
Abstract
Acute rejection (AR) after kidney transplantation, is a common and serious complication that occurs when the recipient’s immune system attacks the graft, and the specific genes and molecular mechanisms underlying the role of mitophagy are still unclear. This study integrated two transcriptomic datasets (GSE129166 and GSE25902) from the GEO database. Thirty differential mitophagy-related genes were identified by intersecting differentially expressed genes, module genes obtained through weighted gene co-expression network analysis and mitophagy-related genes. Functional enrichment analysis uncovered several biological processes and signaling pathways associated with these genes. Four candidate genes including CCND1, ZC3H15, RPL38, and ARPC4, were further identified through Random Forest and Support Vector Machine with recursive feature elimination. Internal, external datasets and a nomogram confirmed they could effectively predict AR. Moreover, these genes significantly correlated with the infiltration of multiple immune cells. Differential expressions of the four genes were also validated in patient’s peripheral blood and AR mice. These four mitophagy-related genes may be novel biomarkers for predicting the occurrence and diagnosis of AR.
Similar content being viewed by others
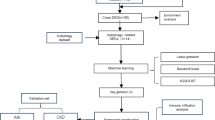


Introduction
End-stage renal disease (ESRD) remains an important global challenge, which is mainly treated with kidney transplantation as the most effective treatment. Since Dr. Murray successfully performed the world’s first kidney transplantation in 1954, the field has witnessed significant progress over the past 70 years, giving millions of ESRD patients a second chance at life. The advances in kidney transplant surgery, widespread use of tissue typing, perioperative antibody induction therapy, and the development of new immunosuppressants have led to a steady decline in acute rejection (AR) rates. However, AR remains the primary threat to long-term graft survival and the leading independent risk factor influencing graft outcomes1. If detected and treated early, AR is often reversible2highlighting the need to investigate the biomarkers and molecular mechanisms driving AR occurrence and development to facilitate early prediction and prevention.
Mitophagy, a selective autophagic process, plays a crucial role in the elimination of dysfunctional mitochondria, sustenance of mitochondrial integrity, and cellular homeostasis3. It mainly recognizes damaged mitochondria through the PINK1/Parkin signaling pathway, then wraps them into autophagosomes, which fuse with lysosomes to degrade them4. This process balances mitochondrial quality and quantity, and ensures cell survival under stress. It is vital for energy metabolism, inflammation, oxidative stress, and cell death5. Mitophagy is regulated by various proteins and is induced by starvation, oxidative stress, hypoxia, and depolarization6. Research has uncovered a potential link between deregulated mitochondria function and mitophagy impairment with the occurrence of diseases such as cancer7cardiovascular disorders8liver diseases9neurological conditions10and metabolic disorders11. In cancer cells, mitophagy maintains energy balance and mitochondrial mass under hypoxic conditions, enhancing their survival in low-oxygen environments. Autophagy also prevents apoptosis by eliminating dysfunctional mitochondria, which can contribute to chemotherapy resistance. Therefore, strategies aimed at modulating mitophagy may be promising therapeutic approaches for improving cancer treatment outcomes7. Studies have also demonstrated that mitochondrial dysfunction characterized by excessive ROS production, altered dynamics, and impaired lysosomal function occurs early in Alzheimer’s disease, accompanied by a significant decrease in mitophagy12. Additionally, Oka et al. noted that the failure to eliminate damaged mitochondrial DNA via mitophagy would trigger Toll-like receptor-mediated inflammation in cardiomyocytes, potentially leading to myocarditis and dilated cardiomyopathy13. Being high-energy-consuming organs, the kidneys have numerous mitochondria to supply their energy demands. Mitophagy participates in chronic kidney disease advancement and kidney repair following acute injury14. However, the specific genes and molecular mechanisms related to mitophagy in AR after kidney transplantation are not well understood, warranting further investigation.
In this study, we identified mitophagy-related genes involved in AR and uncovered candidate genes using two machine learning algorithms. Their prediction potential and diagnostic value in AR were validated using the patient’s peripheral blood and mice models, presenting an improved strategy for predicting and diagnosing AR after kidney transplantation.
Materials and methods
Data collection
Four datasets GSE129166, GSE25902, GSE14328, and GSE1563 with the gene expression profiles of AR (kidney biopsy samples) were retrieved from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). Detailed information of the datasets is presented in Table 1. GSE129166 and GSE25902 were combined to establish the merged dataset; GSE14328 and GSE1563 were selected as the external validation datasets. Mitophagy-associated genes were derived from the GeneCards database (https://www.genecards.org/) using the keyword “mitophagy”.
Differentially expressed genes (DEGs) screening
Data acquisition and probe annotation were downloaded using the “GEOquery” package in R. Two datasets, GSE129166 and GSE25902, were merged using “sva” package, which was utilized to clear batch effects and normalize data among samples. The DEGs between normal and AR specimens were processed with the “limma” package. The screening for the DEGs was conducted using the |log2 fold change (FC)| > 0.5 with P-adjusted < 0.05 (log FC > 0.5, P-adjusted < 0.05 were considered upregulated, whereas those that met the threshold of log FC < -0.5, P-adjusted < 0.05 were regarded as downregulated DEGs). Finally, the “ggplot2” package was employed to construct a volcano plot for the DEGs.
Weighted gene Co-expression network analysis (WGCNA)
In systems biology, WGCNA is applied to reveal modules of closely related genes in gene expression datasets and associate these modules with external data features. WGCNA is widely used for discovering molecular mechanisms, potential therapeutic targets or biomarkers. In our study, the “WGCNA” package was applied to uncover important modules in AR15. Specifically, genes with variance in the top 50% of the merged gene expression matrix were used to construct WGCNA. Sample data preprocessing and sample clustering tree construction were performed to remove outliers. Subsequently, a topological overlap matrix (TOM) was established utilizing the soft threshold (β). Next, we developed a weighted co-expression network model for classifying genes exhibiting consistent expression profiles into gene modules, each with at least 50 genes. Modules that showed a strong correlation with traits were considered the key modules for downstream analyses.
Subsequently, the obtained DEGs, module genes of WGCNA, and mitophagy-related genes were intersected to obtain differential mitophagy-related genes (DMRGs) using the “VennDiagram” package.
Screening candidate genes by RF-RFE and SVM-RFE
The Random Forest (RF) algorithm is often used to classify or assess regression performance by integrating multiple decision trees16. On the other hand, the Support Vector Machine (SVM) achieves data classification or regression by determining the optimal hyperplane that maximizes the margin between classes17. RF-RFE and SVM-RFE feature selection methods were used to combine RF or SVM with recursive feature elimination (RFE) techniques18. They optimize model performance by repeatedly training the RF or SVM model, evaluating the feature importance, and progressively removing less important features. The identified DMRGs were then screened using RF-RFE and SVM-RFE algorithms. The merged dataset was divided into two datasets, the test and training, randomly at the ratio of 7:3. The RF analysis was conducted utilizing the “randomForest” package in R, and SVM analysis was performed with the “e1071” package. RFE was executed with the “caret” package. For both the RF and SVM models, model training and feature selection were performed using 5-fold cross-validation repeated five times. The number of trees for the RF model was optimized to 58 (ntree = 58), while the cost parameter for the SVM model was tuned across a range from 0.0001 to 10. Finally, the “VennDiagram” package was utilized to determine the overlapping genes identified by both methods, which were subsequently considered the key genes.
Functional enrichment analysis
To investigate the biological roles of DMRGs in more detail, functional enrichment analysis was conducted using the “clusterProfiler” package. Gene Ontology (GO) analysis19 was performed to identify molecular functions (MF), biological processes (BP), and cellular components (CC) associated with the DMRGs, utilizing the “org.Hs.eg.db” package for human gene annotations. The analysis employed the Benjamini-Hochberg (BH) method to control the false discovery rate, with a significance threshold set at a P-adjusted < 0.05 and q < 0.05. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis20,21,22,23 was also conducted, converting gene symbols to ENTREZIDs using the “bitr” function from “clusterProfiler” and applying the same adjustment method and cutoffs. Results were visualized using the “enrichplot” and “ggplot2” packages, with the top five enriched terms displayed for GO and significant pathways for KEGG.
Establishment of nomogram model
The nomogram allows the assessment of the relative contributions and risk scores of each variable in a predictive model24. In this study, we designed a prediction nomogram utilizing “rms” package for AR occurrence based on candidate genes, and a calibration curve was generated to evaluate the agreement between predicted and observed probabilities. Additionally, visualize the expression levels of candidate genes in merged datasets of AR and control cohorts with the “ComplexHeatmap” package.
Immune infiltration analysis
The validated leukocyte gene signature matrix (LM22) was employed to deconvolute 22 distinct functional human hematopoietic subsets25. The degree of infiltration of 22 types of immune cells in each sample of the merged dataset was determined with the CIBERSORT algorithm. Subsequently, heatmaps and violin plots were developed using the “ComplexHeatmap” packages, to compare the infiltration scores of 22 immune cell types between the two groups.
The “psych” package was utilized to analyze the correlation between candidate genes and the infiltration of immune cells. The correlation was deemed significant at P < 0.05, and results were displayed using plots generated using the “ggplot2” package.
Subsequently, Gene Set Enrichment Analysis (GSEA) was performed to predict the biological relevance of the candidate genes associated with the AR-related pathways. Expression data from the merged dataset were analyzed using the “clusterProfiler” package, with gene correlations calculated and ranked for each gene. GO and KEGG pathway enrichments were evaluated using the “c5.go.bp.v2023.2.Hs.symbols.gmt” and “c2.cp.kegg_legacy.v2023.2.Hs.symbols.gmt” gene sets, respectively, with a significance threshold set at P < 0.05. Results were visualized using the “enrichplot” and “ggplot2” packages to display the top five enriched pathways.
Validation of candidate genes
The “pROC” package was used to perform ROC analysis, from which the Area Under the ROC Curve (AUC) was calculated to determine the specificity and sensitivity of candidate genes as measures of performance. The 95% confidence intervals for AUC values were calculated to quantify uncertainty. The diagnostic value of the candidate genes was tested using the merged dataset and external datasets (GSE14328 and GSE1563).
Consensus clustering analysis
The Consensus Clustering analysis was performed with the “ConsensusClusterPlus” package on the candidate genes. Briefly, the expression data of candidate genes were collected and standardized. They were then repeatedly clustered through multiple subsampling and K-means or hierarchical clustering algorithms. Next, the frequency of assigning each pair of samples to the same cluster across the overall clustering results was calculated to generate the consensus matrix. The optimal number of clusters was determined based on the consensus index, and the results were visualized by Uniform Manifold Approximation and Projection (UMAP) clustering using the “umap” package.
Additionally, Principal Component Analysis (PCA) was conducted using the “prcomp” function in R to further assess the separation quality of the identified subgroups. The first two principal components (PC1 and PC2) were extracted to visualize the distribution of samples, providing a complementary perspective to the UMAP clustering.
Validation by PBMCs of patients
To validate the role of the four candidate genes in AR after kidney transplantation, we collected eight AR and eight non-AR peripheral blood samples from patients treated at the First Hospital of Jilin University. All participants signed written informed consent, and all procedures were approved by the Ethics Committee of the First Hospital of Jilin University.
The lymphocyte separation medium (Stem Cell, Cat# 07861) was employed to extract peripheral blood mononuclear cells (PBMCs). Subsequently, total RNA was obtained from the cells using Trizol (Thermo Fisher, Cat# 15596026CN) and the RNA concentration was determined with a microplate reader. 1 µg RNA was used to synthesize cDNA with the HiScript II Q RT SuperMix (Novizan, Cat# R223-01). The prepared cDNA (1µL) was mixed with ChamQ Universal SYBR qPCR Master Mix (Novizan, Cat# Q711-03) and subjected to quantitative real-time PCR (qRT-PCR) on the Applied Biosystems StepOnePlus™ Real-Time PCR System Thermal Cycling Block. The mRNA level of target genes was calculated with the 2^−ΔΔCt method, and data were normalized to the expression of β-actin as the housekeeping gene. The primers used for the qRT-PCR reactions are shown in Table 2.
Establishment of animal models and validation of candidate genes
Eight-week-old male Balb/c and C57BL/6J mice were bought from Charles River (Beijing, China). All experiment and handling protocols were approved by the Institutional Animal Care and Use Committee of the First Hospital of Jilin University.
The Balb/c mice were divided into groups as follows: the normal group, in which kidneys were collected from Balb/c donors and transplanted into Balb/c recipients; the AR group, in which the kidneys were extracted from C57BL/6J donors and transplanted into Balb/c recipients. Briefly, the recipient mice were anesthetized with isoflurane delivered through a small animal anesthesia machine. The donor’s left kidney was harvested following internationally reported protocols and transplanted ectopically into the right side of the recipient’s abdomen. Next, the renal artery was anastomosed to the abdominal aorta using an end-to-side patch technique, while the renal vein was anastomosed with the inferior vena cava using the same method. The ureterovesical anastomosis was performed using the insertion technique. Postoperatively, antibiotics were administered via subcutaneous injection to prevent infection. The mice were housed individually in separate cages, and body temperature was maintained using a heating pad.
Mice were euthanized on day 5 post-transplantation via cervical dislocation under deep isoflurane anesthesia, following AVMA Guidelines for the Euthanasia of Animals (2020)26. The transplanted kidney was excised, and prepared for hematoxylin and eosin (H&E) staining to determine whether the AR model was successfully established. In addition, mRNA was extracted from kidney samples from both groups using Trizol subjected to qRT-PCR to quantify the expression levels of four genes. The detailed protocol is described in Sect. 1.10.
Statistical analysis
The statistical analyses were performed using GraphPad Prism (version 8.0.1) and R (version 4.3.3). The Wilcoxon test was applied for non-parametric data, while the unpaired two-tailed Student’s t-test was used for differences in normally distributed data.
Ethics approval and consent to participate
The human study was approved by the Ethics committee of the First Hospital of Jilin University (Approval Number: 24K145-001) and complied with the Declaration of Helsinki. All participants provided written informed consent. The animal study was approved by the Animal Ethics committee of the First Hospital of Jilin University (Approval Number: 20240347), and was carried out in accordance with ARRIVE guidelines. All experiment and handling protocols were performed in accordance with Institutional Animal Care and Use Committee of the First Hospital of Jilin University.
Results
Identification of DEGs
The combination of GSE129166 and GSE25902 datasets using the “sva” package, yielded a merged dataset comprising 256 normal samples and 76 AR samples. Next, we identified 2,616 DEGs, among which 1,257 were upregulated and 1,359 were downregulated based on the selection threshold of |logFC| > 0.5, P-adjusted < 0.05. Volcano plot shown in Fig. 1 displays the differential gene analysis results (Fig. 1).

Identification of DEGs of the AR and normal groups. Volcano plot of all DEGs in the merged dataset (GSE129166 and GSE25902). Blue dots indicate upregulated genes, red dots represent downregulated genes, and black dots denote genes with no significant change in expression.
WGCNA and DMRGs screening
To identify the key genes in AR, we conducted WGCNA using the top 50% genes obtained by variance analysis from the gene expression matrix containing 11,661 genes. Cluster analysis was performed on the 332 samples, with outliers GSM635742 and GSM635823 removed (Fig. 2A). By analyzing the mean connectivity and scale-free topology index, we found that the highest scale-free topology index (R2 = 0.899) and high average connectivity of network nodes were achieved with a soft threshold β of 30 (Fig. 2B). Consequently, using the soft threshold of 30 to establish a scale-free network resulted in the identification of 7 co-expression modules in the WGCNA (Fig. 2C): red, blue, yellow, brown, green, turquoise, and grey. Next, we calculated the correlation between the module eigengenes (ME) of each module and AR, the results were displayed on a correlation heatmap (Fig. 2D). The red module showed the strongest correlation with AR (cor = 0.4008, P = 4.323e − 14), while the yellow module also displayed a positive correlation with AR (cor = 0.2531, P = 3.451e − 06). Additionally, there was a strong association between module membership (MM) and gene significance (GS) within these two modules (red: cor = 0.52, P = 0.00035; yellow: cor = 0.21, P = 0.0058) (Fig. 2E-F). These modules were selected as candidate modules, comprising 214 genes. The gene lists are provided in Supplementary Table S1 for further reference. The intersection of the 2,616 DEGs from the differential gene expression analysis with the 214 key module genes from WGCNA and the 5,150 mitophagy-related genes obtained from the GeneCards database, yielded 30 DMRGs (Fig. 2G, Supplementary Table S2).

WGCNA and DMRGs screening. (A) Clustering analysis of the samples. (B) Measurement of the soft threshold power based on the scale-free fit index (left) and mean connectivity (right). (C) Gene dendrogram with corresponding module colors identified by WGCNA. (D) Heatmap showing the module-trait relationships. (E) The scatter plot showing the correlation between AR and the red module. (F) The scatter plot indicating the association between AR and the yellow module. (G) The Venn diagram representing the intersection of DEGs, WGCNA module genes, and the mitophagy-related genes.
Functional enrichment analysis for DMRGs
The identified 30 DMRGs were subjected to GO and KEGG enrichment analyses to elucidate their biological functions. Notably, the GO analysis revealed significant enrichment in BP, such as “cytoplasmic translation” and “negative regulation of protein localization” (Fig. 3A). CC terms included “ribosomal subunit”, “ribosome”, “large ribosomal subunit”, “cytosolic ribosome”, and “perinuclear endoplasmic reticulum” (Fig. 3A). The enriched MF terms were “structural constituent of ribosome”, “proline-rich region binding”, “cholesterol transfer activity”, “sterol transfer activity”, and “sterol transporter activity” (Fig. 3A). KEGG pathway enrichment analysis identified a significant pathway, “Coronavirus disease - COVID-19” (Fig. 3B).

Function enrichment analysis of DMRGs. (A) Dot plot of GO enrichment analysis for the significant BP, CC, and MF. (B) Dot plot of KEGG pathway enrichment analysis showing the significant pathway.
Identification of candidate genes using two machine learning models
The 30 DMRGs were further screened using RF-RFE and SVM-RFE methods. Initially, the analysis was conducted using the RF algorithm (Fig. 4A). Subsequently, RFE was performed, yielding a ranking of the genes based on their importance (Fig. 4B) and revealing that a combination of 10 gene sets provided the highest classification accuracy (Fig. 4C). Similarly, 11 key genes were selected through SVM-RFE (Fig. 4D). The detailed gene feature rankings and screened genes from RF-RFE and SVM-RFE are provided in Supplementary Table S3 and S4 for reference. By intersecting the genes identified by both RF-RFE and SVM-RFE methods, we identified four common genes as the final candidate genes (Fig. 4E): Cyclin D1 (CCND1), Zinc Finger CCCH-Type Containing 15 (ZC3H15), Ribosomal Protein L38 (RPL38), and Actin Related Protein 2/3 Complex Subunit 4 (ARPC4). These genes were subjected to further analyses.

Identification of candidate genes. (A) Random forest tree. (B) Gene importance ranking based on RF-RFE. (C) RF-RFE feature selection result plot. (D) SVM-RFE feature selection result plot. (E) Venn diagram of the intersection of the genes screened by the RF-RFE and SVM-RFE.
Establishment of the predictive model for AR occurrence
Using the four candidate genes identified through machine learning, we constructed a risk prediction nomogram for predicting the occurrence of AR (Fig. 5A). The individual scores assigned based on the expression levels of each gene were summed up to obtain the total score for assessing the patient’s risk of developing AR. Patients with high ZC3H15 expression and low expression of CCND1, RPL38, and ARPC4 had higher total scores in the nomogram prediction model, indicating an elevated risk of AR. To further evaluate the nomogram’s performance, a calibration curve was generated, as shown in Fig. 5B. The model demonstrated reliable probability predictions, particularly in the low (0.0-0.3) and high (0.6-1.0) predicted probability regions, and a certain calibration bias was seen in the medium probability range (0.3–0.6), where the predicted values slightly exceeded the observed probabilities, suggesting a modest overestimation in this interval. These findings confirmed that the model was effective in predicting AR occurrence.

Construction of a predictive nomogram model and expression levels of candidate genes. (A) Nomogram for assessing the risk of AR occurrence. (B) Calibration curve for the nomogram. (C) Box plot of the expression levels of four candidate genes in normal and AR groups. Data were analyzed using the Wilcoxon test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Simultaneously, the differential expression of four candidate genes was validated in the gene expression dataset and the results were presented in Fig. 5C. Specifically, it was observed that the expression levels of CCND1 and ZC3H15 were significantly upregulated (P < 0.05), whereas that of RPL38 and ARPC4 were significantly downregulated in the AR group compared to the normal group (P < 0.05).
Analysis of immune cell infiltration
To further compare the immune cell infiltration between AR and normal samples, the CIBERSORT algorithm was employed to analyze 22 immune cell types in each sample. The results were visualized on a heatmap shown in Fig. 6A. The analysis of immune cell infiltration (Fig. 6B) revealed significantly increased infiltration of M1 macrophages and gamma-delta T cells, accompanied by significantly decreased infiltration of memory B cells, regulatory T cells (Tregs), and M2 macrophages in AR samples (P < 0.05).

Analysis of Immune Cell Infiltration. (A) Heatmap of immune cell infiltration in normal and AR groups. (B) Box plot of immune cell infiltration in normal and AR groups. Correlation analysis for CCND1 (C), ZC3H15 (D), RPL38 (E), and ARPC4 (F) and immune cell infiltration. Data were analyzed using the Wilcoxon test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, no significance.
Correlations analysis for the four candidate genes and immune cell infiltration revealed significant associations with various immune cells in the tissue microenvironment (Fig. 6C-F). CCND1 (Fig. 6C) and ZC3H15 (Fig. 6D) showed similar correlation patterns, and were significantly positively correlated with M1 macrophages, resting CD4 memory T cells, and resting dendritic cells (P < 0.05), while showing significant negative correlations with neutrophils, monocytes, and resting NK cells (P < 0.05). In contrast, RPL38 (Fig. 6E) and ARPC4 (Fig. 6F) exhibited opposite correlation patterns to CCND1 and ZC3H15, showing significant positive correlations with neutrophils, resting NK cells, and monocytes (P < 0.05), and significant negative correlations with M1 macrophages, resting CD4 memory T cells and resting dendritic cells (P < 0.05).
To investigate the biological pathways associated with the four candidate mitophagy-related genes in AR following kidney transplantation, GSEA was conducted using the merged dataset. The GSEA plots display running enrichment scores and ranked gene lists, revealing the shared and unique pathway enrichments. For GO terms, the four genes (CCND1, ZC3H15, RPL38, and ARPC4) were significantly enriched in “gland development”, “positive regulation of cell adhesion”, “regulation of peptidase activity”, and “response to peptide”. Notably, CCND1 was uniquely enriched in “positive regulation of transferase activity”, ZC3H15 for “cell substrate adhesion”, RPL38 and ARPC4 were enriched in “regulation of apoptotic signaling pathway”. In the KEGG analysis, the four genes were strongly enriched in “focal adhesion”, “lysosome”, and “pathways in cancer”. Furthermore, CCND1 and RPL38 were enriched in “cell cycle”, CCND1, ZC3H15, and ARPC4 for “ribosome”, ZC3H15 and RPL38 were enriched in “neurotrophin signaling pathway”, and ARPC4 was uniquely enriched in “Alzheimer’s disease”. The shared and distinct pathway enrichments are displayed in the corresponding GSEA plots (Fig. 7A-D).

GSEA analysis of candidate genes. GSEA plot for (A) CCND1, (B) ZC3H15, (C) RPL38, and (D) ARPC4.
Validation of candidate genes by ROC curve
To assess the specificity and sensitivity of the four candidate genes in predicting AR occurrence, we performed ROC analysis in the internal dataset (the merged expression matrix of GSE129166 and GSE25902) and external dataset (GSE14328 and GSE1563). The validation results for the internal data are shown in Fig. 8A. Notably, the AUC values for CCND1, ZC3H15, RPL38, and ARPC4 were 0.699, 0.749, 0.744, and 0.731, respectively. In the external dataset, GSE14328, the AUC values for the four genes were 0.719, 0.699, 0.605, and 0.682, respectively (Fig. 8B). In the GSE1563 dataset (Fig. 8C), the AUC values were 0.73, 0.683, 0.714, and 0.873, respectively. These results demonstrated that the four candidate genes could significantly predict AR occurrence.

Validation of the diagnostic value of candidate genes. ROC curves evaluating the diagnostic performance of CCND1, ZC3H15, RPL38, and ARPC4 in (A) the internal dataset (merged expression matrix of GSE129166 and GSE25902), (B) the external dataset GSE14328, and (C) the external dataset GSE1563.
Consensus clustering analysis
The consensus clustering analysis was performed to investigate the classification potential of the four candidate genes in distinguishing biologically distinct subgroups within the merged expression dataset. This aimed to identify the molecular heterogeneity, variations in mitophagy regulation, immune cell dynamics, or mitochondrial function, which are critical to AR pathogenesis.
The analysis revealed the optimal clustering effect for two clusters (Fig. 9A). According to this two-cluster standard, 332 samples were re-clustered, resulting in a strong classification effect (Fig. 9B). To further validate the separation quality of the identified subgroups, we carried out PCA. The PCA results, presented in Fig. 9C, demonstrated a clear separation between the two clusters (Subgroup A and Subgroup B) along the first two principal components (PC1 and PC2), which reinforced the distinct expression patterns observed in the UMAP visualization and enhanced the robustness of the classification.

Consensus Clustering Analysis. (A) Consensus matrix for k = 2, illustrating the optimal clustering of samples. (B) UMAP plot showing the two-dimensional embedding of the re-clustered samples (Subgroup A and Subgroup B). (C) PCA plot demonstrating the separation of Subgroup A and Subgroup B along the first two principal components (PC1 and PC2). Heatmap (D) and box plot (E) of immune cell infiltration between the two re-clustered groups. Data were analyzed using the Wilcoxon test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, no significance.
Further analysis of immune cell infiltration and differences between the two re-clustered groups (Fig. 9D-E) revealed significant differences (P < 0.05) in the infiltration proportions of 19 types of immune cells, including dendritic cells, macrophages, CD8 T cells, plasma cells, memory B cells, CD4 T cells, NK cells, eosinophils, and neutrophils. This analysis confirmed a significant distinction among the reclassified samples, underscoring the classification efficacy of the four candidate genes.
Two clusters were identified by UMAP and PCA, indicating that the expression profiles of the candidate genes could capture the distinct biological states, likely associated with varying degrees of immune response or cellular stress in AR. The significant differences in immune cell infiltration between the subgroups underscore the biological relevance of these genes, implying that they may be involved in the regulation of various immune-mediated processes. These findings provide a foundation for future investigations into the specific mechanisms driving these subgroups, which can potentially guide the implementation of tailored clinical interventions.
Validation of candidate genes
To further validate the expression levels of CCND1, ZC3H15, RPL38, and ARPC4, we first performed qRT-PCR on PBMCs collected from patients with AR and non-AR after kidney transplantation. Consistent with the findings from the GEO database, the mRNA levels of ZC3H15 and CCND1 were markedly elevated, whereas that of RPL38 and ARPC4 were reduced in AR PBMCs compared to non-AR PBMCs (Fig. 10A).

Differential analysis of candidate genes in clinical samples and animal models. (A) qRT-PCR analysis of the expression levels of four candidate genes in PBMCs from normal and AR patients. (B) Representative H&E staining of mouse kidney tissue from the normal and AR groups: (a-b) normal group at 100× and 400× magnification, (c-d) AR group at 100× and 400× magnification. (C) qRT-PCR analysis of four candidate genes in mouse kidney tissue from normal and AR groups. Data were analyzed using the unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, no significance.
We further established a mouse model of AR. On day 5 post-transplantation, H&E staining of the transplanted kidney (Fig. 10B) revealed that the kidneys exhibited typical characteristics of acute T cell-mediated rejection (TCMR), including acute glomerular injury, loss of brush border in the renal tubules, and significant mononuclear cell infiltration in the interstitium. In contrast, the kidneys in the normal group displayed mild and varying degrees of ischemia-reperfusion injury and interstitial edema. We further assessed the mRNA expression levels of the four candidate genes in transplanted kidneys from both groups. As illustrated in Fig. 10C, ZC3H15 and CCND1 were significantly upregulated in the AR group, while RPL38 expression was markedly reduced. Notably, the ARPC4 expression was not significantly different between the two groups, although the expression tended to be lower in the AR group.
Discussion
This study integrated two transcriptomic datasets from the GEO database and identified 2,616 DEGs, among which 1,257 were upregulated and 1,359 were downregulated, through differential expression analysis. The WGCNA further uncovered two modules that were strongly associated with AR. Moreover, the intersection of DEGs, module genes, and mitophagy-related genes revealed 30 DMRGs. KEGG pathway and GO analyses revealed that the DMRGs are primarily involved in “cytoplasmic translation”, “negative regulation of protein localization”, ribosomal components, and the pathway associated with “Coronavirus disease - COVID − 19”. Using RF-RFE and SVM-RFE, we screened and identified four core candidate genes, CCND1, ZC3H15, RPL38, and ARPC4, as the key markers of AR. To evaluate their prediction performance, ROC curves and nomograms were established to facilitate quantitative assessment. Analysis of the genes in internal and external datasets confirmed that the four genes could predict AR occurrence. The AR risk prediction nomogram, comprising the four genes, exhibited good prediction performance on AR occurrence. Additionally, immune cell infiltration analysis demonstrated significantly increased M1 macrophages and gamma-delta T cells in AR samples, and decreased levels of memory B cells, Tregs, and M2 macrophages. Moreover, the CCND1 and ZC3H15 genes were positively correlated with the infiltration levels of M1 macrophages, resting dendritic cells, and resting CD4 memory T cells, while negatively correlated with neutrophils, monocytes, and resting NK cells. However, the other two genes, RPL38 and ARPC4, displayed opposite correlations. Gene expression analysis revealed significant differences: CCND1 and ZC3H15 were upregulated in AR samples, whereas RPL38 and ARPC4 were downregulated relative to the normal controls. Similar expression patterns were observed in PBMCs extracted from clinical AR and non-AR patients, and transplanted kidney tissues from AR and non-AR model mice, confirming that the genes exhibited distinct expression patterns.
Four candidate genes - CCND1, ZC3H15, RPL38, and ARPC4 - were associated with cell cycle regulation, immune modulation, and mitophagy, which aim to preserve mitochondrial integrity during AR. CCND1, encoding cyclin D1, regulates the G1/S phase transition by forming a complex with cyclin-dependent kinases 4 and 6, phosphorylating the retinoblastoma protein, and releasing E2F transcription factors, a mechanism linked to tumorigenesis27 and immune dysregulation in diseases like rheumatoid arthritis28. This implies that it may play a role in modulating immune responses in AR via cell cycle control. Moreover, its regulatory role in cell cycle progression may indirectly influence mitophagy via the PINK1/Parkin pathway, damaging its mitochondrial quality control in AR. ZC3H15, a zinc finger protein, can modulate mRNA stability and translation, influencing cancer cell proliferation and cytokine expression29,30implying that it may play a role in the AR’s immune pathophysiology, and potentially modulates mitophagy by targeting mRNAs of related pathways. RPL38, a ribosomal protein essential for protein synthesis, can regulate the cell cycle and apoptosis, and its overexpression has been shown to contribute to poor prognosis in cancers31indicating that it has a major role in the occurrence of AR via cell homeostasis. It also promotes that ribosomal biogenesis, which is required to enhance mitochondrial protein synthesis, a prerequisite for effective mitophagy31. ARPC4, part of the Arp2/3 complex, drives cytoskeletal dynamics and cell migration, involved in immune cell trafficking in AR, with diverse variants linked to immune-related diseases32,33and may stimulate the formation of autophagosome during mitophagy6. Together, these genes play crucial roles in maintaining mitochondrial homeostasis and activating the immune response in AR, which may worsen graft rejection. Their involvement in both cell cycle regulation and mitophagy underscores their dual functions. Further research is needed to clarify their exact mechanisms and confirm their importance in AR.
The infiltration level of immune cells has been reported to influence AR development. In our study, significant differences in multiple immune cell infiltrations were observed between normal and AR samples. Additionally, four candidate genes were found to be strongly associated with these immune cell infiltrations. γδ T cells modulate the development of AR after kidney transplantation by secreting pro-inflammatory factors such as TNF-α and IFN-γ, which induce rejection response. They also indirectly promote graft rejection by interacting with dendritic cells and macrophages, further escalating the immune attack on the transplanted organ34. Tregs can inhibit AR development after kidney transplantation. Tregs inhibit effector T cell activation and proliferation, thereby reducing immune response to the transplanted kidney. Dysfunction or insufficiency of Tregs is considered an important trigger of AR onset, as weakened Treg function leads to an uncontrolled increase in effector T cell activity, aggravating immune response towards the graft and triggering AR35,36. M1 and M2 macrophages exert opposing effects in AR. Following activation by IFN-γ, M1 macrophages produce inflammation factors, (IL-12, TNF-α, IL-1β, IL-6) that amplify immune responses, leading to inflammation and tissue damage in the transplanted kidney, exacerbating AR. Conversely, M2 macrophages, activated by IL-13 and IL-4 generate anti-inflammatory factors (TGF-β, IL-10) which enhance tissue repair and immune suppression, protecting the transplanted kidney. Notably, the survival of the graft is influenced by the balance of M1 and M2 macrophages, with excessive M1 activation linked to AR and failure, while M2 macrophages improve long-term graft survival37,38. Here, we found increased infiltration of M1 macrophages and gamma-delta T cells in AR, and decreased levels of M2 macrophages and Tregs, which is consistent with previous reports. To provide a molecular basis for these immune dynamics, GSEA was performed using the merged dataset. It was observed that all four genes (CCND1, ZC3H15, RPL38, and ARPC4) were enriched in the “positive regulation of cell adhesion” and “lysosome”, which may support immune cell trafficking and antigen processing, respectively. The enrichment of “response to peptide” across all four genes suggests a potential link to antigen presentation, which is consistent with the increased infiltration level of M1 macrophages and γδ T cells. Furthermore, the unique enrichment of “regulation of apoptotic signaling pathway” in RPL38 and ARPC4, and “cell substrate adhesion” in ZC3H15, can suggest their involvement in immune cell survival and adhesion, consistent with the observed imbalance between M1 and M2 macrophages. Collectively, these findings promote the current understanding of the mechanisms by which these genes contribute to the immune microenvironment in AR.
The enrichment of DMRGs in cytoplasmic translation and ribosomal components is also consistent with the major biological process in AR: the increased protein synthesis demand during immune activation. Cytoplasmic translation is an essential process that facilitates the rapid production of proteins involved in immune responses, such as cytokines and T-cell receptors, which are upregulated during graft rejection39. The presence of “ribosomal subunits”, “large ribosomal subunit”, “cytosolic ribosome”, “perinuclear endoplasmic reticulum”, and “structural constituent of ribosome” suggests their potential dysregulation in protein synthesis machinery, which can damage the immune cell function and contribute to the inflammatory milieu seen in AR40. Furthermore, the perinuclear endoplasmic reticulum enrichment suggests endoplasmic reticulum stress, which is known to induce AR owing to the unfolded protein response, enhanced immune cell activation, and cytokine release41. The notable enrichment of the “Coronavirus disease - COVID-19” pathway provides a novel insight into the mechanisms underlying AR. This pathway may be implicated in AR due to overlapping characteristics of immune dysregulation and cytokine storm, wherein the excessive inflammatory response induced by viral infection mirrors the hyperactive immune processes involved in graft rejection42. The involvement of DMRGs in this pathway suggests a common mitochondrial and autophagic dysfunction that exacerbates AR, which calls for further investigation on the cross-pathway interactions. The molecular functions, including “proline-rich region binding”, “cholesterol transfer activity”, “sterol transfer activity”, and “sterol transporter activity”, suggest a role in lipid metabolism, which can potentially affect the membrane integrity and immune cell signaling during AR. The proline-rich region binding can alter the protein-protein interactions, which are critical for immune regulation, potentially affecting T-cell activation and regulatory T-cell function43. Although these findings provide a solid foundation for understanding AR, the specific mechanisms linking these DMRGs to AR progression need to be investigated through experiments. Future studies should investigate the therapeutic potential of modulating cytoplasmic translation and ribosomal activity, along with the immunological parallels observed in COVID-19, as strategies to reduce the risk of AR.
Our 4-gene signature (CCND1, ZC3H15, RPL38, ARPC4), which was formulated via the integrative transcriptomic analysis, represents a pioneering approach that targets mitophagy-related pathways in AR following kidney transplantation, as benchmarked against established diagnostic panels. In contrast to AlloMap, which monitors immune activation in peripheral blood to rule out acute cellular rejection in heart transplantation with AUCs of 0.70–0.8544, the proposed signature showed comparable diagnostic accuracy (AUC 0.699–0.873) while illuminating mitochondrial dysfunction - a critical yet underexplored driver of AR in kidney allografts. The Banff Human Organ Transplant (B-HOT) panel, comprising 770 genes for biopsy-based molecular diagnosis of rejection45provides a comprehensive immune and injury profile but lacks specificity for mitochondrial processes. Our streamlined panel, validated in both PBMCs and kidney tissue, advances current methodologies by offering applicability in both non-invasive and invasive diagnostic settings. Moreover, it highlights the role of mitophagy in regulating inflammation, oxidative stress, and cell cycle dysregulation in AR. This makes the developed signature as a potential catalyst for precision diagnostics, enabling early detection and formation of tailored interventions that address mitochondrial pathology, thereby reducing graft loss risk. Future studies integrating our panel with multi-omics data are advocated to further dissect the interplay between mitophagy and immune responses, potentially redefining AR management strategies.
Although we performed multiple analyses across various databases, there are specific limitations that should be noted. Although the GEO database offers extensive high-throughput data, AR-related datasets have various limitations, especially in terms of sample size within external validation sets (e.g., only 18 AR samples in GSE14328 and 7 in GSE1563) and clinical PBMC cohorts. This constraint reduces statistical power, potentially affecting the robustness and generalizability of the predictive nomogram and enrichment analyses. As a result, effect sizes may be underestimated due to insufficient representation of biological variability. The small cohort sizes prevented the implementation of a power analysis or downsampling/bootstrapping to evaluate model stability, as these methods require larger samples for reliable estimates. To enhance the validity and applicability of our findings, studies with larger sample sizes, especially for external validation and clinical cohorts, are needed to test the aforementioned statistical methods more extensively. Moreover, there is a need to investigate the mechanisms of these genes in detail to identify key biomarkers for AR treatment and diagnosis.
Nevertheless, the exact mechanisms of mitophagy during AR development are unclear. In this study, we conducted a differential analysis of transcriptomic data from AR and normal kidney transplant tissues in the GEO database, combined with WGCNA and machine learning algorithms. Four mitophagy-related genes strongly linked to AR were uncovered. We further validated these findings by examining their differential expression in PBMCs extracted from clinical AR and non-AR patients, and transplanted kidney tissues from AR and non-AR model mice. Our findings lay the foundation for future identification of biomarkers to facilitate the diagnosis and prognosis prediction of AR.
Data availability
All data generated or analysed during this study are included in this published article.
References
Perrottet, N. et al. Infectious complications and graft outcome following treatment of acute antibody-mediated rejection after kidney transplantation: A nationwide cohort study. Plos One. 16, e0250829. https://doi.org/10.1371/journal.pone.0250829 (2021).
Rodrigo, E., Chedid, M. F., San Segundo, D., San Millán, J. C. R. & López-Hoyos, M. Acute rejection following kidney transplantation: State-of-the-Art and future perspectives. Curr. Pharm. Des. 26, 3468–3496. https://doi.org/10.2174/1381612826666200610184433 (2020).
Ploumi, C., Daskalaki, I. & Tavernarakis, N. Mitochondrial biogenesis and clearance: a balancing act. Febs J. 284, 183–195. https://doi.org/10.1111/febs.13820 (2017).
Han, R., Liu, Y. T., Li, S. H., Li, X. J. & Yang, W. L. PINK1-PRKN mediated mitophagy: differences between and models. Autophagy 19, 1396–1405. https://doi.org/10.1080/15548627.2022.2139080 (2023).
Yang, M. L., Wei, X., Yi, X. & Jiang, D. S. Mitophagy-related regulated cell death: molecular mechanisms and disease implications. Cell. Death Dis. 15, 505. https://doi.org/10.1038/s41419-024-06804-5 (2024).
Pickles, S., Vigié, P. & Youle, R. J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. https://doi.org/10.1016/j.cub.2018.01.004 (2018).
Dong, Y. M. & Zhang, X. Targeting cellular mitophagy as a strategy for human cancers. Front. Cell. Dev. Biol. 12, 1431968. https://doi.org/10.3389/fcell.2024.1431968 (2024).
Atici, A. E., Crother, T. R. & Noval Rivas, M. Mitochondrial quality control in health and cardiovascular diseases. Front. Cell. Dev. Biol. 11, 1290046. https://doi.org/10.3389/fcell.2023.1290046 (2023).
Chen, J. et al. Liver cell mitophagy in metabolic Dysfunction-Associated steatotic liver disease and liver fibrosis. Antioxid. (Basel). 13, 729. https://doi.org/10.3390/antiox13060729 (2024).
Mishra, E., Thakur, M. K. & Mitophagy A promising therapeutic target for neuroprotection during ageing and age-related diseases. Br. J. Pharmacol. 180, 1542–1561. https://doi.org/10.1111/bph.16062 (2023).
Picca, A., Faitg, J., Auwerx, J. & Ferrucci, L. D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 5, 2047–2061. https://doi.org/10.1038/s42255-023-00930-8 (2023).
Hadi, F., Mortaja, M. & Hadi, Z. Calcium (Ca) hemostasis, mitochondria, autophagy, and mitophagy contribute to alzheimer’s disease as early moderators. Cell. Biochem. Funct. 42, e4085. https://doi.org/10.1002/cbf.4085 (2024).
Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure (485, Pg 251, 2012). Nature 490, 292–292. https://doi.org/10.1038/nature11515 (2012).
See, E. J. et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 95, 160–172. https://doi.org/10.1016/j.kint.2018.08.036 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Chaibi, S., Mahjoub, C., Ayadi, W. & Kachouri, A. Epileptic EEG patterns recognition through machine learning techniques and relevant time-frequency features. Biomed. Eng-Biomed Te. 69, 111–123. https://doi.org/10.1515/bmt-2023-0332 (2024).
Noble, W. S. What is a support vector machine? Nat. Biotechnol. 24, 1565–1567. https://doi.org/10.1038/nbt1206-1565 (2006).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19, 432. https://doi.org/10.1186/s12859-018-2451-4 (2018).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Kanehisa, M., Sato, Y. & Kawashima, M. KEGG mapping tools for Uncovering hidden features in biological data. Protein Sci. 31, 47–53. https://doi.org/10.1002/pro.4172 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Iasonos, A., Schrag, D., Raj, G. V. & Panageas, K. S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 26, 1364–1370. https://doi.org/10.1200/Jco.2007.12.9791 (2008).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12, 453–457. https://doi.org/10.1038/Nmeth.3337 (2015).
AVMA Guidelines for the Euthanasia of Animals: 2020 Edition, (2020). https://www.avma.org/resources-tools/avma-policies/avma-guidelines-euthanasia-animals
Wang, J. et al. Aberrant Cyclin D1 splicing in cancer: from molecular mechanism to therapeutic modulation. Cell. Death Dis. 14, 244. https://doi.org/10.1038/s41419-023-05763-7 (2023).
Liu, W. et al. The dual role of CCND1 in heterotopic ossification: A Non-canonical pathway for celecoxib treatment. Heliyon 10, e34936. https://doi.org/10.1016/j.heliyon.2024.e34936 (2024).
Li, Q. et al. ZC3H15 Correlates with a Poor Prognosis and Tumor Progression in Melanoma. Biomed Res Int 2021, 8305299. https://doi.org/10.1155/2021/8305299 (2021).
Qu, X. et al. Macrophage RIPK3 triggers inflammation and cell death via the XBP1-Foxo1 axis in liver ischaemia-reperfusion injury. JHEP Rep. 5, 100879. https://doi.org/10.1016/j.jhepr.2023.100879 (2023).
Goudarzi, K. M. & Lindström, M. S. Role of ribosomal protein mutations in tumor development (Review). Int. J. Oncol. 48, 1313–1324. https://doi.org/10.3892/ijo.2016.3387 (2016).
Huang, S., Li, D., Zhuang, L., Sun, L. & Wu, J. Identification of Arp2/3 complex subunits as prognostic biomarkers for hepatocellular carcinoma. Front. Mol. Biosci. 8, 690151. https://doi.org/10.3389/fmolb.2021.690151 (2021).
Kuroda, Y. et al. A novel variant of ARPC4-related neurodevelopmental disorder. Am. J. Med. Genet. A. 191, 893–895. https://doi.org/10.1002/ajmg.a.63082 (2023).
Sullivan, L. C. et al. The complex existence of γδ T cells following transplantation: the good, the bad and the simply confusing. Clin. Transl Immunol. 8, e1078. https://doi.org/10.1002/cti2.1078 (2019).
Cassano, A., Chong, A. S. & Alegre, M. L. Tregs in transplantation tolerance: role and therapeutic potential. Front. Transpl. 2, 1217065. https://doi.org/10.3389/frtra.2023.1217065 (2023).
Alhosseini, M. N. et al. Therapy with regulatory T-cell infusion in autoimmune diseases and organ transplantation: A review of the strengths and limitations. Transpl. Immunol. 85, 102069. https://doi.org/10.1016/j.trim.2024.102069 (2024).
Lackner, K., Ebner, S., Watschinger, K. & Maglione, M. Multiple shades of Gray-Macrophages in acute allograft rejection. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24098257 (2023).
Ni, B. et al. IL-34 attenuates acute T cell-mediated rejection following renal transplantation by upregulating M2 macrophages polarization. Heliyon 10, e24028. https://doi.org/10.1016/j.heliyon.2024.e24028 (2024).
Rodrigo, E., Chedid, M. F., Segundo, D. S., Millán, J. & López-Hoyos, M. Acute rejection following kidney transplantation: State-of-the-Art and future perspectives. Curr. Pharm. Des. 26, 3468–3496. https://doi.org/10.2174/1381612826666200610184433 (2020).
Das, A. S., Basu, A. & Mukhopadhyay, R. Ribosomal proteins: the missing piece in the inflammation puzzle? Mol. Cell. Biochem. 480, 785–797. https://doi.org/10.1007/s11010-024-05050-9 (2025).
McGirr, T., Onar, O. & Jafarnejad, S. M. Dysregulated ribosome quality control in human diseases. Febs J. 292, 936–959. https://doi.org/10.1111/febs.17217 (2025).
Diao, B. et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front. Immunol. 11, 827. https://doi.org/10.3389/fimmu.2020.00827 (2020).
Spinelli, G. A. et al. Lipid profile changes during the first year after kidney transplantation: risk factors and influence of the immunosuppressive drug regimen. Transpl. Proc. 43, 3730–3737. https://doi.org/10.1016/j.transproceed.2011.08.074 (2011).
Pham, M. X. et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl. J. Med. 362, 1890–1900. https://doi.org/10.1056/NEJMoa0912965 (2010).
Beadle, J. et al. Application of the Banff human organ transplant panel to kidney transplant biopsies with features suspicious for antibody-mediated rejection. Kidney Int. 104, 526–541. https://doi.org/10.1016/j.kint.2023.04.015 (2023).
Acknowledgements
Not applicable.
Funding
This work was funded by the Shandong Provincial Medical and Health Science and Technology Development Plan (202004050936).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. JM and DW: Writing - original draft preparation; JM, XA and HW: Writing - review and editing; YZ and HW: Conceptualization; XA and YZ: Methodology; JM and DW: Formal analysis and investigation; HW: Funding acquisition; HW: Resources; YZ and HW: Supervision. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, J., Wu, D., An, X. et al. Identification of mitophagy-related genes with diagnostic value in acute rejection following kidney transplantation using bioinformatics analysis. Sci Rep 15, 20797 (2025). https://doi.org/10.1038/s41598-025-09143-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09143-3
