
Abstract
The mechanisms underlying mitophagy and mitochondrial dynamics (MD) in cervical cancer (CC), a disease with a high mortality rate, remain poorly understood. This study aimed to assess the prognostic significance of these processes in CC. Mendelian randomization (MR) and 101 machine learning models were employed to identify mitophagy- and MD-associated prognostic genes in CC. A subsequent risk model was developed to stratify patients by risk. Further analyses included functional pathway enrichment, immune infiltration, and single-cell RNA sequencing (scRNA-seq) analysis. The results identified PLOD3, SBK1, and SLC39A10 as prognostic genes for CC. Among these, PLOD3 and SLC39A10 were associated with poor prognosis, while SBK1 was protective. The risk model demonstrated high accuracy, with area under the curve (AUC) values exceeding 0.6. Following this, a prognostic nomogram was constructed incorporating risk score and pathological T stage, achieving high predictive accuracy. Gene Set Enrichment Analysis (GSEA) revealed significant enrichment in pathways such as ECM receptor interaction and olfactory transduction in high-risk groups. Additionally, SBK1 showed the strongest correlation with neutrophil infiltration. Expression pattern alterations of prognostic genes were observed in endothelial cells, T cells, and epithelial cells. In conclusion, a risk model based on mitophagy- and MD-related prognostic genes was established, offering a promising approach for the personalized management of patients with CC.
Similar content being viewed by others

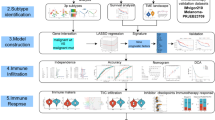
Introduction
Cervical cancer (CC) is a prevalent malignancy with high morbidity and mortality among women1. Persistent infection with high-risk human papillomavirus (HPV) is a key factor in the development of CC, with adolescent HPV vaccination capable of preventing over 90% of cervical precancers and cancers2. However, additional factors, including genetic susceptibility, environmental triggers, and epigenetic mechanisms, also contribute to CC onset. Despite advancements in surgical treatments, chemotherapy, radiation, and targeted therapies, advanced-stage patients continue to face significant challenges such as recurrence, metastasis, and drug resistance1,3. Consequently, there is an urgent need to further investigate the pathogenesis of CC and identify new prognostic genes to better predict outcomes.
Mitochondria, essential dynamic organelles with a double-layered membrane, play a pivotal role in regulating various cellular functions, including metabolism, energy production, calcium ion storage, cell differentiation, proliferation, aging, and apoptosis4. Mitophagy, a cellular protective mechanism, selectively engulfs and degrades dysfunctional mitochondria through autophagy, thereby maintaining cellular homeostasis5. Mitochondrial dynamics (MD) encompasses changes in fission, fusion, mitophagy, and transport, all of which are vital for optimal signal transduction and metabolic function6. Recent studies highlight the significant role of MD and mitophagy in tumor development and progression7. Impaired phagocytic activity has been shown to negatively impact cellular health8. For instance, cisplatin-induced or P53-mediated mitochondrial apoptosis plays a decisive role in the chemosensitivity of gynecological cancer cells5,9. Therefore, exploring the role and prognostic potential of mitochondrial dynamic genes (MDGs) in CC pathogenesis could provide novel targets for prevention, diagnosis, and treatment. Although substantial evidence links mitophagy dysfunction to tumorigenesis, the specific mechanisms of mitophagy-related genes (MRGs) in CC remain unclear7,8. Thus, further investigation into the prognostic significance and regulatory mechanisms of MRGs in CC is warranted.
Recent advancements in genomics have provided substantial evidence of the role genetics plays in the etiology of diseases. Mendelian randomization (MR), an epidemiological approach, leverages genetic variations to assess the impact of exposure factors on outcomes10. MR relies on genome-wide association studies (GWAS) and utilizes genetic variations as instrumental variables (IVs), which are strongly associated with exposure factors, unaffected by confounding variables, and unrelated to the outcome event itself11. Single-cell RNA sequencing (scRNA-seq) technology enables the exploration of cellular heterogeneity, key subpopulations, states, and differentiation trajectories. Its development has made it possible to identify previously undetected disease-related cell populations, functional states, markers, and potential molecular regulators12. Consequently, this study integrates scRNA-seq data and MR to identify genes associated with CC prognosis and to elucidate the molecular mechanisms of MD and mitophagy-related genes in CC.
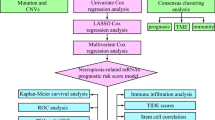
To investigate the relationship between MD, mitophagy, and CC, this study retrieved CC-related genes from public databases and employed bioinformatics approaches, including differential expression analysis, weighted gene co-expression network analysis (WGCNA), univariate Cox regression, MR, and 101 machine learning models, to identify prognostic mitochondrial and MD-related genes in CC and develop a risk model. The model was then validated using an independent dataset. Additionally, independent prognostic analysis, functional enrichment analysis, immune microenvironment assessment, and drug sensitivity analysis were performed, and the expression of prognostic genes was examined at the scRNA-seq level. Finally, the expression of these genes was evaluated in tissue samples. Through these methodologies, this study aims to deepen the understanding of the relationship between MD, mitophagy, and CC, providing valuable scientific evidence and guidance for future clinical practice.
Results
Identification of candidate genes
The differential expression analysis identified 2793 differentially expressed genes (DEGs), with 1764 genes upregulated and 1,029 genes downregulated in the CC group (Fig. 1a,b). Notably, the low score group of MDGs exhibited significantly improved overall survival (OS) (P = 0.0493), whereas the high MRG score group demonstrated substantially better OS (P = 0.0217), suggesting a potential correlation between MD, mitophagy, and the prognosis of individuals diagnosed with CC (Fig. 1c,d). In the weighted gene co-expression network analysis (WGCNA), no outliers were detected, and the soft threshold was determined to be 6 (R2 = 0.8630), which led to the identification of eight co-expression modules (Fig. 1e–g). The MEblue module showed the strongest correlation with MDGs and MRGs, marking it as a key module (MDGs: cor = −0.33, P = 1 × 10–8; MRGs: cor = 0.39, P = 3 × 10–12). As a result, 2821 key module genes were identified (Fig. 1h). These key module genes were intersected with the 2793 DEGs to generate 274 candidate genes associated with MD and mitophagy in CC (Fig. 1i). Gene Ontology (GO) enrichment analysis of these candidate genes revealed significant enrichment in processes such as epithelial tube morphogenesis, distal axon, and anion transmembrane transporter activity (P < 0.05) (Figure S1a,b). Additionally, several Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, including axon guidance, regulation of the actin cytoskeleton, and FcγR-mediated phagocytosis, were notably enriched (P < 0.05) (Figure S1c,d). A protein-protein interaction (PPI) network was constructed using the candidate genes, excluding 123 outlier genes (Figure S2). The final network contained 151 nodes and 197 edges, with an average node degree of 1.44, a local clustering coefficient of 0.331, and an enrichment P-value of 1.12 × 10–8. PXDN and ESR1 exhibited strong interactions with other genes.

Identification of candidate genes. (a,b) Volcano plot and heatmap of differentially expressed genes. (c,d) ssGSEA scores of Kaplan-Meier curves (c MDGs, d MRGs). (e–g) Sample clustering and trait heatmap (e), soft threshold selection (f), identification of co-expression modules (g). (h) Heatmap of relationships between modules and ssGSEA. (i) Venn diagram of candidate genes. All P-values < 0.05.
PLOD3, SBK1, and SLC39A10 were prognostic genes for CC
Univariate Cox regression analysis identified 18 genes related to OS (Fig. 2a). Of these, SH3GLB2, SLC25A14, ROGDI, TMEM116, SLC25A33, SLC25A42, GAD1, E2F1, SBK1, KIAA1211L, and FAM3B were determined to be protective factors for CC (HR < 1, P < 0.05), while PXDN, SBSPON, SHANK2, SLC39A10, FAR2, FUCA2, and PLOD3 were identified as risk factors (HR > 1, P < 0.05).

Identification of prognostic genes. (a) Univariate Cox regression analysis of prognostic genes. (b) Exposure factors for MR analysis. (c) C-index values from 101 machine learning models. All P-values < 0.05.
These 18 genes associated with OS were treated as exposure factors for MR analysis (Table S1). PLOD3, SLC39A10, ROGDI, and SBK1, all showing a significant causal relationship with CC, were identified (P < 0.05 in inverse variance weighted [IVW]) (Fig. 2b, Table S2). PLOD3 (OR (95% CI) = 1.54112 (1.15676–2.05321), P = 0.00313) and SLC39A10 (OR (95% CI) = 1.13572 (1.10467–1.16763), P < 0.001) were identified as risk factors (OR > 1), while ROGDI (OR (95% CI) = 0.93473 (0.89973–0.97108), P < 0.001) and SBK1 (OR (95% CI) = 0.89944 (0.84308–0.95958), P = 0.00133) were protective factors (OR < 1). The slopes of PLOD3 and SLC39A10 in the scatter plot showed positive trends, with the effect sizes of most SNPs in the forest plot exceeding 0. In contrast, ROGDI and SBK1 exhibited negative slopes, with the effect sizes predominantly below 0, confirming the previous findings (Figures S3,S4). The SNP distribution in the funnel plot was symmetrical, consistent with Mendel’s second law (Figure S5). In the heterogeneity test, P-values for ROGDI, SBK1, and SLC39A10 were greater than 0.05, while the P-value for PLOD3 was less than 0.05 (Table S3). However, the IVW method (random-effects model) indicated that heterogeneity did not significantly affect the results, indicating a reliable estimation of the causal effect. Additionally, the P-values for PLOD3, SLC39A10, ROGDI, and SBK1 all exceeded 0.05, suggesting the absence of horizontal pleiotropy, fulfilling the exclusionary assumption that the instrumental variables affect the outcome only through exposure factors (Table S4). Sensitivity analysis, which involved gradually eliminating each SNP, showed no substantial change in the results, confirming the reliability and stability of the MR analysis (Figure S6). The causal direction for all four genes was confirmed as true, and all were retained as causal genes (Table S5). In summary, the results of this study satisfied the core assumptions of MR analysis in terms of relevance, independence and exclusivity, ensuring the validity and reliability of causal inference.

Construction and verification of risk model. (a–f) CC patient risk profile, survival status, Kaplan-Meier analysis, and validation. (g–h) ROC curve and validation. (i,j) Prognostic gene expression heatmap and validation. All P-values < 0.05.

Prognostic nomogram. (a,b) Univariate and multivariate Cox regression analysis. (c) Nomogram diagram. (d) Calibration curve. (e) Decision curve analysis (DCA). (f) Differences in risk scores for clinical features. (g) Prognostic gene expression heatmap in clinical feature groups. All P-values < 0.05.
Subsequently, all four causal genes were used in machine learning to generate 101 consensus models. The StepCox[backward] + RSF model outperformed the others, achieving the highest C-index in the TCGA-CESC training set (C-index = 0.921) and maintaining a C-index above 0.6 in the GSE52903 validation set (C-index = 0.611) (Fig. 2c). Therefore, this model was selected to identify three prognostic genes: PLOD3, SBK1, and SLC39A10.
The accuracy of the risk model was commendable
The risk models constructed for GSE52903 and TCGA-CESC demonstrated exceptional accuracy. High-risk patients exhibited higher mortality rates and significantly reduced OS (P < 0.05) (Fig. 3a–f). In addition, at the 1-, 3-, and 5-year time points, the area under the curve (AUC) values of the job characteristic curves (ROCs) for all subjects in the training set were higher than 0.9, whereas the AUC values in the validation set were all over 0.6 (Fig. 3g,h). Furthermore, the expression of SBK1 was lower in high-risk patients, while the expression levels of PLOD3 and SLC39A10 were higher (Fig. 3i,j).
The predictive accuracy of the prognostic nomogram was higher
After screening, both risk score and pathological T were identified as independent prognostic factors and used to construct a prognostic nomogram (Fig. 4a,b, Table S6). Each factor corresponded to a specific point, and the sum of the points represented the total score (Fig. 4c). Higher total scores were associated with poorer OS. The calibration curve demonstrated a strong agreement between the predicted probabilities of 1, 3, and 5-year OS and the reference line, indicating high predictive accuracy of the nomogram (Fig. 4d). Decision curve analysis showed a notable deviation from the two extreme curves, supporting the robust predictive performance of the nomogram (Fig. 4e). Moreover, the risk score exhibited significant differences among subgroups based on pathological T/M stage and smoking status (Fig. 4f). The heatmap depicted the expression patterns of prognostic genes across various clinical subgroups (Fig. 4g).
Pathways and immune cells related to the prognosis of patients with CC
Gene Set Enrichment Analysis (GSEA) revealed significant enrichment of pathways such as extracellular matrix (ECM) receptor interaction, complement and coagulation cascade, and Nod-like receptor signaling pathway in high-risk samples. In contrast, low-risk samples showed enrichment in pathways related to the metabolism of foreign substances by cytochrome P450, olfactory transduction, and drug metabolism by cytochrome P450, among others (Fig. 5a, Table S7). These pathways may play critical roles in how mitochondrial dynamics influence the prognosis of patients with CC.

Immune cells and pathways associated with prognosis. (a) GSEA enrichment analysis. (b) Analysis of immune cell differences. (c) Correlation analysis between prognostic genes and differential immune cells. (d) Immune checkpoint expression. (e) Drug sensitivity analysis. All P-values < 0.05.
The immune microenvironment significantly impacts cancer prognosis. The infiltration of immune and stromal cells in patients with CC was analyzed, revealing significant differences in three DICs (differential immune cell types) between high-risk and low-risk groups: gammadelta T cells, neutrophils, and regulatory T cells (Fig. 5b). Notably, SBK1 and SLC39A10 were negatively correlated with these three DICs, with the strongest correlation observed between SBK1 and neutrophils (cor = −0.56, P < 0.05) (Fig. 5c). Moreover, significant differences were found in the expression of six immune checkpoint genes (BTLA, CD28, LGALS9, NRP1, CD40LG, and TNFSF18) between high-risk and low-risk groups (P < 0.05) (Fig. 5d).
Drug sensitivity analyses
Additionally, 46 drugs showed significant differences in sensitivity between the high-risk and low-risk groups (Fig. 5e). X681640, vinorelbine, paclitaxel, and other drugs were more effective in the low-risk group, while Z.LLNle.CHO, WO2009093972, WH.4.023, and others were more effective in the high-risk group. These findings provide valuable insights for the personalized treatment of patients with CC.
Endothelial cells, T cells, and epithelial cells were involved in CC prognosis
After quality control, 45,270 cells and 27,613 genes were successfully retained (Figure S7). The top 2000 genes exhibiting the highest variation were selected for principal component analysis (PCA), and the initial 30 principal components (PCs) were chosen based on the scree plot for cluster analysis, resulting in the identification of 26 distinct cell clusters (Fig. 6a–e). Following this, 9 cell types were annotated, including T cells, natural killer (NK) cells, B cells, myeloid cells, endothelial cells, epithelial cells, mast cells, neutrophils, and smooth muscle cells (Fig. 6f). A bubble map was generated to display marker gene expression levels for each specific cell type (Fig. 6g). These 9 cell types were enriched in pathways such as LPS transfer from LBP carrier to CD14, hydroxycarboxylic acid-binding receptors, and NEIL3-mediated resolution of interstrand crosslinks (ICLs) (Fig. 6h). Notably, the expression of three prognostic genes was significantly elevated in endothelial cells, epithelial cells, and smooth muscle cells (Fig. 7a,b). The expression of SBK1 and SLC39A10 was notably different between control and CC samples in endothelial cells and T cells, while PLOD3 and SLC39A10 showed significant differences in epithelial cells. Therefore, these three cell types were identified as key contributors (P < 0.05) (Fig. 7c). Additionally, cell communication analysis revealed numerous interactions, particularly between epithelial cells and endothelial cells (Fig. 7d–f). The interaction between endothelial cells and neutrophils was the most prominent.

Prognostic cells. (a–e) High-variance gene screening (a), cell function enrichment (b), lithotriptic plan (c), PCA showing no clear boundary between samples (d), UMAP clustering (e). (f) Annotated UMAP cluster map. (g) Marker gene expression. (h) Single-cell enrichment analysis. All P-values < 0.05.

Expression of prognostic genes and cellular communication. (a,b) Expression of prognostic genes (PLOD3, SBK1, SLC39A10) in all cell types (a) and differential cells (b). (c) Identification of key cell clusters. (d–f) Cell communication interaction d,e), and number of key cell communications (d). All P-values < 0.05.
In pseudo-time analysis, endothelial cells differentiated into five distinct states, with the temporal progression showing upregulation of prognostic genes from low to high expression levels (Fig. 8a–c). Epithelial cells differentiated into three distinct states, exhibiting a temporal shift from high to low expression of prognostic genes (Fig. 8d–f). Furthermore, as T cells underwent differentiation into five distinct states, SBK1 expression decreased, while PLOD3 and SLC39A10 expression increased (Fig. 8g–i). Dynamic heatmaps were generated to show the expression changes of the top 50 genes and the results of GO enrichment analysis (Figure S8). The observed alterations in the expression patterns of prognostic genes across these three key cell types highlight their involvement in the prognosis of patients with CC.

Pseudo-time analysis for key cells. (a–c) Pseudo-time analysis for endothelial cells. (d–f) Pseudo-time analysis for epithelial cells (d–f). (g–i) Pseudo-time analysis for T cells.
Validation of clinical expression of prognostic genes
Finally, the expression levels of PLOD3 and SLC39A10 were significantly upregulated in CC clinical samples, while SBK1 expression was significantly downregulated (Figure S9). Meanwhile, IHC analysis revealed that PLOD3 and SLC39A10 were significantly overexpressed in cancer tissues (Figure S10).
Discussion
CC is the fourth most common cancer among women worldwide, with treatment outcomes remaining unsatisfactory due to an incomplete understanding of its pathogenesis1. This underscores the urgent need for novel therapeutic approaches. Recent studies have revealed that MD and mitophagy are implicated in numerous tumors, with MD particularly linked to chemotherapy resistance in CC4. This study identified PLOD3, SBK1, and SLC39A10 as prognostic genes associated with MD and mitosis in CC. A risk model was developed, achieving AUC values of 0.9 in the training set (TCGA-CC) and > 0.6 in the validation set (GSE52903). Compared to the traditional Cox regression model based on glycolytic genes by Cai et al.13, which reported AUCs > 0.7 and > 0.6, this study innovatively integrated transcriptomic and single-cell sequencing data. By employing a combination of 101 machine learning algorithms and Mendelian randomization analysis, the model not only improved predictive accuracy but also provided more comprehensive and in-depth prognostic insights. This approach offers a novel alternative for the stratified management of CC patients. Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3 (PLOD3) is an enzyme involved in collagen modification, catalyzing hydroxylation of lysine residues to influence collagen cross-linking and structural stability14,15. Research indicates that PLOD3 plays a significant role in cancer initiation and progression by modulating extracellular matrix (ECM) stability and enhancing tumor cell invasiveness15,16. Furthermore, a close relationship exists between the ECM and mitochondrial function. Mutations in mitochondrial membrane proteins can alter ECM composition, thereby influencing disease severity17. Additionally, ECM stiffness can regulate mitochondrial dynamics through the integrin-cytoskeleton complex, impacting cellular energy metabolism18. These findings suggest that PLOD3 may exert its influence in cancer through the interplay between ECM and mitochondrial function. Solute carrier family 39 member 10 (SLC39A10) belongs to the solute carrier transporter family and is primarily responsible for the transmembrane transport of metal ions. SLC39A10 plays a pivotal role in zinc ion transport and cellular zinc homeostasis, thereby affecting cellular physiology and metabolism19. In gastric cancer, the activation of MAPK/ERK and PI3K/AKT pathways forms a malignant feedback loop with SLC39A10, promoting tumor progression19,20. High expression levels of both PLOD3 and SLC39A10 have been shown to facilitate tumor growth and serve as risk factors for poor prognosis14,21. The present study identified for the first time the differential expression of PLOD3 and SLC39A10 in CC, linking it to MD and mitophagy. The upregulation of PLOD3 and SLC39A10 correlates with poor prognosis, positioning them as risk factors for CC. SBK1, encoding Symbionin-like protein 1, is involved in cell signaling and metabolic regulation, though its precise roles and mechanisms require further investigation22,23. SBK1 exhibits high expression in CC tissues and cells and has been implicated in cervical tumorigenesis by activating the Wnt/β-catenin and Raf/ERK1/2 signaling pathways24. Our study further confirms that SBK1 acts as a protective factor for CC prognosis. RT-qPCR and IHC validation of the expression levels of PLOD3, SLC39A10, and SBK1 in clinical CC samples and adjacent tissues corroborates previous findings. Thus, we hypothesize that PLOD3, SBK1, and SLC39A10 may directly contribute to the initiation and progression of CC. However, this hypothesis requires further validation through functional experiments, such as gene knockout and overexpression studies, to elucidate the specific mechanisms by which these genes operate in CC. Additionally, assessing the expression levels of PLOD3, SBK1, and SLC39A10 in CC patients could not only help evaluate their prognostic value but also identify potential molecular targets for personalized therapy. Future research should integrate functional experiments with clinical validation to comprehensively uncover the biological significance of these genes in CC.
GSEA revealed that the ECM receptor interaction, complement and coagulation cascade, and NOD-like receptor signaling pathway were significantly correlated with CC prognosis. Nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3), a key member of the NOD-like receptor (NLR) family, is a key component of innate immunity. NLRP3 is linked to the progression of various tumors, and studies suggest that its release negatively impacts CC prognosis25, with NLRP3 driving the epithelial-mesenchymal transition (EMT) in CC26. NLRP3 is closely associated with mitochondrial function27. Previous studies demonstrated that zinc ion homeostasis influences mitochondrial stability, which in turn modulates mitophagy and MD28,29. These processes are critically implicated in the pathogenesis and progression of CC30. Based on these findings, we hypothesize that elevated SLC39A10 expression increases zinc ion transport19, leading to mitochondrial homeostasis disruption. This dysregulation subsequently reduces mitophagy activity and elevates cellular NLRP3 levels. The accumulated NLRP3 is proposed to activate the Nod-like receptor signaling pathway, ultimately driving cervical carcinogenesis. Our subsequent work aims to experimentally validate this proposed mechanism. For the low-risk group, GSEA enrichment highlighted the differential expression of cytochrome P450 1B1 between CC and normal cervical tissue, potentially identifying it as a future therapeutic target31, although the metabolism of foreign substances by cytochrome P450 and drug metabolism via cytochrome P450 remain underexplored. As for olfactory transduction, research is currently limited to pan-cancer studies32, warranting further investigation.
Cancer treatment has entered the era of immunotherapy, where the immune microenvironment plays a pivotal role in tumor onset and progression. Identifying suitable and efficient immune targets is crucial for determining the success of immunotherapy. Gammadelta (γδ) T cells, a critical subpopulation of T lymphocytes, have well-established protective roles in cancer33. On one hand, γδ T cells exert direct cytotoxic effects on the tumor microenvironment in CC through dynamic evolution of two-polar cell states. On the other hand, a high level of γδ T cell infiltration shapes an immune-activated microenvironment with cancer-suppressive properties, with infiltration levels positively correlated with prognosis34,35. Recent studies have highlighted the strong correlation between neutrophils and tumor progression, positioning neutrophils as potential therapeutic targets for cancer treatment36. scRNA-seq has mapped the immune cell transcriptome during CC progression, revealing that neutrophils are involved in all stages of the disease, which could inform the development of new therapies aimed at inhibiting CC initiation and progression37. Regulatory T cells, another key subpopulation of T cells, play a pivotal role in immune regulation within the tumor microenvironment. Research has shown that the endogenous stimulator of interferon genes (STING) pathway in T cells upregulates FOXP3 transcription, promoting regulatory T cell induction and immune suppression, thus providing a theoretical basis for immune therapy strategies targeting the STING pathway in CC38. In this study, γδ T cells, neutrophils, and regulatory T cells were identified as significantly differential immune cells, with the highest correlation found between SBK1 and neutrophils, offering a new direction for the search for novel immune treatment targets in CC.
scRNA-seq technology, a cutting-edge tool, enables high-throughput sequencing analysis at the genomic, transcriptomic, and epigenetic levels at the single-cell resolution, revealing gene structures and expression states of individual cells. This technology provides insights into cellular heterogeneity39. In this study, a CC single-cell dataset was introduced and analyzed, leading to the identification of key cell types, including endothelial cells, T cells, and epithelial cells. Differential expression of SBK1, SLC39A10, and PLOD3 was observed in these three cell types. Given the changes in the expression patterns of prognostic genes in these key cell types, it is hypothesized that they are involved in the prognosis of patients with CC, aligning with previous studies’ conclusions37,39.
However, this study has certain limitations. Firstly, the sample sizes of the two datasets used are limited. Future research should validate the findings in larger CC cohorts and incorporate more independent datasets to enhance the generalizability of the results. Secondly, although preliminary validation of some results was conducted through PCR and IHC experiments, the main conclusions are still based on bioinformatics analysis and are predictive in nature. Further functional experiments (such as gene knockout, overexpression, etc.) are necessary to verify the biological functions and mechanisms of the genes. Additionally, we plan to integrate more prognostic factors related to CC progression and treatment response (such as molecular markers) and combine them with clinical variables (such as age, tumor stage, comorbidities, and treatment history) to optimize the model and improve its predictive accuracy. Future research will further explore experimental mechanisms and validate clinical applications, comprehensively assessing the biological significance and clinical translation potential of the model.
Conclusions
In summary, this study aims to comprehensively assess the prognostic value and underlying mechanisms of MDGs and MRGs in CC. DEGs associated with MDGs and MRGs were identified through transcriptomic analysis, followed by univariate Cox regression, MR analysis, and 101 machine learning models to identify three prognostic genes related to MDGs and MRGs in CC. A prognostic model was constructed and validated using clinical samples, and single-cell correlation analysis was performed based on the identified prognostic genes. The results suggest that PLOD3, SBK1, and SLC39A10 may serve as potential targets for CC treatment, offering new avenues for therapeutic strategies. For instance, in patients exhibiting high expression of PLOD3 or SLC39A10, or low expression of SBK1, additional treatment modalities may be considered alongside conventional therapy. These could include immunotherapy, targeted therapies, or an appropriately adjusted increase in chemotherapy dosage. Furthermore, eligibility criteria for radiotherapy or chemotherapy regimens might be cautiously expanded to accommodate clinical indications.
Methods
Data collection
The TCGA-CESC dataset was retrieved from the University of California, Santa Cruz (UCSC) Xena website (http://xena.ucsc.edu/), which includes 293 CC tissue samples with survival data and 3 control tissue samples. The GSE63514 (GPL570) dataset, consisting of 28 CC and 24 control tissue samples, and the GSE52903 (GPL6244) dataset, which includes 55 CC tissue samples with survival data, were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds). The scRNA-seq dataset, E-MTAB-11,948, was sourced from the ArrayExpress database (https://www.ebi.ac.uk/biostudies/arrayexpress), including 3 CC and 3 control tissue samples. A total of 23 MDGs and 36 MRGs were obtained from the literature40,41. The Integrative Epidemiology Unit (IEU) Open GWAS database (https://gwas.mrcieu.ac.uk/) was used to access gene expression quantitative trait loci (eQTL) data and CC-related datasets (ebi-a-GCST90018817), containing 24,138,337 single nucleotide polymorphisms (SNPs) from 909 patients with CC and 238,249 control individuals of European descent.
Differential expression analysis
The limma package (v 3.54.0)42 was employed to identify DEGs between the 28 CC and 24 control tissue samples in the GSE63514 dataset (adj.P < 0.05, |log2FoldChange (FC)| > 1). Data visualization was performed using the ggplot2 package (v 3.4.4)43 and ComplexHeatmap package (v 2.14.0)44.
Weighted gene co-expression network analysis (WGCNA)
The GSVA package (v 1.46.0)45 was utilized to calculate MDGs and MRGs scores in CC samples from TCGA-CESC using the single-sample gene set enrichment analysis (ssGSEA) algorithm. Based on the optimal cut-off values of these scores, the samples were classified into high and low score groups. Kaplan-Meier (KM) survival curves, generated using the survival package (v 3.5-3)46, were employed to compare survival between the high and low score groups, with statistical significance determined by the Log-Rank test (P < 0.05). Subsequently, WGCNA was performed based on MDG and MRG scores using the WGCNA package (v 1.71)47. Clustering analysis was conducted on CC samples to exclude potential outliers. The soft threshold (β) was chosen to ensure a mean connectivity approaching 0, with the R2 value greater than 0.85. A scale-free network was constructed, and genes were categorized into several modules assigned different colors (minModuleSize = 100, deepSplit = 4, mergeCutHeight = 0.1). Key modules, defined by strong correlations (|cor| > 0.3, P < 0.05) with MDG and MRG scores, were identified, and genes within these modules were considered key module genes.
Enrichment analysis and protein-protein interactions (PPI) network of candidate genes
DEGs and key module genes were intersected to identify candidate genes related to MD and mitophagy. These candidate genes were input into the clusterProfiler package (v 4.7.1.003)48 for GO and KEGG49,50,51 enrichment analyses to explore related functions and pathways. GO analysis included molecular function (MF), biological process (BP), and cellular component (CC). Protein-protein interaction (PPI) data for candidate genes were obtained from the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org/) (confidence > 0.4), and the PPI network was visualized using Cytoscape software (v 3.9.1)52.
Univariate Cox regression and Mendelian randomization (MR) analyses
In the TCGA-CESC dataset, univariate Cox regression analysis was performed using the survival package (v 3.5-3)46 to identify genes associated with OS in CC samples (hazard ratio (HR) ≠ 1, P < 0.05).
To determine causal genes, the TwoSampleMR package (v 0.6.1)53 was employed for MR analysis, using genes related to OS as exposure factors and CC as the outcome. MR analyses relied on three assumptions: (1) Instrumental variables were correlated with exposure factors. (2) Instrumental variables were not correlated with outcomes. (3) Instrumental variables influenced outcomes directly through exposure factors without interference from confounders. To ensure significant correlation between SNPs and OS-related genes, SNPs were filtered with a threshold of P < 5 × 10− 8. Redundant SNPs were removed based on the linkage disequilibrium (LD) coefficient (clump = TRUE, r2 = 0.001, kb = 10), and snp with LD coefficients were selected to avoid potential bias caused by LD and to ensure the independence of the instrumental variable (IV). In addition, the strength of each IV was assessed using the F-statistic and weak IVs with F < 10 were excluded. The F-statistic was calculated as follows: \(\:\text{F}=\frac{{\text{R}}^{2}}{\text{K}}\times\:\frac{\text{N}-\text{K}-1}{1-{\text{R}}^{2}}\), \(\:{R}^{2}={\sum\:}_{i=1}^{k}\left[2\times\:{\beta\:}_{i}^{2}\times\:{MAF}_{i}\times\:\left(1-{MAF}_{i}\right)\right]\). \(\:{\beta\:}_{i}^{2}\) is the amount of effect of IV on exposure, MAFi is the minor allele frequency, R2 is the extent to which IV explains exposure, K is the number of IVs, and N is the sample size. Exposure factors with SNP < 3 were also excluded to further ensure the reliability of the instrumental variables. Finally, the harmonise_data function was used to harmonise effect alleles and effect sizes to ensure data consistency. Together, these steps ensured the significance, independence, and strength of the instrumental variables, thereby improving the accuracy and reliability of the analyses. The harmonise_data function was used to harmonize effect alleles and effect sizes. The mr function combined five algorithms—MR Egger54, Weighted Median55, IVW56, Simple Mode57, and Weighted Mode58—to perform MR analysis. Causal genes with significant relationships to CC were identified using the IVW algorithm (P < 0.05). An odds ratio (OR) greater than 1 indicated a risk factor, while an OR less than 1 indicated a protective factor. Scatter, forest, and funnel plots were generated to visually represent the MR analysis results.
The robustness of the MR analysis was assessed through sensitivity analyses, including heterogeneity, horizontal pleiotropy, and Leave-One-Out (LOO) analyses, using the mr_heterogeneity, mr_pleiotropy_test, and mr_leaveoneout functions from the TwoSampleMR package, respectively.
Finally, MRsteiger analysis was performed on exposure factors significantly associated with CC. Exposure factors identified as TRUE were retained as causal genes.
Machine learning
The construction of a risk model involved integrating 10 machine learning algorithms: Least Absolute Shrinkage and Selection Operator (LASSO), Random Survival Forest (RSF), Elastic Network (Enet), Stepwise Cox Regression (Stepwise Cox), Ridge, CoxBoost, Partial Least Squares Regression (PLSR), Supervised Principal Components (SuperPC), Generalized Boosted Modeling (GBM), and Support Vector Machine-Recursive Feature Elimination (SVM-RFE), generating 101 prediction models. In TCGA-CESC, a leave-one-out cross-validation (LOOCV) framework was used to develop these models. Usually a C-index > 0.6 is considered an acceptable or discriminatory threshold, therefore, the C-index in GSE52903 and TCGA-CESC was calculated for each model and the model with the highest C-index in the training set (TCGA-CC) and a C-index greater than 0.6 in the validation set (GSE52903) was selected to obtain prognostic genes59,60. Specifically, the randomForestSRC package (v 3.2.2)61 was used for RSF, the glmnet package (v 4.1-4)62 for Enet, LASSO, and Ridge, the survival package for Stepwise Cox, the CoxBoost package (v 1.4)63 for CoxBoost, the plsRcox package (v 1.7.7)64 for PLSR, the SuperPC package (v 1.12)65 for SuperPC, the gbm package (v 2.1.8.1)66 for GBM, and the survivalsvm package (v 0.0.5) for SVM-RFE.
Appraisal and verification of the risk model
The risk model was constructed by integrating the expression levels (expr) of prognostic genes with their corresponding coefficients (coef) derived from the selected machine learning model: \(\:\text{R}\text{i}\text{s}\text{k}\text{s}\text{c}\text{o}\text{r}\text{e}\:=\:\sum\:_{\text{i}\:=\:1}^{\text{n}}\text{c}\text{o}\text{e}\text{f}\left({\text{g}\text{e}\text{n}\text{e}}_{\text{i}}\right)\text{*}\text{e}\text{x}\text{p}\text{r}\left({\text{g}\text{e}\text{n}\text{e}}_{\text{i}}\right)\). In TCGA-CESC, patients with CC were categorized into high- and low-risk groups based on the risk score cutoff. Kaplan-Meier survival analysis, implemented via the survminer (v 0.4.9)67 and survival packages, assessed differences in OS between these groups. Time-dependent ROC curves at 1, 3, and 5 years were generated using the survivalROC package (v 1.0.3.1)68. Additionally, the expression patterns of prognostic genes across risk groups were visualized. The model’s robustness was further validated using analogous analyses in GSE52903.
Independent prognostic analysis
A prognostic nomogram integrating risk scores with five pathological indicators (age, smoking, pathological T/M/N) was developed to predict OS in TCGA-CESC. Smoking status was categorized as follows: individuals with a lifetime cigarette consumption of fewer than 100 were classified as “1”, current smokers (including daily and occasional smokers) as “2”, former smokers who quit for over 15 years as “3”, and those who quit within the past 15 years as “4”. These six variables underwent univariate Cox regression analysis (P < 0.05), proportional hazards (PH) assumption testing (P ˃ 0.05), and multivariate Cox regression analysis (P < 0.05) to identify independent prognostic factors, which were incorporated into the nomogram using the rms package (v 6.5-0)69. Model performance was assessed via calibration curves generated by the rms package and decision curves constructed using the rmda package (v 1.6)70. To examine associations between pathological indicators and risk scores, Wilcoxon tests in the rstatix package were applied across different pathological subgroups in TCGA-CESC (P < 0.05). Furthermore, prognostic gene expression patterns were analyzed across these subgroups.
Gene set enrichment analysis (GSEA)
To elucidate functional pathways associated with risk stratification, GSEA was conducted using the clusterProfiler package (v 4.7.1.003). Differential expression analysis between high- and low-risk groups in TCGA-CESC was performed with DESeq2 (v 1.38.0)71, and log2FC values of DEGs were ranked in descending order for GSEA (P adj < 0.05). The reference gene set “c2.cp.kegg.v7.4.symbols.gmt” was sourced from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb).
Immune infiltration analysis
The immune microenvironment was characterized by evaluating enrichment scores of 28 immune cell types72 and two stromal cell types using the ssGSEA algorithm. Differentially enriched immune cells (DICs) were identified via Wilcoxon tests (P < 0.05). Additionally, Spearman correlation analysis, implemented in the psych package (v 2.2.9)73, assessed associations between DICs and prognostic genes. Wilcoxon tests were further employed to analyze the expression of 48 common immune checkpoints74 between risk groups in TCGA-CESC (P < 0.05).
Drug-sensitivity analysis
To assess chemotherapy response across different risk groups in TCGA-CESC, the 50% inhibitory concentration (IC50) values of 138 chemotherapy and targeted therapy drugs from the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/) were calculated for patients with CC using the “pRRophetic” package (v 0.5)75. Differences in drug IC50 values between risk groups were visualized with box plots generated using the ggplot2 package (P < 0.05).
The scRNA-seq analysis
To explore prognostic gene profiles at the single-cell level, we systematically analysed single-cell RNA sequencing (scRNA-seq) data. Data preprocessing and quality control were done using the Seurat software package (v5.1.0)76. Get the gene count of each cell by scRNA$nFeature_RNA, set minGene = 200 and maxGene = 6000 to filter out cells with gene count below 200 or above 6000. Meanwhile, the UMI count of each cell was obtained by scRNA$nCount_RNA, and maxUMI = 20,000 was set to exclude cells with UMI counts over 20,000 to exclude potentially abnormal cells. In addition, the percentage of mitochondrial genes was calculated, and only cells with a percentage of mitochondrial genes less than 10% were retained to ensure data quality. The data were standardized using the LogNormalize method, and the 2000 most variable genes were selected with the FindVariableFeatures function. Then, linear dimension reduction techniques were employed to obtain optimal linear dimensions for cell clustering. Detailedly, scree plots were generated based on the results of principal component analysis (PCA), and the number of principal components was determined by selecting the points where the curves tended to flatten out, in order to preserve the data features, reduce dimensionality, and remove the noise, and improve the efficiency of the clustering analysis. In UMAP cell clustering, a moderate resolution (resolution = 0.5) was used to balance the clustering fineness and accuracy, avoiding the omission of important cell subgroups or excessive subdivision of similar cells, and optimising the interpretability and practicality of the clustering results. Marker genes identified from the literature77 were used for cell annotation (Figure S11).
To explore the functions associated with annotated cell types, functional enrichment analyses were performed using the ReactomeGSA package (v 1.12.0)78.
Violin plots generated by the ggplot2 package illustrated the expression levels of prognostic genes across all cell types, with cell types exhibiting two or more differentially expressed prognostic genes designated as key cell types (P < 0.05).
Receptor-ligand interactions within annotated cell types were evaluated to infer cell-to-cell communication using the CellChat package. Interactions were explored using CellChatDB.human (http://www.cellchat.org/) as the reference.
Additionally, the Monocle algorithm was applied for pseudo-time analysis to examine the developmental trajectory of key cell types and their differentiation direction. Prognostic gene expression during differentiation was analyzed, and the top 50 genes were selected based on q-values for GO enrichment analysis.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Prognostic gene expression was evaluated in clinical tissue samples using RT-qPCR. Tissue specimens were obtained from the 960th Hospital of the Joint Logistics Support Force, comprising 5 CC and 5 paracancerous tissue samples. Informed consent was obtained from all participants through signed consent forms, and the study received ethical approval from the Medical Ethics Committee of the 960th Hospital of the Joint Logistics Support Force (2024 Research Ethics Review No. 089). The study was conducted in accordance with the guidelines of the Declaration of Helsinki. RNA was extracted using TRIzol reagent (Ambion, Austin, USA), and cDNA synthesis was performed using the SureScript First-strand cDNA synthesis kit (Servicebio, Wuhan, China). RT-qPCR was conducted with the 2xUniversal Blue SYBR Green qPCR Master Mix (Servicebio). Primer sequences are provided in Table S8. The RT-qPCR amplification protocol involved an initial denaturation at 95 °C for 1 min, followed by 40 cycles consisting of denaturation at 95 °C for 20 s, annealing at 55 °C for 20 s, and extension at 72 °C for 30 s. Data were analyzed using the 2−ΔΔCt method, with GAPDH serving as the internal reference gene for normalization.
Immunohistochemistry (IHC)
For IHC, the tissue source was identical to that used for PCR analysis. Given that the expression of SBK1 in cervical cancer tissues has already been confirmed, this experiment focused on verifying the expression of PLOD3 and SLC39A10. For each gene, three samples of cancerous tissue and paracancerous tissue were collected for analysis. In the IHC procedure, tissues were sectioned into 5 μm slices, followed by deparaffinization with xylene and rehydration through a graded ethanol series. Antigen retrieval was performed using sodium citrate buffer under microwave heating. Subsequently, the slices were treated with 3% H2O2 for 25 min and 3% bovine serum albumin (BSA) for 30 min. The sections were then incubated overnight at 4 °C with primary antibodies (goat-derived). Afterward, the sections were washed three times with phosphate-buffered saline (PBS) and incubated with secondary antibodies (HRP-conjugated goat anti-rabbit IgG) for 50 min. Visualization was achieved using the 3,3’-diaminobenzidine (DAB) substrate kit (Servicebio) for 1 min, followed by counterstaining with hematoxylin (Servicebio). The stained sections were examined under a bright-field microscope. Hematoxylin-stained nuclei appeared blue, while DAB-positive staining exhibited brownish-yellow coloration.
Statistical analysis
R software version 4.2.2 was employed for data processing and analysis. Differences across groups were assessed using the Wilcoxon rank-sum test, with P-values below 0.05 considered statistically significant. The methodological steps are provided in Table S9.
Data availability
The original data presented in the study are openly available in (TCGA-CESC) at (http://xena.ucsc.edu/), (GEO) at (https://www.ncbi. nlm.nih.gov/gds), (STRING) at (https://string-db.org/), KEGG at (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb), (GDSC) at (https://www.cancerrxgene.org/), CellChat at (http://www.cellchat.org/), scRNA-seq at (https://www.ebi.ac.uk/biostudies/arrayexpress), GWAS at (https://gwas.mrcieu.ac.uk/).
Abbreviations
- CC:
-
Cervical cancer
- MD:
-
Mitochondrial dynamics
- MR:
-
Mendelian randomization
- PLOD3 :
-
Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3
- SBK1:
-
Encodes symbionin-like protein 1
- SLC39A10:
-
Solute carrier family 39 member 10
- scRNA-seq:
-
Single-cell RNA sequencing
- GSEA:
-
Gene set enrichment analysis
- AUC:
-
Area under the curve
- MDGs:
-
Mitochondrial dynamic genes
- MRGs:
-
Mitophagy related genes
- OS:
-
Overall survival
- DEGs:
-
Differential expression genes
- GWAS:
-
Genome-Wide association studies
- WGCNA:
-
Weighted gene co-expression network analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Yang, X. et al. Decreased expression of CLCA2 and the correlating with immune infiltrates in patients with cervical squamous cell carcinoma: A bioinformatics analysis. Taiwan. J. Obstet. Gynecol. 60, 480–486 (2021).
Perkins, R. B., Wentzensen, N., Guido, R. S. & Schiffman, M. Cervical cancer screening: A review. JAMA 330:547–558 (2023).
Chen, P., Zhang, W., Chen, Y., Zheng, X. & Yang, D. Comprehensive analysis of aberrantly expressed long noncoding rnas, micrornas, and mRNAs associated with the competitive endogenous RNA network in cervical cancer. Mol. Med. Rep. 22 (1), 405–415 (2020).
Kong, B. et al. Prohibitin 1 interacts with p53 in the regulation of mitochondrial dynamics and chemoresistance in gynecologic cancers. J. Ovarian Res. 15, 70 (2022).
Wang, S. et al. The mitophagy pathway and its implications in human diseases. Signal. Transduct. Target. Ther. 8 (1), 304 (2023).
Chen, W., Zhao, H. & Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal. Transduct. Target. Ther. 8 (1), 333 (2023).
Lu, Y. et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule Pharmacological regulation. Theranostics 13 (2), 736–766 (2023).
Picca, A. et al. Mitophagy in human health, ageing and disease. Nat. Metab. 5 (12), 2047–2061 (2023).
Han, C. Y. et al. Nuclear HKII-P-p53 (Ser15) interaction is a prognostic biomarker for chemoresponsiveness and glycolytic regulation in epithelial ovarian cancer. Cancers (Basel). 13 (14), 3399 (2021).
Wang, M. et al. Coffee consumption and prostate cancer risk: Results from National health and nutrition examination survey 1999–2010 and Mendelian randomization analyses. Nutrients 13 (7), 2317 (2021).
Fang, Z., Song, M., Lee, D. H. & Giovannucci, E. L. The role of Mendelian randomization studies in Deciphering the effect of obesity on cancer. J. Natl. Cancer Inst. 114 (3), 361–371 (2022).
Liu, Z. et al. Single-cell transcriptional profiling reveals aberrant gene expression patterns and cell States in autoimmune diseases. Mol. Immunol. 165, 68–81 (2024).
Cai, L. et al. Identification and validation of a six-gene signature associated with Glycolysis to predict the prognosis of patients with cervical cancer. BMC Cancer. 20 (1), 1133 (2020).
Gong, S. et al. A human pan-cancer system analysis of procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3 (PLOD3). Int. J. Mol. Sci. 22 (18), 9903 (2021).
Chen, Y. et al. PLOD3 contributes to HER-2 therapy resistance in gastric cancer through FoxO3/Survivin pathway. Cell. Death Discov. 8 (1), 321 (2022).
Dong, W. et al. To investigate the tumor promotion role of PLOD3 in colorectal cancer and its potential as a prognostic biomarker and therapeutic target. Sci. Rep. 15 (1), 5371 (2025).
Piñeiro-Llanes, J. et al. Matrix produced by diseased cardiac fibroblasts affects early myotube formation and function. Acta Biomater. 152, 100–112 (2022).
Cai, L., Shi, L., Peng, Z., Sun, Y. & Chen, J. Ageing of skeletal muscle extracellular matrix and mitochondria: Finding a potential link. Ann. Med. 55 (2), 2240707 (2023).
Ren, X. et al. SLC39A10 promotes malignant phenotypes of gastric cancer cells by activating the CK2-mediated MAPK/ERK and PI3K/AKT pathways. Exp. Mol. Med. 55 (8), 1757–1769 (2023).
Guan, X. et al. Dual Inhibition of MYC and SLC39A10 by a novel natural product STAT3 inhibitor derived from Chaetomium globosum suppresses tumor growth and metastasis in gastric cancer. Pharmacol. Res. 189, 106703 (2023).
Ma, Z. et al. SLC39A10 upregulation predicts poor prognosis, promotes proliferation and migration, and correlates with immune infiltration in hepatocellular carcinoma. J. Hepatocell Carcinoma. 8, 899–912 (2021).
Hu, Z. W. et al. CD69 and SBK1 as potential predictors of responses to PD-1/PD-L1 Blockade cancer immunotherapy in lung cancer and melanoma. Front. Immunol. 13, 952059 (2022).
Guo, Q. et al. A novel risk model of three gefitinib-related genes FBP1, SBK1 and AURKA is related to the immune microenvironment and is predicting prognosis of lung adenocarcinoma patients. Aging (Albany NY). 15 (18), 9633–9660 (2023).
Chen, X. et al. SH3 domain-binding kinase 1 promotes proliferation and inhibits apoptosis of cervical cancer via activating the Wnt/β-catenin and Raf/ERK1/2 signaling pathways. Mol. Carcinog. 62 (8), 1147–1162 (2023).
Fernandes, F. P. et al. Cervical carcinoma induces NLRP3 inflammasome activation and IL-1ß release in human peripheral blood monocytes affecting patients’ overall survival. Clin. Transl. Oncol. 25 (11), 3277–3286 (2023).
Sui, H. et al. LRRC75A-AS1 drives the epithelial-mesenchymal transition in cervical cancer by binding IGF2BP1 and inhibiting SYVN1-mediated NLRP3 ubiquitination. Mol. Cancer Res. MCR-23-0478 (2024).
Gupta, S., Cassel, S. L., Sutterwala, F. S. & Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev., 329(1), e13410 (2025).
Rensvold, J. W. et al. Defining mitochondrial protein functions through deep multiomic profiling. Nature 606 (7913), 382–388 (2022).
Ma, T. et al. A pair of transporters controls mitochondrial Zn2+ levels to maintain mitochondrial homeostasis. Protein Cell. 13 (3), 180–202 (2022).
Kong, B. et al. Prohibitin 1 interacts with p53 in the regulation of mitochondrial dynamics and chemoresistance in gynecologic cancers. J. Ovarian Res. 15 (1), 70 (2022).
Alshammari, F. O. F. O., Al-Saraireh, Y. M., Youssef, A. M. M., Al-Sarayra, Y. M. & Alrawashdeh, H. M. Cytochrome P450 1B1 overexpression in cervical cancers: Cross-sectional study. Interact. J. Med. Res. 10(4), e31150 (2021).
Hua, Y., He, Z. & Zhang, X. A pan-cancer analysis based on weighted gene co-expression network analysis identifies the biomarker utility of lamin B1 in human tumors. Cancer Biomark. 34 (1), 23–39 (2022).
Mensurado, S., Blanco-Domínguez, R. & Silva-Santos, B. The emerging roles of γδ T cells in cancer immunotherapy. Nat. Rev. Clin. Oncol. 20 (3), 178–191 (2023).
Li, J. et al. Multiomics profiling reveals the benefits of gamma-delta (γδ) T lymphocytes for improving the tumor microenvironment, immunotherapy efficacy and prognosis in cervical cancer. J. Immunother. Cancer 12(1), e008355 (2024).
Dong, J. et al. γδ T cell-mediated cytotoxicity against patient-derived healthy and cancer cervical organoids. Front. Immunol. 14, 1281646 (2023).
Que, H., Fu, Q., Lan, T., Tian, X. & Wei, X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim. Biophys. Acta Rev. Cancer. 1877 (5), 188762 (2022).
Li, C. & Hua, K. Dissecting the single-cell transcriptome network of immune environment underlying cervical premalignant lesion, cervical cancer and metastatic lymph nodes. Front. Immunol. 13, 897366 (2022).
Ni, H. et al. T cell-intrinsic STING signaling promotes regulatory T cell induction and immunosuppression by upregulating FOXP3 transcription in cervical cancer. J. Immunother. Cancer 10(9), e005151 (2022).
Li, C. et al. Single-cell transcriptomics reveals cellular heterogeneity and molecular stratification of cervical cancer. Commun. Biol. 5 (1), 1208 (2022).
Zhang, L. et al. MTFR2-dependent mitochondrial fission promotes HCC progression. J. Transl. Med. 22 (1), 73 (2024).
Jia, Y. et al. Integrative analysis of the prognostic value and immune microenvironment of mitophagy-related signature for multiple myeloma. BMC Cancer. 23 (1), 859 (2023).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47 (2015).
Wang, J. et al. PROS1 shapes the immune-suppressive tumor microenvironment and predicts poor prognosis in glioma. Front. Immunol. 13, 1052692 (2023).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32 (18), 2847–2849 (2016).
Hänzelmann, S., Castelo, R. & Guinney, J. G. S. V. A. Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Lei, J. et al. Clinicopathological characteristics of pheochromocytoma/paraganglioma and screening of prognostic markers. J. Surg. Oncol. 128 (4), 510–518 (2023).
Langfelder, P. & Horvath, S. W. G. C. N. A. An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Wu, T. et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb). 2 (3), 100141 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. K. E. G. G. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504 (2003).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32 (5), 377–389 (2017).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40 (4), 304–314 (2016).
Burgess, S. et al. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 30 (7), 543–552 (2015).
Chen, X. et al. Depression and prostate cancer risk: A Mendelian randomization study. Cancer Med. 9 (23), 9160–9167 (2020).
Hu, J. et al. Reverse causal relationship between periodontitis and shortened telomere length: Bidirectional two-sample Mendelian random analysis. Front. Immunol. 13, 1057602 (2022).
Huang, L. et al. Assessment of a radiomic signature developed in a general Nsclc cohort for predicting overall survival of alk-positive patients with different treatment types. Clin. Lung Cancer. 20 (6), e638–e651 (2019).
Ashfaq, A. et al. Survival analysis for pediatric heart transplant patients using a novel machine learning algorithm: A UNOS analysis. J. Heart Lung Transpl. Off. Publ. Int. Soc. Heart Transpl. 42 (10), 1341–1348 (2023).
Zhao, P., Zhen, H., Zhao, H., Huang, Y. & Cao, B. Identification of hub genes and potential molecular mechanisms related to radiotherapy sensitivity in rectal cancer based on multiple datasets. J. Transl. Med. 21 (1), 176 (2023).
Sasikumar, D. et al. Caging and photo-triggered uncaging of singlet oxygen by excited state engineering of electron donor-acceptor-linked molecular sensors. Sci. Rep. 12 (1), 11371 (2022).
Rong, F. et al. Machine learning for prediction of outcomes in cardiogenic shock. Front. Cardiovasc. Med. 9, 84968 (2022).
Wang, Z. et al. Identification of a ferroptosis-related gene signature predicting recurrence in stage II/III colorectal cancer based on machine learning algorithms. Front. Pharmacol. 14, 1260697 (2023).
Zhang, P. et al. Integrating multiple machine learning methods to construct glutamine metabolism-related signatures in lung adenocarcinoma. Front. Endocrinol. (Lausanne). 14, 1196372 (2023).
Li, K. et al. Efficient gradient boosting for prognostic biomarker discovery. Bioinformatics 38 (6), 1631–1638 (2022).
Liu, T. T. et al. Identification of cdk2-related immune forecast model and Cerna in lung adenocarcinoma, a pan-cancer analysis. Front. Cell. Dev. Biol. 9, 682002 (2021).
Heagerty, P. J., Lumley, T. & Pepe, M. S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 56 (2), 337–344 (2000).
Xu, J. Q. et al. A nomogram for predicting prognosis of patients with cervical cerclage. Heliyon 9 (11), e21147 (2023).
Kerr, K. F., Brown, M. D., Zhu, K. & Janes, H. Assessing the clinical impact of risk prediction models with decision curves: Guidance for correct interpretation and appropriate use. J. Clin. Oncol. 34 (21), 2534–2540 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550 (2014).
Charoentong, P. et al. Pan-cancer Immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint Blockade. Cell. Rep. 18 (1), 248–262 (2017).
Robles-Jimenez, L. E. et al. Worldwide traceability of antibiotic residues from livestock in wastewater and soil: A systematic review. Anim. (Basel) 12 (1), 60 (2021).
Wu, J. et al. A risk model developed based on tumor microenvironment predicts overall survival and associates with tumor immunity of patients with lung adenocarcinoma. Oncogene 40 (26), 4413–4424 (2021).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE. 9 (9), e107468 (2014).
Hao, Y. H. et al. Integrated analysis of multimodal single-cell data. Cell 184 (13), 3573–3587 (2021).
Shiau, C. K. et al. High throughput single cell long-read sequencing analyses of same-cell genotypes and phenotypes in human tumors. Nat. Commun. 14 (1), 4124 (2023).
Griss, J. et al. ReactomeGSA - efficient multi-omics comparative pathway analysis. Mol. Cell. Proteom. 19 (12), 2115–2125 (2020).
Acknowledgements
Thanks to Zhen Mingwan for her help in the submission process.
Author information
Authors and Affiliations
Contributions
Jiankui Li and Juan Li participated in the project design, Xi Chen was mainly responsible for the statistical analysis of data, and Jiankui Li was responsible for the writing of the manuscript. All authors agreed of the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All participants provided informed consent by completing and signing consent forms, and the ethical approval was granted by the Medical Ethics Committee of the 960th Hospital of the Joint Logistics Support Force (2024 Research Ethics Review No. 089).
Consent for publication
All authors have approved the manuscript for submission and publication.
Duplicate publication policy
All authors confrm that the content of the manuscript has not been published, or submitted for publication elsewhere.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Chen, X. & Li, J. Exploration and validation of the prognostic value of mitophagy and mitochondrial dynamics-related genes in cervical cancer. Sci Rep 15, 24950 (2025). https://doi.org/10.1038/s41598-025-09310-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-09310-6

{kind=link}