognosis of gliomas remains dismal, characterized by high recurrence rates, limited survival durations, and treatment-associated morbidities13. Notably, glioblastoma (GBM) exhibits a 5-year survival rate below 10%, reflecting its aggressive biological behavior. In pediatric populations, diffuse midline gliomas—exemplified by diffuse intrinsic pontine glioma (DIPG)—demonstrate even greater lethality, with clinical cohorts reporting a median overall survival of merely 11 months, a 5-year survival rate under 1%, and near-universal mortality approaching 100%14. Therefore, it is crucial to explore new diagnostic and treatment approaches for glioma.
Lysine acetylation is a dynamic post-translational modification that regulated the functions and subcellular localization of target proteins through structural alterations, thereby influencing key physiological processes such as cell proliferation and transcriptional regulation15. In glioma, this process not only drives oncogenic signaling pathways through epigenetic regulation (such as chemotherapy resistance related to histone deacetylase HDAC activity), but also has a deep interaction with the histone methylation modification network, jointly shaping the malignant phenotype of the tumor16,17. Recurrent histone mutations—including H3K27M, H3G34V/R, and H3K36M—disrupt epigenetic homeostasis by altering histone methylation patterns, thereby driving oncogenic transcriptional programs in gliomas18,19. Notably, the H3K27M mutation, a hallmark of diffuse midline gliomas such as DIPG, induces global hypomethylation at H3K27 residues while concurrently modulating lysine acetylation dynamics through cross-talk with histone acetyltransferases (HATs) and deacetylases (HDACs). This dual perturbation of methylation and acetylation networks creates a permissive chromatin state that sustains tumor proliferation across pediatric and adult glioma subtypes20.
This dual perturbation of methylation and acetylation is not an independent event but jointly drove the initiation and progression of glioma through coordinated regulation of chromatin accessibility and transcriptional programs21. Specifically, hyperacetylation at oncogenic promoters synergizes with histone mutation-driven epigenetic reprogramming to amplify tumor growth signals, while hypoacetylation states correlate with therapy resistance via DNA repair pathway suppressio22,23. Therapeutic targeting of acetylation regulators, such as HDAC inhibitors, has shown promise in restoring epigenetic balance and overcoming treatment refractoriness in preclinical models24. These findings position lysine acetylation not merely as a parallel epigenetic mechanism but as an integral component of the histone modification network governing gliomagenesis and therapeutic vulnerability.
In this study, we employed integrated bioinformatics and statistical approaches to systematically identify lysine acetylation-related differentially expressed genes (KAc-DEGs) in gliomas. Through multi-omics analysis of public glioma datasets, five pivotal genes (CD79B, STXBP4, DDHD1, FKBP1B, and TRAM2) demonstrating significant associations with tumor progression and clinical outcomes were identified. These molecular markers were subsequently incorporated into a novel prognostic model, which was rigorously validated using an independent cohort of glioma specimens through immunohistochemical analysis. Furthermore, we quantified tumor mutational burden (TMB) patterns in TCGA datasets to investigate their potential correlation with therapeutic responsiveness. Our findings not only elucidate the critical role of lysine acetylation-associated genes in glioma pathogenesis but also provide clinically actionable biomarkers for patient stratification. This investigation aims to address current knowledge gaps in glioma etiology by identifying molecular targets with diagnostic and therapeutic potential, ultimately facilitating the development of precision medicine strategies to improve clinical outcomes and quality of life for glioma patients.
Results
6767 differentially expressed genes (DEGs) and 2890 module genes were identified
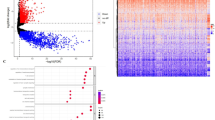
There were 6767 DEGs were found between glioblastoma multiforme (GBM) and normal samples in The Cancer Genome Atlas Glioblastoma Multiforme (TCGA-GBM) and included 3400 up-regulated and 3367 down-regulated genes (Fig. 1A,B). All samples were in clusters, so it is not necessary to remove outlier (Supplementary Figure 1). Scale-free network was constructed with optimal soft threshold set to 6 (Fig. 1C). Finally, 11 gene modules were obtained (Fig. 1D). Furthermore, correlation analysis demonstrated that blue module had significantly highest correlation (cor = 0.7267338, p value < 0.05) with lysine acetylation-related sSingle-sample gene set enrichment analysis (ssGSEA) score. Thus, 2890 genes involved in this module were used for further research (Fig. 1E).

Identification of lysine acetylation-related genes in glioma. (A) Volcano plot: Upregulated genes are shown in red, and downregulated genes are shown in blue. (B) Heat map of differential genes. Red represents higher relative expression while blue denotes lower relative expression. (C) Analysis of the scale-free fit index and the mean connectivity for 1–20 soft-thresholding powers (β). (D) Gene modules identified by WGCNA. E. The correlation of gene modules with clinical traits. The ordinate represents different modules and the abscissa represents different groups. The first line in the module is the correlation coefficient, and the parentheses are p value.
Enriched analysis and protein–protein interaction (PPI) network analysis of differentially expressed lysine acetylation-related genes (DE-LARGs)
A total of 313 DE-LARGs were obtained by taking the intersection of 6767 DEGs and 2890 module genes (Fig. 2A). Based on DE-LARGs, totally 458 terms were enriched in GO-biological processes (BP), such as axon development, gland development. A total of 45 terms were enriched in GO-cellular component (CC), such as cell leading edge, collagen-containing extracellular matrix. Totally 49 terms were enriched in GO-molecular functions (MF), such as GTPase regulator activity (Fig. 2B,C). In addition, 18 kyoto encyclopedia of genes and genomes (KEGG) pathways were enriched (p value < 0.05), including focal adhesion, PI3K-Akt signaling pathway, which were relative with Glioma and lysine acetylation (Fig. 2D,E). Finally, the PPI network that included 215 nodes which associated with 414 edges was constructed. Up-regulated genes were AR, CCND1, PXDN, FLNA, NOTCH2 and so on. Down-regulated genes were following: MN1, SH3TC2, SH3TC2, PTK2, ZEB2, etc. (Fig. 2F). Multiple topological analysis methods were used to evaluate the importance of nodes and screen out the core gene modules. The top 10 genes were selected. These genes have high connectivity in the network, suggesting that they may play a key role in lysine acetylation related pathways and the progression of glioma (Supplementary Figure 2).

Enriched analysis and PPI network analysis of DE-LARGs. (A) The Venn plot of the intersection gene. 313 intersection gene was obtained. (B) GO functional analysis of intersecting genes. The horizontal coordinate represents the number of genes enriched in the corresponding pathway, and the vertical coordinate represents the enriched GO function. (C) GO functional annotation of intersection genes. The circles from the outside to the inside represent: the first layer, the ID of the GO function, showing the three functions of BP, CC and MF respectively; In the second layer, the color depth represents the significance size, and the length width and number represent the number of genes enriched in the function. The third layer represents the number of up-down-regulated genes enriched in this function, and the color is used to distinguish the up-down-regulated genes. The innermost color piece and color represents different functions, on behalf of the way to the size of the RichFactor (RichFactor refers to the transcription of differentially expressed the transcription of the median in the GO items this number with all comments transcription in the GO in this entry the transcription of the ratio of the total, the greater the RichFactor, the greater the degree of enrichment. (D) KEGG molecular pathway of intersecting genes. The circles from the outside to the inside represent: the first layer, the KEGG molecular pathway; In the second layer, the color depth represents the significance size, and the length width and number represent the number of genes enriched in the function. The third layer represents the number of up-down-regulated genes enriched in this function, and the color is used to distinguish the up-down-regulated genes. The innermost color block, where the color represents the different function, and the size represents the RichFactor of the pathway (RichFactor refers to the ratio of the number of differentially expressed transcripts located in that entry to the total number of all annotated transcripts located in that entry, the larger the RichFactor,the greater the degree of enrichment. (E) KEGG pathway enrichment bubble map of intersection gene. (F) PPI network of intersection DEGs using STRING tool and Cytoscape software. Nodes symbolize genes, lines depict reciprocal connections, and colors indicate gene expression levels: red for upregulation and blue for downregulation.
Identification of 5 prognosis genes
Among 215 genes obtained from PPI network, 5 genes (CD79B, STXBP4, DDHD1, FKBP1B and TRAM2) were identified as prognosis genes by univariate Cox, Least absolute shrinkage and selection operator (LASSO), multivariate Cox and multiple stepwise regression analysis (Fig. 3A–E). Optimal cutoff was calculated aimed to 5 genes. Subsequently, samples of GBM were divided into high and low risk groups through optimal cutoff. After log Rank test, Kaplan–Meier (KM) curve were plotted, these 5 genes had significantly different expression in two groups. CD79B, FKBP1B, TRAM2 had longer OS in low risk group (Fig. 3F1, F4, F5), but STXBP4 and DDHD1 had opposite result (Fig. 3F2, F3).

5 prognosis genes were identified as by univariate Cox, LASSO, multivariate Cox and multiple stepwise regression analysis and Kaplan–Meier analysis of prognostic genes. (A) Forest plot of univariate Cox analysis. Through univariate Cox analysis, 17 genes were considered to be statistically significant for patient survival. (B) Prognostic genes cross-validation of LASSO regression. The x-axis is log(λ) and the y-axis is degrees of freedom. The dashed lines show the λ values for the smallest error mean and within 1 standard deviation. The top number shows the number of genes. Red dots show error values for each λ, with bars representing confidence intervals. (C) Plot of LASSO coefficient profiles. The x-axis is the log value of λ, the y-axis is the regression coefficient. The curve represents the relationship between the regression coefficient and λ, with the number at the top indicating the variables whose coefficient remains non-zero when λ is chosen. (D) Forest plot of multivariate Cox regression analysis. 9 model genes (SPAG4, BHLHE40, CD79B, STXBP4, DDHD1, FABP5, FKBP1B, TRAM2, PXDN) were identified using multivariate COX regression analysis. (E) The forest map of multivariate stepwise regression analysis. 5 model genes (CD79B, STXBP4, DDHD1, FKBP1B, TRAM2) were identified using multivariate stepwise regression analysis. F. Kaplan–Meier analysis of 5 prognostic progno genes. The K-M curve shows survival time and probability, with red representing high-risk and blue representing low-risk groups. The bottom half displays the risk list with remaining samples at different survival times. From left to right are CD79B,STXBP4, DDHD1,FKBP1B,TRAM2.
Characterization of riskscore to predict the prognosis of GBM
Riskscore = CD79B × (0.597) + STXBP4 × (− 1.047) + DDHD1 × (− 0.519) + FKBP1B × (0.391) + TRAM2 × (0.611). All patients was separated into high (n = 87) and low risk (n = 80) groups by optimal cutoff (1.139165) in training set (Fig. 4A). OS of two groups was analyzed (Fig. 4B). KM curve showed OS of low risk groups was significantly longer. Area under the curve (AUC) at 1, 2, 3 years were 0.67, 0.73, 0.86 respectively. These results demonstrated that risk model had a high accuracy (Fig. 4C–E). In validation set, among 137 samples, 59 patients was categorized to high risk patients and 78 were low risk patients relied on the optimal cutoff (2.588629) (Fig. 4F,G). KM curve showed longer OS in low risk group and AUC at 1, 2, 3 years were 0.61, 0.66, 0.66 (Fig. 4H,I), this result indicated that the model had good predictive effects. The heat map of the validation set risk model showed that CD79B,STXBP4, DDHD1,FKBP1B and TRAM2 were expressed differently in the high and low risk groups (Fig. 4J).

Characterization of riskscore to predict the prognosis of GBM. (A) The risk score of training set (The data was derived from TCGA). The cut-off value was 1.139165, classifying patients as either high risk (n = 87) or low risk (n = 80). (B) Survival distribution of GBM patients. In the training set, mortality increased as risk scores increased. (C) Kaplan–Meier estimates of survival curves. The K-M curve shows survival time and probability, with the top half representing high and low-risk groups, and the bottom half showing the number of samples remaining at different survival times. (D) Time-dependent ROC curves analysis. The x-axis shows FPR (specificity) and the y-axis shows TPR (sensitivity) in a graph. Higher AUC values indicate greater prediction accuracy, with curves closer to the upper left corner being more accurate. AUC values for 1, 2, and 3 years are all > 0.6, indicating high accuracy. (E) Risk model gene heat map. Red indicates high gene expression and blue indicates low gene expression. (F) Survival distribution of GBM patients in the validation set. (G) The risk score of validation set (The data was derived from CGGA). Mortality increased as risk scores increased. The optimal cut-off value of 2.588629 divided the sample into high and low groups, with 59 patients classified as high risk and 78 patients classified as low risk. (H) Kaplan–Meier estimates of survival curves in the validation set. (I) Time-dependent ROC curves analysis of validation set.The AUC at 1, 2, and 3 years was over 0.6, showing effective prognostic diagnosis efficiency. (J) Risk model gene heat map of validation set.
Prognosis genes could predict the OS
Univariate Cox analysis showed only riskscore had correlation with prognosis for patient survival (Fig. 5A). Therefore, prognosis genes were used to construct nomogram which predicted the survival of patients at 1, 2 and 3 years (Fig. 5B). Results of receiver operating characteristic (ROC) and calibration curves showed that the values of AUC were 0.67,0.74 and 0.86 in 1,2 and 3 years, respectively (Fig. 5C). Decision Curve (DCA) demonstrated that the model was valuable and reliable (Fig. 5D–F).

Construction of predictive nomogram. (A) The results of univariate Cox analysis show that only risk scores were associated with survival outcomes. (B) Construct nomograms to predict 1-year, 2-year, and 3-year prognosis of patients with GBM. (C) ROC of prognosis model 1, 2, 3 years. (D–F) DCA curves of prognosis model at 1, 2 and 3 years. The x-axis is ThresholdProbability and the y-axis is net benefit rate.
Different expression of TME score
Potential mechanism of difference in OS was recognized by GSVA, and the result showed 84 significant pathways (p.adj < 0.05). From these pathways, graft versus host disease, allograft rejection were up-regulated in high risk group. While non homologous end joining, homologous recombination were down-regulated (Fig. 6A,B). ESITIMATE results indicated that all TME scores had a significantly higher expression in high risk group (Fig. 6C). These three scores all had a significantly positive correlation with riskscore and Cor > 0.3 (Fig. 6D).

Results of GSVA enrichment analysis. (A) The most significant 20 molecular pathways. (B) Heat map of GSVA enrichment analysis. Red indicates high gene expression and blue indicates low gene expression. (C) The ESTIMATE, immune, and matrix scores were significantly higher in the high-risk group compared to the low-risk group. (D) The correlation between ESTIMATE, immune, and matrix scores with risk score was strong, with a correlation coefficient above 0.3.
Difference of immune expression of different group
Among 28 immune cell types, 23 types involved monocyte, macrophage, etc. had differential expression (p.adj < 0.05) (Fig. 7A). Correlation analysis showed riskscore and differential immune cells had significantly positive correlation (Fig. 7B,C). Immune function pathway analysis showed these pathways were higher expressed in high risk groups except MHC class I and Type I IFN Response (Fig. 7D).

Results of immune microenvironment analysis. (A) The difference between high and low risk risk groups. Risk scores were significantly correlated with various immune cells. (B) Heat map of correlation between immune cells and risk score prognostic genes. (C) Scatter plot of correlation between immune cells and risk score. (D) The enrichment scores of immune function pathways were different between high and low risk groups. (E) Volcanic map of TIDE analysis of immune checkpoint results. A total of 28 immune checkpoints were significant. (F) Volcanic map of TIDE analysis of chemokine results. There were 29 significant results for chemokines.
The significant results of immune checkpoints and chemokines showed that among 28 immune checkpoints, 23 immune checkpoints were up-regulated (IDO1, CD70, PDCD1LG2, CD48, TNFRSF9, etc.) and 5 (TNFRSF25, CD244, TNFSF9, CD200 and HHLA2) were down-regulated. For chemokines, 25 (CXCL13, CXCL10, CXCL9, CCL23, etc.) were up-regulated, 4 (CX3CL1, CCL28, CCR9 and CCL1) were down-regulated (Fig. 7E,F). And the correlation about immune checkpoints and chemokines was shown in Supplementary Figure 3, 4.
Analysis of mutant genes expression
Mutant landscape showed that mutations frequency was higher in high risk group (PTEN: low: 39%, high: 43%. TTN: low: 27%, high: 34%.) (Fig. 8A,B). As shown in the Fig. 8C, the OS of patients in high TMB was significantly better (P < 0.05). Then TIDE algorithm was used to further investigated patients sensitivity of immunetherapy response of two groups. From this study, lower dysfunction score in low risk group was significant (Fig. 8D). Furthermore, differential IC50 of anti-tumor drugs of different groups was analyzed. Among 64 drugs which had significant difference between two groups (Fig. 8E), 4 drugs (PAC.1, OSI.906, WH.4.023, BMS.536924) were selected for further research (Fig. 8F). The correlation analysis showed that PAC.1 and OSI.906 had negative correlation with riskscore. WH.4.023 and BMS.536924 had positive correlation with riskscore (Fig. 8G).

Analysis results of mutant genes expression. (A) Mutations of the top10 gene in the GBM high risk group. In the high-risk group, PTEN and TTN genes had mutation frequencies of 43% and 34%, respectively. (B) Mutations of the top10 gene in the GBM low risk group. In the low-risk group, PTEN and TTN genes had mutation frequencies of 39% and 27%, respectively. Highly mutated genes PTEN and TTN had higher mutation frequencies in the high-risk group compared to the low-risk group, while TP53 and EGFR had higher mutation frequencies in the low-risk group compared to the high-risk group, showing a correlation between risk score and gene mutation frequency. (C) KM analysis of different risk-mutation groups. The KM curve shows survival time and probability on the upper part, with total survival time on the x-axis and survival probability on the y-axis. The lower part shows the number of samples remaining in different groups at different survival times. C1. KM analysis found a significant correlation between TMB score and patient prognosis. C2. Survival rates differed significantly among the HighRisk_HTMB, HighRisk_LTMB, LowRisk_HTMB, and LowRisk_LTMB groups, with the HighRisk_LTMB group having the lowest survival rate. (D) Differences in TIDE, Dysfunction and Exclusion scores between high and low risk groups. (E) Variations in drug sensitivity between high and low risk groups. (F) By analyzing four drugs (PAC-1, OSI-906, WH-4.023, BMS-536924), genes associated with risk scores and drugs showed significant correlations above 0.3. (G) Heat map of the correlation between risk scores, prognostic genes, and drug response.
Regulatory network and ceRNA network analysis of prognosis genes
The lncRNA, miRNA and TFs were predicted to construct ceRNA and TF-mRNA regulatory networks respectively. For ceRNA regulatory network, 5 mRNAs (TRAM2, DDHD1, CD79B, FKBP18 and STXBP4), 24 miRNAs and 26 lncRNAs were included (Fig. 9A). Blue point indicated mRNA, orange square stood for miRNA and green circle meant lncRNA. For TF-mRNA, the network included 79 nodes which connected with 106 edges. CD79B, DDHD1, STXBP4 and TRAM2 were all regulated by AR (Fig. 9B).

Regulatory network and ceRNA network analysis of prognosis genes. (A) The ceRNA regulatory network was built using the relationships between lncRNA-miRNA-mRNA in the miRNet database. (B) TF-mRNA regulatory network. TRRUST database was utilized to predict regulatory biomarker TFs and identify TF-biomarker relationships.
3 prognosis genes were highly expressed in GBM
In this study, TRAM2, STXBP4 and CD79B had up-regulated expression, while the expression of FKBP1B and DDHD1 was down-regulated in GBM. The corresponding protein of prognosis genes (Supplementary Table 2) was analyzed. The results showed that corresponding protein of CD79B, STXBP4 and FKBP1B had varying degrees of staining intensity in Glioma (Fig. 10A–C). Strength of STXBP4 was the highest (Fig. 10D). The signal of CD79B and STXBP4 were detected in cell nucleus. However, the protein of DDHD1 and TRAM2 was not detected in Glioma (Fig. 10E,F).

Prognosis genes were highly expressed in GBM. (A) Expression of prognostic genes in training sets. (B) Immunohistochemical expression of CD79B corresponding protein. (C) Immunohistochemical expression of STXBP4 corresponding protein. (D) Immunohistochemical expression of DDHD1 corresponding protein. (E) Immunohistochemical expression of FKBP1B corresponding protein. (F) Immunohistochemical expression of TRAM2 corresponding protein. CD79B, STXBP4, and FKBP1B proteins show varying staining intensity in glioma samples, with STXBP4 having the highest intensity. CD79B and STXBP4 exhibit nuclear detection signals, while DDHD1 and TRAM2 show no protein level signal in glioma.
Validation of the prognosis gene expression level by IHC
In order to further verify the expression level of the prognosis gene (CD79B, STXBP4, DDHD1, FKBP1B and TRAM2), we used IHC to compare gene expression levels in the glioma tissue and adjacent tissue. The IHC results showed that the expression level of the biomarkers exhibited significant difference. The expression levels of 5 prognosis gene were significantly higher in glioma tissue than that in the superficial temporal adjacent tissue (Fig. 11).

The expression level of corresponding protein of prognostic gene was detected by IHC. The corresponding protein staining intensity of prognostic genes (CD79B, STXBP4, DDHD1, FKBP1B and TRAM2) in tumor tissues was significantly higher than that in normal tissues.
Discussion
Glioma, a prevalent and aggressive cancer of the central nervous system, presents limited therapeutic interventions. Acetylation has been shown to modulate tumor growth by influencing immune response and regulating cellular processes such as transcription and DNA repair. The process of lysine acetylation is known to play a pivotal role in the pathogenesis of glioma, impacting cellular function and facilitating tumor progression25. Lysine acetylation affects glioma cell growth and differentiation by controlling important genes and proteins. Dysregulation can cause unchecked cell proliferation and tumor development. It also influences the invasion and spread of glioma cells by regulating genes and proteins related to migration and adhesion, potentially increasing their ability to metastasize in the brain. Higher levels of lysine acetylation in certain genes or proteins are associated with more severe forms of glioma. The complex impact of lysine acetylation on glioma involves the intricate interplay of numerous genes, proteins, and signaling pathways. Consequently, further investigation into the precise mechanisms underlying lysine acetylation in glioma is imperative to elucidate its contributions to the initiation, progression, and malignancy of glioma, and to propose novel therapeutic approaches. This study used bioinformatics analysis and IHC verification to investigate the key genes associated with glioma and lysine acetylation.
The differential genes between glioma and normal samples were intersected with genes associated with lysine acetylation in glioma, followed by GO and KEGG enrichment analyses on the identified genes. The results of the enrichment analyses revealed that the intersecting genes were involved in biological processes such as axon development, gland development, epithelial tube morphogenesis, regulation of supramolecular fiber organization, and axonogenesis. The pathways under consideration exhibit a specific association with glioma and lysine acetylation. Glioma, a malignant tumor, is linked to biological processes including axon development, cell leading edge formation, and the presence of a collagen-containing extracellular matrix. The cellular components predominantly influenced by the intersecting genes include the cell leading edge, collagen-containing extracellular matrix, nuclear speck, extrinsic component of the membrane, and postsynaptic specialization. The predominant molecular functions exhibited by the intersecting genes include GTPase regulator activity, nucleoside-triphosphatase regulator activity, ubiquitin-like protein transferase activity, ubiquitin-protein transferase activity, and molecular adaptor activity. These pathways are potentially implicated in the pathogenesis and progression of glioma. Furthermore, lysine acetylation—a key post-translational modification—exerted regulatory effects on glioma progression through pathways such as molecular adaptor activity and ubiquitin-mediated protein degradation, which were directly associated with its dynamic modification of target proteins26.
Through the utilization of various statistical methods including univariate COX regression, LASSO regression, multivariate COX regression, and stepwise regression, a study identified five genes (CD79B, STXBP4, DDHD1, FKBP1B, and TRAM2) that are significantly correlated with survival outcomes in glioma patients. Specifically, CD79B, a key component of the B-cell receptor complex, is suggested to potentially impact immune system functionality27. Alterations in CD79B expression within glioma tumors may consequently influence the dynamic interplay between the tumor and the immune system, ultimately shaping tumor progression and patient prognosis. STXBP4, a constituent of the synaptotagmin-binding protein family, plays a role in neurotransmitter release. In the context of glioma, it may contribute to aberrant neural cell signaling, thereby impacting tumor proliferation and infiltration28. The gene DDHD1 is implicated in lipid metabolism and its role may be linked to the maintenance of cell membrane integrity and signal transduction. Alterations in DDHD1 within glioma cells could impact tumor cell metabolic pathways, thereby influencing their proliferation and viability29. FKBP1B, a member of the FK506-binding protein family, is commonly associated with immune modulation and cellular stress responses. In the context of glioma, FKBP1B may play a role in modulating the stress response of tumor cells or affecting their responsiveness to therapeutic interventions30. TRAM2 is recognized to play a role in immune system functions. In the context of glioma, TRAM2 may modulate immune evasion strategies employed by tumor cells or impact the interaction between immune cells and tumor cells. Research has demonstrated that TRAM2 enhances the cancerous advancement of glioma via the PI3K/AKT/mTOR pathway31. The findings from the Kaplan–Meier and receiver operating characteristic analyses were statistically significant and indicated robust predictive capabilities. Furthermore, the validation results from an external dataset were deemed satisfactory, underscoring the prognostic relevance of the identified five genes in glioma. The study utilized univariate COX regression analysis to determine the association between risk score and survival prognosis in patients, leading to the development of a prognostic correlation model. Subsequent Kaplan–Meier (KM) and receiver operating characteristic (ROC) tests were conducted, followed by validation through a calibrated model.
To elucidate the mechanisms contributing to survival disparities, a functional enrichment analysis was conducted on cohorts categorized as high and low risk. This analysis elucidated the molecular pathways impacted, offering insight into potential therapeutic targets and bolstering the rationale for drug development. The findings revealed disparities in molecular pathways between high and low risk groups, including glycolipid biosynthesis ganglion series, cytokines interacting with cytokine receptors, ubiquitin-mediated protein degradation, propionic acid metabolism, and lysine degradation. Researchers have recently studied the role of various biological processes in glioma, including glucolipid biosynthesis, cytokine and receptor interaction, protein degradation, propionic acid metabolism, and lysine degradation. Glycolipid is crucial for cell membrane function and plays a key role in glioma cell growth, differentiation, and signaling. Abnormal activation of glycolipid synthesis in glioma cells is linked to tumor progression, invasion, and blood vessel formation32. Cytokine and receptor interactions are important in gliomas, impacting tumor growth, invasion, and immune escape. Understanding these mechanisms can improve glioma treatment strategies33. The ubiquitin mediated protein degradation pathway is crucial in glioma, as abnormalities can cause tumor cell malignancy and drug resistance34. Propionic acid metabolism is disrupted in glioma, impacting energy supply and tumor cell growth and invasion35. Lysine degradation is disrupted in gliomas, potentially causing metabolic disorders and promoting tumor growth, ultimately advancing the disease36. Overall, glioma development is linked to glucolipid biosynthesis, cytokine interactions, protein degradation, propionic acid metabolism, lysine degradation, and other processes.
Studying the tumor microenvironment is crucial for understanding tumor development, immune escape, drug resistance, prognosis, and treatment strategies. It can provide insights into the pathological mechanisms of tumors and offer new approaches for treatment and prognosis assessment. Different algorithms were used to analyze the tumor microenvironment, including ESTIMATE, immune cell differences, immune functional pathways, TIDE and other aspects, and obtained significant results. There were significant differences in 23 immune cells. By constructing the correlation of prognostic genes, risk scores and different immune cells among sample groups, the results showed that risk scores and different immune cells were positively correlated and significant. Immune cells are integral to the pathogenesis, progression, and therapeutic interventions of glioma. The findings of this study demonstrate the involvement of specific immune cell types in the lysine acetylation process within glioma. Activated B cells, known for their role in antibody production and humoral immunity, have been identified as potential contributors to anti-tumor immune responses in gliomas by generating antibodies against tumor-associated antigens37. Nevertheless, gliomas may employ mechanisms to evade immune surveillance by impairing the functionality of activated B cells. Activated CD8 T cells can directly kill tumor cells, including glioma cells, but gliomas can evade immune attack by downregulating immune checkpoints or releasing immunosuppressive factors38. Activated dendritic cells play a crucial role in initiating anti-tumor immune responses in gliomas by presenting tumor antigens to T cells. CD56dim NK cell: An important immune cell that can directly kill tumor and virus-infected cells, potentially playing a role in anti-tumor immune response in glioma39. Central memory CD4 and CD8 T cells have long-lasting memory capabilities, quickly activating and mounting a robust immune response upon re-encountering antigens40. In gliomas, they may play a role in ongoing immune surveillance and targeting of glioma cells. Effector memory T cells can quickly kill tumor cells or release cytokines to regulate the immune response in gliomas41. Macrophages play a dual role in the glioma microenvironment by removing tumor debris and potentially aiding in immune evasion42.
Studying the connection between immune checkpoints and gliomas is a complex and evolving field. Some immune checkpoint molecules have been well-studied, while others are still being explored. Upregulated immune checkpoints like CD70 in gliomas may enhance T cell and NK cell activation, potentially boosting immune response against tumors43. Additionally, elevated levels of CD44 in gliomas have been linked to increased aggressiveness and migration, making it a potential therapeutic target44. Further research is needed to fully understand their roles in glioma development. Upregulation of PD-1 in gliomas inhibits T cell activation and cytotoxic effects, contributing to immune escape. Targeting PD-1 with immunotherapy may be effective in treating glioma. PD-L1 expression in gliomas is linked to immune escape. Blocking PD-L1 and PD-1 interaction can restore T cell antitumor activity, offering a new treatment approach for glioma45. Down-regulation of TNFRSF25 and CD244 in gliomas may impact immune response and tumor progression by affecting TNF signaling pathways and T cell activation, respectively46,47.
Chemokines are significant contributors to the pathogenesis, progression, and therapeutic strategies of glioma. Through their ability to orchestrate the migration of immune and non-immune cells to the tumor microenvironment, chemokines exert a profound influence on the immune response within the glioma. The findings of this study underscore the pivotal role of chemokines in glioma biology. Chemokines in glioma: CXCL13 promotes B cell migration, CXCL10 is elevated in higher grade tumors and associated with poor prognosis, and CXCL9 attracts T cells and natural killer cells to enhance anti-tumor immune response48. In gliomas, up-regulation of CXCL9 activates the immune system to fight the tumor, while up-regulation of chemokines like CCL23, CCL24, and CCL20 recruits tumor-associated macrophages and enhances tumor angiogenesis and aggressiveness. Up-regulation of receptors like CXCR2, CCR2, and CXCR4 increases sensitivity to chemokines and promotes cell migration and invasion in glioma cells. Reduced levels of chemokines like CX3CL1 in glioma can hinder immune cell interactions, impacting immune surveillance and anti-tumor response. Downregulation of CCL28, CCR9, and CCL1 may also decrease the recruitment of specific immune cells to glioma cells, affecting the immune balance in the tumor microenvironment. The relationship between chemokines and glioma was complex and multifaceted, with different chemokines having unique mechanisms and potential interactions49. Studying the relationship between chemokines and glioma can help us understand the disease better and find new treatment targets. Future research should focus on how chemokines affect the progression and prognosis of glioma.
To investigate variances in immunochemotherapy drugs among high and low-risk groups and offer a basis for experimental research, we utilized the R package “pRRophetic” to identify the four drugs with the most significant distinctions between the aforementioned groups: PAC.1, OSI.906, WH.4.023, and BMS.536924. PAC-1 targets the PC-3 protein, which is abundant in cancer cells and plays a key role in apoptosis signaling. Clinical trials have shown PAC-1 to have anti-cancer effects on various types of cancer and to have synergies with approved anticancer drugs50. OSI.906 (Linsitinib) is a selective inhibitor of IGF-1R and dual inhibitor of IGF-1 and insulin receptor with oral activity. It inhibits the proliferation of various tumor cell lines, including non-small cell lung cancer and colorectal cancer cells51. WH.4.023 is a selective inhibitor of Lck and Src kinases with strong inhibitory activity52. BMS-536924 is a selective inhibitor of IGF-1R and IR kinases. BMS-536924 is a potent inhibitor of IGF-1R and IR with an IC50 value of 100 nM and 73 nM, respectively. It also shows moderate inhibitory activity against Mek, Fak, and Lck, but minimal activity against Akt1 and MAPK1/253. This compound has anticancer properties.
Glioma is a complex brain tumor that involves multiple molecular networks in its malignant progression. Studies have shown that miRNA and lncRNA play a crucial role in the growth and spread of glioma cells. Specifically, TRAM2 promotes glioma progression through the PI3K/AKT/mTOR pathway31, but more research is needed on the interaction between specific miRNAs, lncRNAs, and TRAM2 or its downstream genes. By utilizing bioinformatics tools such as miRNet and TRRUST databases, we predict the miRNAs, lncRNAs, and transcription factors (TFs) that interact with glioma-associated genes and construct interaction networks of these regulatory elements. These predictions provide important clues to reveal the molecular regulatory mechanisms of gliomas and may provide new targets for future diagnostic and therapeutic strategies. However, these predictions still need to be validated experimentally to confirm their accuracy and biological significance. Future studies will focus on validating the specific roles of these miRNAs, IncRNAs, and TFs in gliomas and exploring how their interactions affect glioma progression. These studies are expected to provide new ideas and methods for the diagnosis and treatment of glioma, and bring better treatment results and quality of life for patients.
In summary, this study comprehensively evaluated the expression levels and prognostic value of genes associated with lysine acetylation score in gliomas through bioinformatics analysis. We have successfully identified potential biomarkers and therapeutic targets, providing a new reference and basis for the prevention, diagnosis and treatment of glioma patients. However, we are also aware that there are still some limitations in this study, and further exploration of the function and regulatory mechanism of these genes is still needed in vitro experiments and clinical drug trials, in order to provide more accurate and effective strategies for the treatment and prognosis assessment of glioma.
Materials and methods
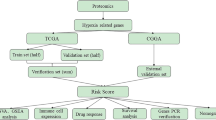
Samples were divided into disease and control groups based on TCGA cohort data. Differential expression analysis and WGCNA were sequentially performed to identify key modules and genes. Lysine acetylation-related differential genes were obtained, and GO and KEGG analyses were conducted. A PPI network was constructed, and univariate Cox analysis was performed. The Lasso regression was used to build a risk model, which was then validated to obtain prognostic genes. A series of analyses on key genes were conducted. The specific research process is shown in Fig. 12.

Schematic workflow of the study. IHC: Immunohistochemistry.
Data sources
TCGA-GBM dataset was downloaded as training set from University of California Santa Cruz (UCSC) database (https://xenabrowser.net/datapages/), involved 168 GBM and 5 control samples. Glioma related dataset was obtained from Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/) as validation set. A total of 137 GBM patients of gene expression profiles and clinical survival information were included. Lysine acetylation-related genes (LARGs) was acquired from published literature54 (Supplementary Table 1).
Differentially expressed analysis
The differentially expresssed grenes (DEGs) between GBM and control samples were analyzed through DESeq2 package (version 1.34.055) in TCGA-GBM. The screening criterias were p.adj < 0.05 and |log2FoldChangeFC|> 1. Then top 10 DEGs were displayed in volcanic map and heatmap through ggplot2 (version 3.3.556).
Weighted gene co-expression network analysis (WGCNA)
Initially, a hierarchical clustering tree is constructed to screen and identify outlier samples, ensuring the quality and consistency of the data. In the context of The Cancer Genome Atlas Glioblastoma Multiforme (TCGA-GBM) dataset, single-sample Gene Set Enrichment Analysis (ssGSEA) scores for Large Abundance of Gene Sets (LARGs) are calculated, serving as the traits for analysis. Subsequently, the Weighted Gene Co-expression Network Analysis is conducted on all the samples using the WGCNA package (version 1.70-357). The selection of the soft threshold is crucial as it ensures that the gene interactions within the network are maximally consistent with scale-free network properties, which is a fundamental assumption of the WGCNA methodology. Following the construction of the gene co-expression network, Pearson correlation analysis is performed to explore the relationship between the ssGSEA scores and each of the identified modules. Genes that are found within modules that are significantly correlated with the LARGs traits are then considered as LARGs-related module genes, providing insights into the functional gene sets associated with the disease or biological process of interest.
Fnrichment analysis of intersection genes
Differentially expressed-LARGs (DE-LARGs) was obtained through taking the intersection of DEGs and WGCNA module genes. Next, DE-LARGs were processed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis to find out relative functions and pathways by clusterProfiler package (version 4.2.258). Interaction of DE-LARGs on protein level was predicted by STRING database (https://cn.string-db.org/, confidence = 0.3). After removing genes with no interaction, the protein–protein interaction (PPI) network was visualized using Cytoscape (v 3.7.159) software. Additionally, the cytoHubba plugin for Cytoscape (v 3.7.1) was employed to perform key topological analysis, including degree centrality, betweenness centrality, closeness centrality, maximum clique centrality (MCC), and density of maximal neighborhood component (DMNC), to evaluate the importance of nodes in the network and identify key genes and subnetworks.
Construction and verification of risk model
Patients (overall survival) OS-associated genes from DE-LARGs were identified. Univariate Cox regression analysis was performed to find out hub genes in training set. Then LASSO regression analysis was to obtain crucial genes. After that the results of PH hypothesis test were used to construct multivariate Cox regression model. Finally, multiple stepwise regression analysis was carried out to find prognosis genes for forming risk model. Riskscore for each GBM patients was calculated by the formula: RiskScore =\(\sum_{i=1}^{n}({\text{Exp}}_{i}\times {\text{Coe}}_{i})\) (Expi indicated the expression level for each prognosis genes, and \({\text{Coe}}_{i}\) indicated the corresponding Cox regression coefficient). Optimal cutoff was calculated by prognosis genes in training set. GBM patients were separated into high and low risk groups based on optimal cutoff. The accuracy of the model was appraised by Kaplan–Meier (KM) survival analysis and receiver operating characteristic (ROC) curve. Finally, risk model verified in validation set by KM survival analysis and ROC curve.
Construction of predictive nomogram
Firstly, univariate Cox regression analysis of riskscore, age and gender was conducted to screen independent prognosis factors. Secondly, the nomogram was built based on prognosis genes. OS of GBM patients were predicted at 1, 2 and 3 years. Finally, accuracy was evaluated by ROC and Decision Curve Analysis (DCA) respectively.
Gene set variation analysis (GSVA) enrichment analysis
The c2.cp.kegg.v7.1.symbols.gmt dataset was downloaded from Molecular Signatures Database (MSigDB) as background genes. GSVA score of pathway in TGCA-GBM was calculated. Difference of biological functions from different groups were compared based on the GSVA score.
Estimation of tumor microenvironment (TME)
Stromal score and immune score were calculated by Single-sample gene set enrichment analysis (ssGSEA) respectively. Then these two scores were combined to predict the purity of tumor. Based on the RNA-seq of TCGA, Estimation of stromal and immune cells in malignant tumour tissues using expression data (ESTIMATE) algorithm was processed by IOBR package (version 0.99.960) to evaluate TME score in GBM. TME score was divided into two groups relied on immune score. Non-parametric tests were used to find out whether statistical difference was existed among these scores from different groups.
Immune function and tumor immune dysfunction and exclusion (TIDE)
Expression features among 28 immune cell types was calculated through ssGSEA algorithm by GSVA package (version 1.42.061). Differentially infiltrated level of immune cell was analyzed by Wilcoxon test. Relation of riskscore and prognosis genes with immune cells was calculated and analyzed.
Immune function pathways including checkpoint, parainflammation etc. Enrichment score of immune function pathway from different groups was evaluated by ssGSEA. Difference was then analyzed by wilcox test in training cohort.
TIDE score was calculated in training set. Wilcox test counted the difference of TIDE score between different groups. In order to evaluate potential influence of riskscore in immunetherapy response, immune checkpoints and chemokines were analyzed from different groups in training set.
The landscape of mutant
Somatic mutation data acquired from TCGA, and then analyzed by maftools package (version 2.6.0562). In TCGA-GBM, tumor mutation burden (TMB) score of each patient was calculated. GBM patients were divided into high and low TMB groups by median of TMB score. Then survival of GMB patients was analyzed by KM survival curve. Two different groups were combined. OS of GMB among HighRisk HTMB, HighRisk LTMB, LowRisk HTMB and LowRisk LTMB was analyzed.
Immunochemotherapeutic drugs analysis
The inhibitory concentration 50 (IC50) of 138 drugs in each tumor sample of TCGA-GBM in different groups was examined by “pRRophetic” package (version 0.563). The difference of IC50 between two groups was analyzed by Wilcoxon test. Finally, the correlation of riskscore and prognosis genes with drugs was analyzed.
Construction of regulatory network
The lncRNAs and miRNAs which regulated mRNAs was predicted in miRNet database. Then ceRNA regulatory network was built. Furthermore, this study used TRRUST database (http://www.grnpedia.org/trrust) to search transcription factors (TFs) which regulated mRNAs. Corresponding TF-mRNA interaction was obtained.
Expression analysis of prognosis genes
To identify the expression of prognosis genes between GBM and control samples in training set, differential expressed analysis was used. Moreover, corresponding protein of prognosis genes was retrieved by human protein atlas (HPA). Expression, location, intensity, distribution of protein in GBM tissues and cells were recognized.
Validation of the gene expression level by IHC
The samples from 10 glioma tissue and adjacent tissue were used for IHC to verify the expression level of prognosis genes. The written informed consent was obtained from all participants prior to enrollment into the study. And study protocols were approved by the Ethics Committee of the University of South China, based on the ethical principles for medical research involving human subjects of the Helsinki Declaration. All procedures complied with the Declaration of Helsinki. Experimental procedure: The tissues were embedded in paraffin wax and sectioned. Primary antibody incubation: Tissue sheet baked at 64℃ for 1 h, followed by dewaxing in xylene and hydration in alcohol series. Rinse with PBS three times for 5 min each. Antigen repair: Boil citric acid buffer in pressure cooker, add slide, cook for 3 min, cool to room temperature, rinse with PBS 3 times for 5 min each. Blocking: Incubate with 3% H2O2 for 20 min at room temperature to deactivate peroxidase, rinse with PBS 3 times for 5 min each. Incubate 5% bovine serum albumin V at 37℃ for 30 min. Dilute primary antibody with 2% bovine serum albumin V according to instructions, add drops to slide, and refrigerate at 4℃ overnight. Secondary antibody incubation: The slides were rewarmed at 37℃ for 30 min, washed with PBS, and then treated with reaction enhancement solution and enzyme-labeled sheep anti-mouse/rabbit IgG polymer. Each step included 3 washes with PBS for 5 min each. DAB color development involves adding DAB dye drops to tissue blocks for staining and observing under a microscope. The dye solution is rinsed with PBS three times for 5 min each. Hematoxylin restaining includes dyeing the slide in hematoxylin for 5 min, rinsing with distilled water, differentiating in alcohol hydrochloric acid solution for 10–15 s, and returning to blue in tap water for at least 15 min. Dehydration process includes steps of 70% alcohol for 2 min, 80% alcohol for 3 min, 95% alcohol , 100% alcohol I for 5 min, and finally 100% alcohol II. Transparent: xylene I and xylene II for 10 min each, seal with neutral gum, then scan.
Statistical analysis
The R software was used to process and analyze the data. The P value < 0.05 was considered statistically significant.
Data availability
The datasets generated and analyzed during the current study are available from the first author, Min Zhou. Please contact the first author at 2015020049@usc.edu.cn for data access.
References
Scott, A. J. et al. Metabolomic profiles of human glioma inform patient survival. Antioxid. Redox Signal. 39(13–15), 942–956 (2023).
Kadiyala, P. et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Invest. 131(4), e139542 (2021).
Taher, M. M. et al. EGFRvIII expression and isocitrate dehydrogenase mutations in patients with glioma. Oncol. Lett. 20(6), 384 (2020).
Pratt, D. et al. High-grade glioma with pleomorphic and pseudopapillary features (HPAP): A proposed type of circumscribed glioma in adults harboring frequent TP53 mutations and recurrent monosomy 13. Acta Neuropathol. 143(3), 403–414 (2022).
Shaw, R. et al. MGMT in TMZ-based glioma therapy: Multifaceted insights and clinical trial perspectives. Biochim. Biophys. Acta Mol. Cell Res. 1871(3), 119673 (2024).
Venneti, S. et al. Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discov. 13(11), 2370–2393 (2023).
Kashino, G. et al. VEGF affects mitochondrial ROS generation in glioma cells and acts as a radioresistance factor. Radiat. Environ. Biophys. 62(2), 213–220. https://doi.org/10.1007/s00411-023-01021-8 (2023).
Brown, C. E. et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: A phase 1 trial. Nat. Med. 30(4), 1001–1012 (2024).
Aili, Y. et al. The role of exosomal miRNAs in glioma: Biological function and clinical application. Front. Oncol. 11, 686369 (2021).
Liu, L. et al. LncRNA INHEG promotes glioma stem cell maintenance and tumorigenicity through regulating rRNA 2’-O-methylation. Nat. Commun. 14(1), 7526 (2023).
Kang, X. et al. Progresses, challenges, and prospects of CRISPR/Cas9 gene-editing in glioma studies. Cancers (Basel) 15(2), 396 (2023).
Fauvet, C., Villain, M. & Gatignol, P. Repeated awake surgery and quality of life in patients with diffuse glioma: a systematic review and meta-analysis. Neurosurg. Rev. 46(1), 156 (2023).
Wu, Y. et al. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 22(1), 96 (2023).
Perrone, M. G. et al. Diffuse intrinsic pontine glioma (DIPG): Breakthrough and clinical perspective. Curr. Med. Chem. 28(17), 3287–3317 (2021).
Śledzińska, P. et al. Current and promising treatment strategies in glioma. Rev. Neurosci. 34(5), 483–516 (2022).
Kikuchi, M. et al. GAS41 promotes H2A.Z deposition through recognition of the N terminus of histone H3 by the YEATS domain. Proc. Natl. Acad. Sci. USA 120(43), 3120 (2023).
Pienkowski, T. et al. Glioma and post-translational modifications: A complex relationship. Biochim. Biophys. Acta Rev. Cancer 1878(6), 189009 (2023).
Yu, N. et al. H3G34-mutant diffuse hemispheric glioma with osseous metastases: a case report and literature review. CNS Oncol. 12(2), CNS95 (2023).
Yang, J., Qiu, Q. & Chen, L. Histone lysine-to-methionine mutation as anticancer drug target. Adv. Exp. Med. Biol. 1283, 85–96 (2021).
Saratsis, A. M. et al. H3K27M mutant glioma: Disease definition and biological underpinnings. Neuro Oncol. 26(Supplement_2), S92–S100 (2024).
Panditharatna, E. et al. BAF complex maintains glioma stem cells in pediatric H3K27M glioma. Cancer Discov. 12(12), 2880–2905 (2022).
Trejo-Solís, C. et al. Metabolic roles of HIF1, c-Myc, and p53 in glioma cells. Metabolites 14(5), 249 (2024).
Chen, Y. et al. Tumor-associated monocytes promote mesenchymal transformation through EGFR signaling in glioma. Cell Rep. Med. 4(9), 101177 (2023).
Noll, A. et al. Therapeutic HDAC inhibition in hypermutant diffuse intrinsic pontine glioma. Neoplasia 43, 100921 (2023).
Fernández-Coto, D. L. et al. Dynamics of mitochondrial proteome and acetylome in glioblastoma cells with contrasting metabolic phenotypes. Int. J. Mol. Sci. 25(6), 3450 (2024).
Gao, Y. et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 22(9), 1064–1075 (2020).
Tkachenko, A., Kupcova, K. & Havranek, O. B-cell receptor signaling and beyond: The role of Igα (CD79a)/Igβ (CD79b) in normal and malignant B cells. Int. J. Mol. Sci. 25(1), 10 (2023).
Vargas, R. E. et al. Elucidation of WW domain ligand binding specificities in the Hippo pathway reveals STXBP4 as YAP inhibitor. EMBO J. 39(1), e102406 (2020).
Kitamura, C. et al. Expression of lysophosphatidylinositol signaling-relevant molecules in colorectal cancer. Anticancer Res. 41(5), 2349–2355 (2021).
Niu, H. et al. The significance of the redox gene in the prognosis and therapeutic response of glioma. Am. J. Clin. Oncol. 47, 259–270 (2024).
Gao, X. et al. TRAM2 promotes the malignant progression of glioma through PI3K/AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 586, 34–41 (2022).
Dong, W. et al. Glioma glycolipid metabolism: MSI2-SNORD12B-FIP1L1-ZBTB4 feedback loop as a potential treatment target. Clin. Transl. Med. 11(5), e411 (2021).
Barthel, L. et al. Glioma: Molecular signature and crossroads with tumor microenvironment. Cancer Metastasis Rev. 41(1), 53–75 (2022).
Hu, Y. et al. S100 Calcium Binding Protein A16 Promotes Cell Proliferation by triggering LATS1 ubiquitin degradation mediated by CUL4A ligase to inhibit Hippo pathway in Glioma development. Int. J. Biol. Sci. 19(7), 2034–2052 (2023).
Huang, R. et al. Collagen Type III Alpha 1 chain regulated by GATA-Binding Protein 6 affects Type II IFN response and propanoate metabolism in the recurrence of lower grade glioma. J. Cell Mol. Med. 24(18), 10803–10815 (2020).
Li, Y. et al. Lysine methylation promotes NFAT5 activation and determines temozolomide efficacy in glioblastoma. Nat. Commun. 14(1), 4062 (2023).
Lee-Chang, C. et al. Activation of 4–1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J. Exp. Med. 218(1), e20200913 (2021).
Naulaerts, S. et al. Multiomics and spatial mapping characterizes human CD8+ T cell states in cancer. Sci. Transl. Med. 15(691), 1016 (2023).
Pellegatta, S. et al. ABCC3 expressed by CD56dim CD16+ NK cells predicts response in glioblastoma patients treated with combined chemotherapy and dendritic cell immunotherapy. Int. J. Mol. Sci. 20(23), 5886 (2019).
Campian, J. L. et al. Long-acting recombinant human interleukin-7, NT-I7, increases cytotoxic CD8 T cells and enhances survival in mouse glioma models. Clin. Cancer Res. 28(6), 1229–1239 (2022).
Zhao, Q. et al. FGL2-targeting T cells exhibit antitumor effects on glioblastoma and recruit tumor-specific brain-resident memory T cells. Nat. Commun. 14(1), 735 (2023).
Khan, F. et al. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Invest. 133(1), e163446 (2023).
Seyfrid, M. et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. J. Immunother. Cancer 10(1), e003289 (2022).
Du, Z. et al. Association of glioma CD44 expression with glial dynamics in the tumour microenvironment and patient prognosis. Comput. Struct. Biotechnol. J. 20, 5203–5217 (2022).
Yan, T., Kong, Z. & Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: clinical studies, challenges and potential. Hum. Vaccin Immunother. 17(2), 546–553 (2021).
Marques, R. F. et al. Digital expression profile of immune checkpoint genes in medulloblastomas identifies CD24 and CD276 as putative immunotherapy targets. Front Immunol. 14, 1062856 (2023).
So, T. & Ishii, N. The TNF-TNFR family of co-signal molecules. Adv. Exp. Med. Biol. 1189, 53–84 (2019).
Lin, Y. T. et al. CXCL13 is a biomarker of anti-leucine-rich glioma-inactivated protein 1 encephalitis patients. Neuropsychiatr. Dis. Treat. 15, 2909–2915 (2019).
Qiao, S. et al. Cytokines/chemokines and immune checkpoint molecules in anti-leucine-rich glioma-inactivated 1 encephalitis. Neurol. Sci. 44(3), 1017–1029 (2023).
Schlein, L. J. et al. Immunohistochemical characterization of procaspase-3 overexpression as a druggable target with PAC-1, a procaspase-3 activator, in canine and human brain cancers. Front Oncol. 9, 96 (2019).
Quail, D. F. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352(6288), aad3018 (2016).
Kafita, D. et al. High ELF4 expression in human cancers is associated with worse disease outcomes and increased resistance to anticancer drugs. PLoS ONE 16(4), e0248984 (2021).
Zhou, Q. BMS-536924, an ATP-competitive IGF-1R/IR inhibitor, decreases viability and migration of temozolomide-resistant glioma cells in vitro and suppresses tumor growth in vivo. Onco Targets Ther. 8, 689–697 (2015).
Deng, S. Z. et al. Integrative analysis of lysine acetylation-related genes and identification of a novel prognostic model for oral squamous cell carcinoma. Front Mol. Biosci. 10, 1185832 (2023).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 550 (2014).
Gustavsson, E. K. et al. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 38(15), 3844–3846 (2022).
Langfelder, P. & Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46(11), i11 (2012).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2(3), 100141 (2021).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Zeng, D. et al. IOBR: Multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front Immunol. 12, 687975 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Wang, Y. et al. Potential immune biomarker candidates and immune subtypes of lung adenocarcinoma for developing mRNA vaccines. Front Immunol. 12, 755401 (2021).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9), e107468 (2014).
Funding
This work was supported by grants from the Key Project of Hunan Provincial Health Commission of China (20201912) and the Clinical Innovation Guidance Program project of Hunan Provincial Science and Technology Department (2021SK517714).
Author information
Authors and Affiliations
Contributions
Min Zhou completed the analysis and experiment,wrote the main manuscript text and prepared figures. Bing Wang and Richu Liang guided part of the experimental ideas and support. Xinping Luan guided the experiment and the financial support throughout the whole process. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Informed consent was obtained by the patient and his family in the case, and the protocol was approved by the ethical committee University of South China (No. 2023767). All procedures complied with the Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, M., Wang, B., Liang, R. et al. Role of lysine acetylation-related genes in the diagnosis and prognosis of glioma. Sci Rep 15, 20762 (2025). https://doi.org/10.1038/s41598-025-07738-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07738-4
