
Abstract
Hyperhomocysteinemia can cause severe damage to kidney. Ferroptosis represents a critical mechanism in the initiation and development of kidney disorders. We focus on the β-catenin/GPX4 signaling pathway to explore how homocysteine influences ferroptosis regulation in renal tubular epithelial cells. C57BL/6J mice were administered drinking water with high level of homocysteine to establish a hyperhomocysteinemia model. In the cell experiments, HKC-8 cells were exposed to homocysteine for a duration of 12 h. Active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 were detected using Western blotting; Biochemical assays were conducted to measure lipid ROS, Fe2+, and GSH; GPX4 and β-catenin were detected through immunohistochemistry and immunofluorescence techniques; Mitochondrial damage was examined using transmission electron microscopy; ChIP analysis, coupled with dual-luciferase reporter gene assays, was employed to investigate the relationship between β-catenin protein and GPX4 gene promoter. Our findings revealed that homocysteine disrupted β-catenin signaling, inhibited GPX4 expression in renal tubular epithelial cells, subsequently promoted ferroptosis. Overexpression of β-catenin or GPX4 inhibited ferroptosis induced by homocysteine, and β-catenin regulated GPX4 expression in renal tubular epithelial cells. Further assays demonstrated that GPX4 acted as a target gene of β-catenin. In conclusion, homocysteine elicits ferroptosis in renal tubular epithelial cells by disrupting β-catenin signaling and inhibiting its target gene, GPX4.
Similar content being viewed by others


Introduction
Hyperhomocysteinemia, as a common metabolic disorder, is characterized by elevated levels of homocysteine (homocysteine > 15 µmol/L) in the blood. The prevalence of hyperhomocysteinemia in the population ranges from 5 to 30%. The condition is more common in older adults, patients with chronic diseases such as diabetes and kidney disease, and individuals with malnutrition or vitamin deficiencies1. The high global prevalence of hyperhomocysteinemia underscores its significance as a critical issue in public health research.
Homocysteine is a critical intermediate in the metabolic cycle of methionine and cysteine, predominantly existing in a protein-bound form2. Elevated homocysteine levels are implicated in renal injury mechanisms, including oxidative stress, endothelial dysfunction, and inflammation3,4. Furthermore, declining renal function impairs the clearance of homocysteine, leading to elevated blood homocysteine levels. This interaction establishes a vicious cycle between hyperhomocysteinemia and renal impairment, which in turn aggravates kidney injury and further deteriorates renal function5. Preliminary experiments revealed that homocysteine disrupted β-catenin signaling and induced a reduction of glutathione peroxidase 4 (GPX4) expression in the kidneys. β-catenin and GPX4 cooperatively regulate tumor progression and chemotherapy response via ferroptosis, suggesting a synchronized role in cancer development and therapeutic resistance6,7. As a key negative regulator of ferroptosis, GPX4 is a crucial enzyme which inhibits lipid peroxidation and reduces lipid reactive oxygen species (ROS) through the utilization of glutathione (GSH). Ferroptosis is a form of regulated cell death characterized by iron-dependent lipid peroxidation, driven by intracellular Fe2+ accumulation8. Ferroptosis is strongly associated with the damage caused by homocysteine9,10. In addition, GPX4 is likely a key intermediary molecule that counteracts the damaging effects of homocysteine11. Therefore, ferroptosis may represent one of the key mechanisms by which homocysteine induces renal damage. This may provide a novel potential target for preventing and treating homocysteine-associated kidney diseases.
The β-catenin signaling at normal baseline levels is essential for maintaining proper cellular energy metabolism12. In response to stressors such as inflammation and injury, intracellular β-catenin signaling is activated, promoting cell proliferation and repair. This response assists the organism in resisting external stimuli13,14. Hence, the homeostasis of β-catenin signaling is crucial for preserving normal renal function; disruption of β-catenin signaling can initiate and exacerbate the progression of kidney diseases.
Based on the literature evidence and preliminary findings, this study aims to assess whether homocysteine induces ferroptosis in renal tubular epithelial cells, specifically exploring the role of the β-catenin/GPX4 pathway in this process.
Results
Homocysteine impaired β-catenin signaling and promoted ferroptosis in renal tubular epithelial cells
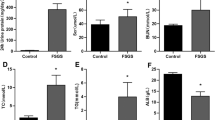
To investigate the impact of hyperhomocysteinemia on the kidney, we developed a mouse model of hyperhomocysteinemia by administering 1.8 g/L homocysteine in the drinking water. Western blot analysis revealed that elevated homocysteine levels significantly reduced the levels of both active β-catenin and β-catenin (Fig. 1A,C). Concurrently, hyperhomocysteinemia markedly reduced GPX4 expression, increased MDA (Malondialdehyde) and Fe2+ levels in the kidney, while no significant changes were noted in the concentrations of ferritin heavy chain 1 (FTH1) and GSH (Fig. 1D,H). Transmission electron microscopy analysis showed that renal tubular cells of mice with hyperhomocysteinemia exhibited mitochondrial membrane rupture and cristae reduction, indicating potential involvement of ferroptosis (Fig. 1I). These findings indicated that elevated homocysteine levels triggered ferroptosis in the kidneys. To assess the impact of elevated homocysteine on renal tubular epithelial cells, HKC-8 cells were exposed to 500 µmol/L homocysteine. The results demonstrated that homocysteine suppressed β-catenin signaling and GPX4 expression in HKC-8 cells, while increasing intracellular MDA and Fe2+ levels (Fig. 1J,Q). Similarly, homocysteine induced ferroptosis-like changes in HKC-8 cells, including mitochondrial membrane disruption and reduction in mitochondrial cristae (Fig. 1R). These findings suggested that elevated homocysteine levels triggered ferroptosis in renal tubular epithelial cells.

Homocysteine impaired β-catenin signaling and promoted ferroptosis in renal tubular epithelial cells. The hyperhomocysteinemia model was established by administering drinking water containing 1.8 g/L homocysteine to the mice. In the in vitro experiments, HKC-8 cells were treated with homocysteine at a concentration of 500 µmol/L. (A) Western blotting was employed to evaluate the expression levels of active β-catenin, β-catenin, GPX4, and FTH1 in the kidneys of mice. (B–E) Quantitative analysis was conducted to determine the levels of active β-catenin, β-catenin, GPX4, and FTH1 in the kidneys of mice. (F–H) Biochemical assays were performed to measure the levels of Fe2+, MDA, and GSH in mouse kidney tissues. (I) The morphology of mitochondria in renal tubular epithelial cells was examined using transmission electron microscopy. Yellow arrows highlighted mitochondrial damage, including the rupture of the mitochondrial membrane and the reduction of mitochondrial cristae. Scale bar, 300 nm. (J) Western blotting was employed to evaluate the expression levels of active β-catenin, β-catenin, GPX4, and FTH1 in HKC-8. (K–N) Quantitative analysis was conducted to determine the levels of active β-catenin, β-catenin, GPX4, and FTH1 in HKC-8. (O–Q) Biochemical assays were performed to measure the levels of Fe2+, MDA, and GSH in HKC-8. (R) Mitochondrial morphology in HKC-8 cells was examined using electron microscopy. Yellow arrows highlighted mitochondrial damage, including the rupture of the mitochondrial membrane and the reduction of mitochondrial cristae. Scale bar, 300 nm. *P < 0.05 vs. the controls (n = 6). Hcy, homocysteine; TEM, transmission electron microscopy.
Fer-1 alleviated renal ferroptosis triggered by hyperhomocysteinemia without altering β-catenin and GPX4 levels
To determine if hyperhomocysteinemia induces renal ferroptosis, we administered the ferroptosis inhibitor ferrostatin-1 (Fer-1) to mice with hyperhomocysteinemia. Western blot showed that Fer-1 had no significant impact on the homocysteine-induced reduction of β-catenin and GPX4 (Fig. 2A and D). However, Fer-1 effectively countered the effects of homocysteine, reducing MDA and Fe2+ levels in mouse kidneys and decreasing kidney injury molecule-1 (KIM-1) (Fig. 2F and H). In addition, homocysteine had no effect on the levels of FTH1 and GSH (Fig. 2E and I). Furthermore, immunohistochemical staining of renal tissues showed that homocysteine markedly downregulated the expression of β-catenin and GPX4 in renal tubular epithelial cells, while Fer-1 had no significant effect on the expression of these proteins (Fig. 2J). Hence, Fer-1 alleviated renal ferroptosis triggered by hyperhomocysteinemia without altering β-catenin and GPX4 levels.

Fer-1 alleviated renal ferroptosis triggered by hyperhomocysteinemia without altering β-catenin and GPX4 levels. The ferroptosis inhibitor Fer-1 (2 mg/kg/day, i.p.) was administered to mice with hyperhomocysteinemia, and its therapeutic efficacy was evaluated. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in the kidneys of mice from indicated groups. (B–F) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in Figure (A) was performed. (G–I) Biochemical determination of the levels of Fe2+, MDA, and GSH in the kidney tissues of mice from indicated groups was conducted. (J) Immunohistochemical analysis of β-catenin and GPX4 in the renal tissues was performed. Scale bar, 20 μm. *P < 0.05 vs. the controls; #P < 0.05 vs. hyperhomocysteinemia mice (n = 6). Hcy, homocysteine.
Fer-1 selectively attenuated homocysteine-induced ferroptosis in renal tubular epithelial cells without affecting β-catenin signaling and GPX4 expression levels
In vitro experiments, Fer-1 was administered to homocysteine-stimulated HKC-8 cells. Fer-1 did not markedly affect the homocysteine-triggered downregulation of β-catenin and GPX4 (Fig. 3A,D). However, Fer-1 effectively mitigated the deleterious impacts of homocysteine, significantly reducing MDA and Fe2+ accumulation in renal tubular epithelial cells (Fig. 3G,H). Additionally, Fer-1 substantially lowered the levels of KIM-1, highlighting its protective role against homocysteine-induced renal tubular injury (Fig. 3F). In addition, homocysteine had no effect on the levels of FTH1 and GSH in HCK-8 cells (Fig. 3E and I). Subsequent immunofluorescence analysis demonstrated that homocysteine conspicuously reduced levels of β-catenin and GPX4 expression, while Fer-1 did not notably alter the expression levels of these proteins in HKC-8 cells (Fig. 3J).

Fer-1 selectively attenuated homocysteine-induced ferroptosis in HKC-8 cells without affecting β-catenin signaling and the levels of GPX4. The ferroptosis inhibitor Fer-1 (10 µmol/L) was used to treat homocysteine-stimulated HKC-8 cells, and its therapeutic effect was evaluated. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in HKC-8 cells from indicated groups. (B–F) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in Figure (A) was performed. (G–I) Biochemical determination of the levels of Fe2+, MDA, and GSH in HKC-8 cells from indicated groups was conducted. (J) Immunofluorescence was employed for the detection of β-catenin and GPX4 in HKC-8 cells. Scale bar, 25 μm. *P < 0.05 vs. the controls; #P < 0.05 vs. homocysteine stimulation alone (n = 6). Hcy, homocysteine.
Restoration of β-catenin signaling enhanced GPX4 expression and inhibited ferroptosis induced by hyperhomocysteinemia in the kidneys
To explore the involvement of β-catenin signaling in homocysteine-induced renal ferroptosis, mice with hyperhomocysteinemia were infected with the adeno-associated virus vector expressing β-catenin. β-catenin was notably increased in the kidneys of mice after infection with β-catenin-expressing adeno-associated virus (Fig. 4A,C). The upregulated β-catenin signal augmented the expression of GPX4, concurrently downregulating MDA, Fe2+ and KIM-1 levels in the kidneys of mice with hyperhomocysteinemia (Fig. 4D,E,H,I). SLC7A11 and ACSL4 are two key genes that play critical roles in ferroptosis regulation. SLC7A11 inhibits ferroptosis through its involvement in an antioxidant pathway, while ACSL4 promotes ferroptosis by enhancing lipid peroxidation. Hyperhomocysteinemia decreased the expression level of SLC7A11 while concurrently upregulating ACSL4 expression. In contrast, β-catenin overexpression antagonized the effects of hyperhomocysteinemia by upregulating SLC7A11 expression and downregulating ACSL4 expression (Fig. 4F,G). Nevertheless, β-catenin overexpression had no effect on the levels of GSH in kidney (Fig. 4J). In addition, the immunohistochemical results demonstrated that overexpression of β-catenin elevated GPX4 levels in the renal tubular epithelial cells of mice with hyperhomocysteinemia (Fig. 4K). Further, transmission electron microscopy analysis revealed that renal tubular epithelial cells of hyperhomocysteinemic mice exhibited mitochondrial membrane disruption, mitochondrial cristae reduction and mitochondrial shrinkage, indicating ferroptosis-associated alterations; while the overexpression of β-catenin markedly attenuated this mitochondrial damage (Fig. 4L). These findings suggested that upregulation of β-catenin signaling enhanced GPX4 expression and markedly suppressed renal ferroptosis induced by hyperhomocysteinemia.

Restoration of β-catenin signaling enhanced GPX4 expression and inhibited ferroptosis induced by hyperhomocysteinemia in the kidneys. Adeno-associated virus expressing β-catenin was used to infect mice with hyperhomocysteinemia, and the effect of β-catenin overexpression on renal tubules was analyzed. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, KIM-1, SLC7A11, and ACSL4 in the kidneys of mice from indicated groups. (B–G) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, KIM-1, SLC7A11, and ACSL4 in Figure (A) was performed. (H–J) Biochemical determination of the levels of Fe2+, MDA, and GSH in the kidney tissues of mice from indicated groups was conducted. (K) Immunohistochemical analysis of β-catenin and GPX4 in the renal tissues was performed. Scale bar, 20 μm. (L) Mitochondrial morphology in renal tubular epithelial cells was observed using electron microscopy. Yellow arrows highlighted mitochondrial damage, including mitochondrial membrane rupture, reduction of mitochondrial cristae and mitochondrial shrinkage. Scale bar, 300 nm. *P < 0.05 vs. the controls; #P < 0.05 vs. hyperhomocysteinemia mice (n = 6). Hcy, homocysteine; AAV, adeno-associated virus; TEM, transmission electron microscopy.
Upregulation of β-catenin signaling enhanced GPX4 expression and markedly suppressed ferroptosis induced by elevated homocysteine levels in renal tubular epithelial cells
In vitro experiments revealed that the β-catenin signal was conspicuously upregulated in HKC-8 cells transfected with pcDNA3.1-β-catenin (Fig. 5A,C). Elevated β-catenin signaling notably boosted GPX4 and SLC7A11 expression in HKC-8 cells exposed to high homocysteine levels, while simultaneously reducing ACSL4, MDA, Fe2+, and KIM-1 concentrations (Fig. 5A,J). Nevertheless, β-catenin overexpression had no effect on the levels of FTH1 and GSH in HKC-8 cells (Fig. 5E andK). In addition, immunofluorescence staining results indicated that overexpression of β-catenin significantly upregulated GPX4 levels in renal tubular epithelial cells exposed to high homocysteine (Fig. 5L). Furthermore, transmission electron microscopy revealed that high homocysteine levels led to mitochondrial membrane disruption and mitochondrial shrinkage in renal tubular epithelial cells, indicating morphological changes linked to ferroptosis. Notably, overexpression of β-catenin mitigated these mitochondrial damage (Fig. 5M). Lipid ROS levels were assessed using the fluorescent probe BODIPY581/591 C11. Furthermore, the results revealed that homocysteine stimulation markedly increased lipid ROS levels in HKC-8 cells, whereas β-catenin overexpression significantly reduced lipid ROS levels (Fig. 5N). These results suggested that enhancing β-catenin signaling upregulated GPX4 expression and inhibited ferroptosis induced by homocysteine in renal tubular epithelial cells.

Upregulation of β-catenin signaling enhanced GPX4 expression and markedly suppressed ferroptosis in HKC-8 cells induced by elevated homocysteine levels. The β-catenin plasmid was transfected into homocysteine-treated HKC-8 cells, and the effect of β-catenin overexpression on the renal tubular epithelium was analyzed. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, FTH1, KIM-1, SLC7A11, and ACSL4 in HKC-8 cells from indicated groups. (B–H) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, FTH1, KIM-1, SLC7A11, and ACSL4 in Figure (A) was performed. (I–K) Biochemical determination of the levels of Fe2+, MDA, and GSH in HKC-8 cells from indicated groups was conducted. (L) Immunofluorescence was employed for the detection of β-catenin and GPX4 in cells. Scale bar, 25 μm. (M) Mitochondrial morphology in HKC-8 cells was examined using transmission electron microscopy. Yellow arrows highlighted mitochondrial damage, including mitochondrial membrane rupture, reduction of mitochondrial cristae and mitochondrial shrinkage. Scale bar, 300 nm. (N) Lipid ROS levels were assessed using the fluorescent probe BODIPY581/591 C11 in each group. Scale bar, 25 μm. *P < 0.05 vs. the controls; #P < 0.05 vs. homocysteine stimulation alone (n = 6). Hcy, homocysteine; pcDNA, pcDNA3.1; TEM, transmission electron microscopy.
Upregulation of GPX4 expression was capable of inhibiting ferroptosis induced by homocysteine in renal tubular epithelial cells without affecting β-catenin signaling
The aforementioned findings suggested that GPX4, serving as a downstream effector of β-catenin, exerts a key effection in the pathophysiology of homocysteine-induced ferroptosis in renal tubular epithelial cells. To investigate the role of GPX4 in homocysteine-induced ferroptosis, HKC-8 cells were transfected with pcDNA3.1-GPX4. Western blot analysis revealed that GPX4 did not influence the β-catenin signaling suppressed by homocysteine (Fig. 6A,C); nevertheless, the augmented expression of GPX4 conspicuously antagonized the effects of homocysteine, reducing intracellular levels of MDA, Fe2+, and KIM-1 (Fig. 6D,H). Nevertheless, GPX4 overexpression had no effect on the levels of FTH1 and GSH in HKC-8 cells (Fig. 6E and I). Therefore, elevating GPX4 expression inhibited homocysteine-induced ferroptosis in renal tubular epithelial cells.

Upregulation of GPX4 expression is capable of inhibiting homocysteine-induced ferroptosis in renal tubular epithelial cells without affecting β-catenin expression levels. The GPX4 plasmid was transfected into homocysteine-treated HKC-8 cells, and the effect of GPX4 overexpression on HKC-8 cells was analyzed. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in HKC-8 cells from indicated groups. (B–F) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 in Figure (A) was performed. (G–I) Biochemical determination of the levels of Fe2+, MDA, and GSH in HKC-8 from indicated groups was conducted. *P < 0.05 vs. the controls; #P < 0.05 vs. homocysteine stimulation alone (n = 6). Hcy, homocysteine; pcDNA, pcDNA3.1.
As a target gene of β-catenin, enhanced GPX4 expression counteracted the ferroptosis induced by downregulated β-catenin signaling
The results above indicated that β-catenin modulated homocysteine-induced ferroptosis through GPX4 in renal tubular epithelial cells. To elucidate the relationship between β-catenin and GPX4, we altered β-catenin expression by transfecting HKC-8 with pcDNA3.1-β-catenin and β-catenin siRNA. β-catenin overexpression led to an increase in both protein and mRNA levels of GPX4 in cells. Conversely, silencing β-catenin suppressed GPX4 expression in cells (Fig. 7A,F). Intervention experiments on β-catenin expression indicated that GPX4 was a target gene of β-catenin. Moreover, ChIP assay, combining dual luciferase reporter gene assay, confirmed the interaction between β-catenin and the GPX4 gene promoter, confirming that GPX4 acted as a target gene of β-catenin (Fig. 7G,H). To examine the roles of β-catenin and GPX4 in ferroptosis, HKC-8 cells were subjected to transfection of β-catenin siRNA and pcDNA3.1-GPX4. Silencing β-catenin reduced GPX4 expression in HKC-8 cells and elevated intracellular levels of KIM-1, Fe2+, and MDA; Overexpression of GPX4 attenuated the effects of β-catenin knockdown, decreasing intracellular levels of KIM-1, Fe2+, and MDA (Fig. 7I,O).

As a target gene of β-catenin, enhanced GPX4 expression counteracted the ferroptosis induced by downregulated β-catenin signaling. β-catenin siRNA and pcDNA-β-catenin were transfected into HKC-8 cells to assess the effect of β-catenin on GPX4 expression. (A) Western blotting was used to assess the expression of active β-catenin, β-catenin, and GPX4 in HKC-8 cells from indicated groups. (B–D) Quantitative analysis of the levels of active β-catenin, β-catenin, and GPX4 in Figure (A) was performed. *P < 0.05 vs. the controls (n = 6). (E) The mRNA levels of GPX4 in each group were analyzed using RT-PCR. (F) The mRNA levels of GPX4 in figure (E) were quantitatively analyzed. *P < 0.05 vs. the controls (n = 6). (G) ChIP analysis of the interaction between β-catenin and the promoter region of the GPX4 gene. (H) Dual-luciferase reporter gene assay was used to analyze the regulatory effect of β-catenin on GPX4. *P < 0.05 vs. the controls (n = 4). β-catenin siRNA and GPX4 plasmid were co-transfected into HKC-8 cells to assess their interaction. (I) Western blotting was used to assess the expression of active β-catenin, β-catenin, GPX4, and KIM-1 in HKC-8 cells from indicated groups. (J–M) Quantitative analysis of the levels of active β-catenin, β-catenin, GPX4, and KIM-1 in Figure (I) was performed. (N–O) Biochemical determination of the levels of Fe2+ and lipid MDA in HKC-8 cells from indicated groups was conducted. *P < 0.05 vs. the controls; #P < 0.05 vs. β-catenin siRNA transfection (n = 6). Sc-siR, scramble siRNA; β-catenin siR, β-catenin siRNA; pcDNA, pcDNA3.1.
Discussion
Elevated homocysteine levels are strongly linked to a higher risk of chronic kidney disease15. Folic acid and B vitamins demonstrate therapeutic efficacy against hyperhomocysteinemia; however, genetic variability and concomitant diseases can compromise treatment efficacy in some patients16. It is increasingly urgent to elucidate the mechanism of homocysteine-induced renal injury and develop effective renoprotective therapies. Our study reveals that homocysteine induces kidney injury via ferroptosis in renal tubular epithelial cells, mediated through the β-catenin/GPX4 pathway (Fig. 8).

A graphical abstract of the mechanism underlying homocysteine-induced ferroptosis in renal tubular epithelial cells.
The detrimental effects of homocysteine are closely linked to GPX4 inactivation, with reactive oxygen species (ROS) playing a pivotal mediating role17,18. Ferroptosis is characterized by augmented oxidative stress within mitochondria and increased lipid peroxidation, marked by elevated levels of ROS in cells19. Therefore, the damage mechanism of homocysteine shares a similar molecular basis with the pathophysiological process of ferroptosis. Our study reveals that homocysteine suppresses GPX4 expression in renal tubular epithelial cells. In the ferroptosis signaling pathway, GPX4 reduces lipid ROS by utilizing glutathione, alleviates oxidative stress in mitochondria, and consequently suppresses ferroptosis20. Additionally, our research demonstrates that homocysteine promotes ferroptosis in renal tubular epithelial cells. Furthermore, overexpression of GPX4 in renal tubular epithelial cells significantly inhibits homocysteine-induced ferroptosis. Thus, it is likely that homocysteine promotes ferroptosis in renal tubular epithelial cells by inhibiting GPX4 expression.
Simultaneously, our study found that homocysteine inhibits β-catenin signaling in renal tubular epithelial cells. In normal cells, β-catenin signaling is sustained at a low baseline level, yet it is essential for preserving cellular physiological functions and balance of energy metabolism21. Under conditions of inflammation or oxidative stress, activation of the β-catenin signal exerts a crucial role in facilitating cell survival and repair22,23. Encouragingly, overexpression of β-catenin enhances GPX4 expression, thereby inhibiting homocysteine-induced ferroptosis. Further ChIP experiments and dual-luciferase reporter gene assays confirm that β-catenin transcriptionally regulates the target gene GPX4, thereby conferring resistance to ferroptosis.
Previous studies have demonstrated that β-catenin directly regulates GPX4 expression at the transcriptional level, which is consistent with the findings of the present study6. Additionally, β-catenin also exerts an indirect regulatory effect on GPX4 expression through TP63, contributing to the modulation of ferroptosis7. The β-catenin/GPX4 axis plays a critical role in homocysteine-induced ferroptosis, a process mediated by redox dysregulation. Nuclear factor erythroid 2-related factor 2 (Nrf2) and the cystine/glutamate antiporter system (System Xc−) are key regulators of cellular redox homeostasis and are mechanistically linked to GPX4-dependent ferroptosis24. Accumulating evidence indicates that the Nrf2/System Xc−/GPX4 signaling axis represents a central pathway governing ferroptotic cell death25. However, the precise mechanisms underlying homocysteine-triggered ferroptosis remain incompletely understood, suggesting the involvement of additional regulatory factors that warrant further experimental investigation.
The accumulation of ferrous ions represents a hallmark change during cellular ferroptosis. This study found that homocysteine can lead to elevated levels of divalent iron ions in epithelial cells of the renal tubules. That finding warrants further investigation to elucidate its underlying mechanisms and implications.
The research findings establish that ferroptosis represents a critical pathogenic mechanism underlying homocysteine-induced nephrotoxicity. Furthermore, this study elucidates the specific mechanism through which homocysteine promotes ferroptosis. The findings offer new perspectives on the mechanisms by which hyperhomocysteinemia induces renal injury.
Conclusion
Homocysteine induces ferroptosis in renal tubular epithelial cells by disrupting the β-catenin signaling pathway and suppressing the expression of its target gene, GPX4. The findings demonstrate that ferroptosis represents a critical mechanism by which homocysteine induces renal injury, suggesting a potential preventive strategy for kidney damage associated with hyperhomocysteinemia.
Methods
Animal models
Male C57BL/6J mice (8 weeks old, weighing 15–20 g) under specific pathogen-free (SPF) conditions were obtained from Hunan Silaike Jingda Laboratory Animal Co. Ltd (Changsha, China). Laboratory mice were assigned into indicated groups randomly (n = 6): (1) the control group; (2) mice model of hyperhomocysteinemia (Homocysteine was introduced into the drinking water at a level of 1.8 g/L); (3) model mice received Fer-1 treatment (Model mice received intraperitoneal administration of Fer-1 at a dosage of 2 mg/kg/day); (4) mice received Fer-1 treatment (Mice received intraperitoneal administration of Fer-1 at a dosage of 2 mg/kg/day) (5) hyperhomocysteinemia model of mice infected with AAV-β-catenin (5 × 1011 adeno-associated virus-β-catenin were suspended in 100 µl saline and administered via the tail vein two weeks prior to the establishment of hyperhomocysteinemia model); (6) hyperhomocysteinemia model of mice infected with AAV-control. After the 4th week, mice were humanely euthanized via subcutaneously injecting pentobarbital sodium (30 mg/kg). All animal experiments had been approved by the Animal Ethics Committee at the University of South China, located in Hengyang, China. All methods were carried out in accordance with relevant guidelines and regulations and reported in accordance with ARRIVE guidelines.
Cell culture
Human proximal tubular epithelial (HKC-8) cells were purchased from the Chinese National Infrastructure of Cell Line Resource (NICR). HKC-8 cells were cultivated in DMEM/F-12 medium (Gibco, USA) enriched with 10% bovine serum. HKC-8 cells underwent serum-starvation for 12 h prior to a variety of treatments. Cells were exposed to homocysteine (500 µmol/L). Fer-1 (10 µmol/L) were administered as the treatments, and the incubation time was 12 h. Then, cells were collected for immunofluorescence staining, electron microscope examination, and western blot, respectively.
Cell transfection with plasmids or SiRNAs
HKC-8 cells were cultivated on 6-well plates and prepared for transfection after the cell confluence reaching approximately 80%. HKC-8 were transfected using lipofectamine with pcDNA3.1-β-catenin (2.5 µg) or pcDNA3.1-GPX4 (2.5 µg). A total of 75 pmol of small interfering RNA targeting β-catenin (β-catenin-siRNA) was incubated with liposome for 20 min at room temperature to generate liposome complex. The incubation of cells and liposome complex lasted for 6 hours. Opti-MEM (Gibco, USA) and Lipofectamine 3000 (Invitrogen, USA) were utilized for the transfection of plasmids and siRNAs. The siRNA sequences were as follows: the β-catenin-siRNA sequence 5′-GCCUCUGAUAAAGGCAACUTT-3′, and the β-catenin scramble sequence 5′-CAGUACUUUUGUGUAGUACAA-3′.
Western blot
Western blotting was performed for protein quantification following standard procedures. The following primary antibodies were employed in the study: active β-catenin (#19807, Cell Signaling Technology), β-catenin (610154; BD Biosciences), GPX4 (Ab125066, Abcam), KIM-1 (Ab233720, Abcam), FTH1 (Ab183781, Abcam) and β-actin (AA128, Beyotime). The levels of protein expression were normalized using β-actin.
Immunohistochemical and immunofluorescent staining
The preparation of paraffin sections was conducted following the protocol as previously described26. HKC-8 cells were seeded on the coverslips. Both tissue and cells sections were immunostained for target proteins with primary antibodies for β-catenin (610154; BD Biosciences, CA) and GPX4 (Ab125066; Abcam). Images of immunohistochemical staining were acquired on a bright-field microscope (Nikon). All micrographs of immunofluorescent staining were acquired on a Leica fluorescence microscope.
GSH (glutathione) assay
The levels of GSH were tested with a kit (#A006-2, Jiancheng, Nanjing). The levels of GSH were tested by a spectrophotometer at 420 nm. The GSH levels were normalized to the total protein concentration.
Labile iron assay
Labile iron concentrations in cells and kidney tissue were assessed with a kit (Abcam, #ab83366), following the instructions of manufacturer.
Lipid ROS assay
The concentration of lipid ROS was tested by BODIPY581/591 C11, a fluorescent probe (D3861, Invitrogen, USA). HKC-8 cells were seeded in 6-well plates and transfected with pcDNA3.1-β-catenin, then stimulated with homocysteine, and incubated with BODIPY581/591 C11 at a concentration of 10 µM for 30 min in darkness. HKC-8 cells were visualized on the fluorescence microscope (Leica, Wetzlar, Germany). Malondialdehyde (MDA) was quantified via thiobarbituric acid to detect lipid ROS and MDA assay kit was purchased from Beyotime (Nanjing, China).
Chromatin immunoprecipitation (ChIP)
The pcDNA3.1-β-catenin was utilized to transfected HKC-8 cells. After 48 hours, cells underwent being fixed in 4% formaldehyde buffer to facilitate protein-DNA crosslinking. ChIP was carried out by utilizing the SimpleChIP Plus Kit (Cat. 9005, Cell Signaling). Cells were lysed using an ultrasonic disruptor at 30% amplitude, with 10-second pulses interspersed by 30-second cooling intervals on ice (15 cycles total). Chromatin fragments (150 µg) were then immunoprecipitated with 5 µg of target-specific antibody. The antibody against β-catenin (ab32572, abcam), RNAP II (RNA polymerase II), and rabbit IgG were added for incubation overnight at 4 ℃. Subsequently, protein A-agarose was added and incubated for 1 h. After washing, the purified DNA was used for PCR amplification. The primer pair designed for the GPX4 gene promoter included: forward primer 5′- AAGCCAGGTTTCCTTGTGTG-3′ and reverse primer 5′-ATGCCTGTGACTGTACATGC-3′.
RT-PCR
Total RNA in cells was extracted using TRIzol reagent. 2 µg of RNA were added as templates to a 20 µl reverse transcription reaction system (RK20433, ABclonal) to synthesize the first strand cDNA. RT-PCR was conducted on an Applied Biosystems (2720, America). RT-PCR was conducted following the protocol as previously described26. The primer pair designed for GPX4 PCR was listed as follows: sense primer 5′-CAACCAGTTCGGGAGGCAGGAG-3′ and antisense primer 5′-TGGGCTGGACTTTCATCCATTTC-3′.
Dual luciferase reporter gene assay
Dual luciferase reporter gene assay kit and reporter gene plasmids (pGL6-TA and pRL-SV40-C) were purchased from Beyotime Biotechnology. Both the pGL6-TA-GPX4 promoter and pRL-SV40-C were co-transfected into HEK293T cells along with pcDNA3.1-β-catenin. After 48 h of transfection, the cells were subjected to lysis and centrifugation to obtain the supernatant. Samples were loaded into a 96-well microplate, and fluorescence intensity was determined on a luminometer (Bio Tek, Synergy, USA). Luciferase activity was evaluated by the intensity of firefly fluorescence, which was standardized by the intensity of Renilla fluorescence.
Statistical analyses
All data were expressed as mean ± SEM in our study. Statistical analysis was perform with GraphPad Prism (La Jolla, CA). The comparison among groups was implemented with one-way ANOVA. And statistical analyses between two groups was carried out by Student-Newman-Kuels test. P < 0.05 was a significant difference of statistic.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author upon reasonable request.
References
Yang, B. et al. Prevalence of hyperhomocysteinemia in china: a systematic review and meta-analysis. Nutrients. 7, 74–90 (2014).
Wu, D. F. & Yin, R. X. Homocysteine, hyperhomocysteinemia, and H-type hypertension. Eur. J. Prev. Cardiol. 31, 1092–1103 (2024).
Zhang, M. et al. Hyperhomocysteinemia exacerbates ischemia-reperfusion injury-induced acute kidney injury by mediating oxidative stress, DNA damage, JNK pathway, and apoptosis. Open. Life Sci. 16, 537–543 (2021).
Pushpakumar, S., Kundu, S. & Sen, U. Hydrogen sulfide protects hyperhomocysteinemia-Induced renal damage by modulation of Caveolin and eNOS interaction. Sci. Rep. 9, 2223 (2019).
Angelini, A. et al. The link between homocysteine, folic acid and vitamin B12 in chronic kidney disease. G Ital. Nefrol. 38, 2021–vol2024 (2021).
Wang, Y. et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell. Death Differ. 29, 2190–2202 (2022).
Guo, S. et al. The malignancy suppression and ferroptosis facilitation of BCL6 in gastric cancer mediated by FZD7 repression are strengthened by RNF180/RhoC pathway. Cell. Biosci. 13, 73 (2023).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149, 1060–1072 (2012).
Wang, Z., Wang, B. & Jin, X. Amentoflavone attenuates homocysteine-induced neuronal ferroptosis-mediated inflammatory response: involvement of the SLC7A11/GPX4 axis activation. Brain Res. Bull. 215, 111005 (2024).
Li, X. et al. Linking homocysteine and ferroptosis in cardiovascular disease: insights and implications. Apoptosis. 29, 1944–1958 (2024).
Shi, J., Chen, D., Wang, Z., Li, S. & Zhang, S. Homocysteine induces ferroptosis in endothelial cells through the systemXc(-)/GPX4 signaling pathway. BMC Cardiovasc. Disord. 23, 316 (2023).
Balatskyi, V. V. & Palchevska, O. L. β-Catenin regulates cardiac energy metabolism in sedentary and trained mice. Life (Basel). 10, 357 (2020).
Xu, Z. H. et al. Hypoxia-inducible factor protects against acute kidney injury via the Wnt/β-catenin signaling pathway. Am. J. Physiol. Ren. Physiol. 322, F611–f624 (2022).
Ming, W. H. et al. The crosstalk of Wnt/β-catenin signaling and p53 in acute kidney injury and chronic kidney disease. Kidney Res. Clin. Pract. 43, 724–738 (2024).
Chen, W. et al. Association of hyperhomocysteinemia and chronic kidney disease in the general population: a systematic review and meta-analysis. BMC Nephrol. 24, 247 (2023).
Christen, W. G., Ajani, U. A., Glynn, R. J. & Hennekens, C. H. Blood levels of homocysteine and increased risks of cardiovascular disease: causal or casual? Arch. Intern. Med. 160, 422–434 (2000).
Majumder, A. Targeting homocysteine and hydrogen sulfide balance as future therapeutics in cancer treatment. Antioxid. (Basel). 12, 1520 (2023).
Zhang, X. et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 160, 552–565 (2020).
Yu, Y. et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell. Death Discov. 7, 193 (2021).
Fujii, J. & Imai, H. Oxidative metabolism as a cause of lipid peroxidation in the execution of ferroptosis. Int. J. Mol. Sci. 25, 7544 (2024).
Balatskyi, V. V. et al. Cardiac-specific β-catenin deletion dysregulates energetic metabolism and mitochondrial function in perinatal cardiomyocytes. Mitochondrion. 60, 59–69 (2021).
Jiang, Z. & Zhou, W. A protective role of canonical Wnt/β-Catenin pathway in pathogenic bacteria-induced inflammatory responses. Mediat. Inflamm. 2024, 8869510 (2024).
Vallée, A., Vallée, J. N. & Lecarpentier, Y. Potential role of Cannabidiol in parkinson’s disease by targeting the WNT/β-catenin pathway, oxidative stress and inflammation. Aging (Albany NY). 13, 10796–10813 (2021).
Yang, R. et al. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine. 122, 155135 (2024).
Yuan, Y., Zhai, Y., Chen, J., Xu, X. & Wang, H. Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules. 11, 923 (2021).
Zhao, Y., Lei, Y., Li, Y., Zhang, J. & Tang, H. Wnt/β-catenin signalling mediates cardiac hypertrophy in type 4 cardiorenal syndrome. Nephrology. 26, 549–560 (2021).
Funding
This work was funded by Natural Science Foundation of Hunan Province (2021JJ40476 & 2021JJ40506 & 2022JJ30528), Scientific Research Project of Hunan Provincial Health Commission (202203014395), Scientific Research Project of Hunan Provincial Department of Education (21B0414), the Scientific Research Fund Project of Hunan Provincial Health Commission (Grant no.C202303019182), National Key Clinical Specialty Scientific Research Project (Grant no. Z2023006) and National Natural Science Foundation of China (82100499).
Author information
Authors and Affiliations
Contributions
.Yanping Lei: investigation, funding acquisition, methodology, visualization, data curation, writing-original draft. Hengjing Hu: funding acquisition, writing-review and editing. Huifang Tang: writing-review and editing. Lewu Xu: funding acquisition, writing-review and editing, supervision. Rui Liu: methodology, investigation. Yue Zhao: supervision, funding acquisition, project administration. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study protocol was reviewed and approved by the Animal Care and Use Committee of University of South China (Ethics approval number: USC2023XS087).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lei, Y., Hu, H., Tang, H. et al. Homocysteine induces ferroptosis in renal tubular epithelial cells via β-catenin/GPX4 signaling pathway. Sci Rep 15, 23953 (2025). https://doi.org/10.1038/s41598-025-09221-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-09221-6
