
Abstract
With the need for clean sustainable energy and low-waste practices rising, battery-free technologies that run on renewable ambient energy sources are an attractive solution to these environmental concerns. Herein, Graphene oxide (GO) and GO-PEDOT: PSS water-based inks were formulated from inexpensive precursor materials following user-friendly, up-scalable methods. GO-based moisture-active films were deposited on flexible lightweight substrates using inkjet printing to fabricate Moisture Energy Generators (MEG), devices that convert ambient moisture energy variation into a voltage output. The performance of the fabricated MEGs was evaluated using a custom-made automated humidity chamber, yielding an average voltage output of Vo=183 (± 1.76) mV for pure GO and Vo=194 (± 0.97) mV for GO-PEDOT: PSS, in the relative humidity range of 45–95%. We support that this study may provide fertile ground for the development of low-cost energy harvesters for self-powered portable and wearable technologies, minimising the need for batteries and reducing electronic waste.
Similar content being viewed by others


Main
One of the main environmental topics of the 21 st century is replacing conventional polluting fossil fuels with clean, sustainable renewable energy sources. In the effort to meet this demand, novel technologies have risen that harvest ambient environmental energy, such as solar and wind energy that would otherwise go unused. Another important ecological concern is the electronic waste created by the dependence of portable devices on batteries, creating a need for self-powered technologies. Ambient humidity is an abundant source of waste energy that can be exploited through Moisture Energy Generators (MEGs). These devices exploit the energy harvested from environmental humidity or other moisture sources and convert it into clean electrical power. The key element in these harvesters is a functional, hydrophilic material that adsorbs water molecules, which triggers a mechanism through which chemical energy induces an ion conductivity that is converted into electrical energy.
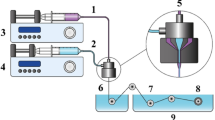
In this work, we developed a fully printed lightweight, flexible and portable MEG as a promising and inexpensive avenue for self-powered, battery-free devices (Fig. 1). The moisture adsorbing layer is based on Graphene Oxide (GO), which is rich in oxygen-containing groups making it highly hydrophilic and granting it a high water uptake1. High-quality GO can be synthesised in large quantities, with good reproducibility, following a simple chemical protocol and using inexpensive materials. GO is also readily dispersible in water, meaning that non-toxic, environmentally friendly GO printable inks can be developed, with no need for toxic and hazardous organic solvents. Printable GO-based inks are formulated through Liquid Phase Exfoliation (LPE) of previously synthesised bulk graphite oxide; a user-friendly, economical and scalable ink production method. The GO-based MEGs were manufactured on thin, flexible substrates using inkjet printing, which offers a streamlined and scalable fabrication technique as well as the ability for simultaneous printing of multiple devices. The flexibility, minimal thickness, and near-weightless characteristics of these GO-based MEGs hold the potential to generate energy for self-powered wearable electronics, such as biometric monitors, biosensors, and skin patches2,3,4,5. GO-based MEGs have been fabricated and studied in recent research, with the GO moisture-absorbing layer developed in the form of films, foams, aerogels, and through laser processing6,7,8,9,10,11,12,13. In this article, the GO-based inks are deposited via inkjet printing, which offers a user-friendly, inexpensive and scalable fabrication technique14.

Graphical representation summarising the operation of the inkjet-printed GO-based MEGs.
The mechanism through which a GO-based MEG generates electricity has been studied recently and found to be based on the release and diffusion of free-moving hydrogen cations or proton (H+) upon water molecule adsorption6,15. When water is adsorbed, H+ dissociate from the surface carboxyl groups (-COOH) within the GO structure. These H+ then migrate due to an oxygen content gradient while the -COO− groups remain immobile. This results in a voltage difference between the two electrodes and the generation of an electric field. Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT: PSS) was investigated as a second component in a GO-polymer hybrid ink for enhanced MEG performance16,17,18. PEDOT: PSS was elected for (a) its hygroscopicity, to enhance the moisture adsorbing ability of the active layer, and (b) its high conductivity and charge transfer ability, to potentially increase the generated output voltage19,20,21.
The performance of the fabricated GO- and GO-PEDOT: PSS-based nanogenerators was evaluated using a custom-made automated humidity testing chamber. This setup allows for the observation of moisture-induced electrical responses under controllable and repeatable humidity conditions. The devices’ voltage output (Vo) was measured in the Relative Humidity (RH%) range of 45-95%, with results revealing an average of Vo=183 (± 1.76) mV for pure GO and Vo=194 (± 0.97) mV for GO-PEDOT: PSS. These results hold potential for developing low-cost ambient energy harvesters for self-powered portable and wearable technologies, minimising the need for batteries.
Results and discussion
Firstly, the quality and chemical composition of the synthesised graphite oxide were investigated, as a high oxygen-group content of the GO layer is crucial to its moisture adsorption ability and its humidity-induced voltage efficiency. For this purpose, Attenuated Total Reflectance Infrared (ATR-IR) spectroscopy, Raman spectroscopy, X-ray Photoelectron spectroscopy (XPS) and X-ray Diffraction (XRD) were performed on the graphite oxide powder to determine the degree of oxidation and the nature of the oxygen groups formed.
The recorded transmittance spectra of the synthesised graphite oxide and the graphite precursor are presented and analysed in Fig. 2a22. The graphite powder has no distinctive peaks, except for a low-intensity signal at 1587.3 cm−1, which corresponds to the stretching vibrations of the aromatic C = C bonds23. After oxidation, the produced graphite oxide exhibits multiple peaks associated with C-O bonds, specifically at 1039.6 cm−1, 1724.3 cm−1 and 3168.9 cm−1, attributed to the C-O, C = O and O-H stretching modes, respectively24. The peak at 1346.2 cm−1 corresponds to the O-H bending vibrations of alcohol groups, while the low-intensity peak at 2796.6 cm-1 is attributed to the C-H stretching mode25,26. The peaks at 977.9 cm−1 and 1213.2 cm−1 coincide with the C-O-C stretching vibrations of the epoxide groups27. These observations provide ample evidence of successful chemical oxidation.

(a) ATR-IR spectra, (b) Raman spectra and (c) XRD patterns of graphite oxide powder synthesised chemically via a modified Hummers’ method and its graphite precursor.
The Raman spectrum of graphite (Fig. 2b) exhibits the characteristic G and D bands at 1580.9 cm−1 and 1342.4 cm−1, respectively28. The G peak arises due to the first-order scattering of the E2g phonons at the centre of the Brillouin zone, which results from the in-plane vibrational mode of the sp2-carbon atoms. The D peak is a result of the breathing mode of the A1g symmetry phonons around the K or K’ points of the Brillouin zone. The G peak for the pure graphite powder is very intense and narrow, while the D peak is very weak and broad, which aligns with the literature29. The 2D and 2D’ bands, which appear due to second-order Raman scattering, are detected at 2719.7 cm−1 and 3246.1 cm−1, respectively30. The 2D peak in graphene-related materials is associated with the number of layers, with the I2D value decreasing and the peak becoming broader as we shift from monolayer graphene to a bulk graphite structure31. The asymmetrical shape of the 2D peak and its relatively low intensity compared to that of monolayer graphene are characteristic of bulk graphite and agree with previous reports30,32. Two additional low-intensity peaks visible at 2448.8 cm−1 and 2991.5 cm−1 and attributed to the D + D” and D + D’ Raman modes, respectively, occur due to combination scattering33. For the as-prepared graphite oxide powder, the G and D bands shift to higher wavenumbers, appearing at 1596.3 cm−1 and 1347.9 cm−1, respectively, while the G peak becomes broader34. The increased intensity of the D band is expected, as it is a Raman mode induced by the disorder associated with structural imperfections and the disruption of the sp2 system within the graphite flakes35. As a result, the ID/IG ratio increases from 0.10 for graphite to 0.99 for graphite oxide, which reveals an increase in defect density, which is a well-accepted indication of successful oxidation36. The D + D’ becomes broader upon oxidation, with its centre red-shifting from 2991.5 cm−1 to 2922.5 cm−1. Furthermore, the 2D peak blueshifts from 2719.7 cm−1 to 2741.1 cm−1 and becomes broader and weaker compared to that for pure graphite powder. The shift in position alongside the broadening of these peaks, constitutes further evidence of functionalisation with oxygen groups37,38.
The recorded XRD patterns of the graphite oxide and graphite reference are presented in Fig. 2c. The strong, sharp diffraction peak at 26.4o corresponds to the (002) Bragg’s plane, while those at 44.5o (101), 54.5o (004) and 59.9o are attributed to the (101), (004) and (110) orientations, respectively39. The interplanar spacing of the (002) is d(002) = 0.34, according to Bragg’s equation (Eq. 1), which is indicative of a highly ordered carbon structure40. For graphite oxide, the (002) diffraction peak disappears, while a broad and low-intensity peak appears at 10.5o. This peak corresponds to the (001) orientation and is characteristic of graphite oxide structures41. The d-spacing increases to d(001) = 0.84 due to the introduction of oxygen functional groups between the GO layers24,40,42.
Where:
n = diffraction order Positive integer, the order.
λ = XRD radiation wavelength (Å).
d = interplanar distance (Å).
θ = Bragg angle (o).
XPS was employed to investigate the level of oxidation and the purity of the synthesised graphite oxide. The resulting XPS survey spectrum is featured in Fig. 3a, wherein the C1s peak (286.5 eV), O1s peak (532.5 eV), and the O-KVV Auger peak (976 eV) are indicated observed43,44. No signals from other elements were detected. The atomic concentration (%) of carbon and oxygen was calculated from the C1s and O1s peaks and was found to be 61.1% and 38.9%, respectively, while the C/O ratio is 1.57. These results are on par with XPS results for similarly synthesised graphite oxide samples, and confirm a high level of oxidation, while also verifying the purity of the synthesised graphite oxide41,45,46. Deconvolution of the C1s peak (Fig. 3b) reveals five carbon components: sp2 C-C (284.6 eV), sp3 C-C (285.5 eV), C-OH (286.8 eV), C = O (288.4 eV), and -COOH (289.5 eV), the %content of which are listed in Table 147,48. Results show that the synthesised graphite oxide is rich in hydroxyl groups, while a significant amount of carbonyl and carboxyl groups are also present within its chemical structure.

(a) XPS survey spectrum and (b) deconvolution of C1s peak of as-prepared graphite oxide powder.
The graphite oxide powder was exfoliated via ultrasonication in ultrapure water (LPE) to prepare an aqueous GO dispersion. A GO-PEDOT: PSS blend was then formulated from this aqueous GO dispersion. Both processes are described in the “Experimental Procedures” section. The chemical structures of GO and PEDOT: PSS are featured in Fig. 4a. The UV-Visible (UV-Vis) absorption spectra of both the pure GO aqueous dispersion and the GO-PEDOT: PSS blend are displayed in Fig. 4b. GO exhibits a maximum absorption peak at 229 nm, which arises from the π→π* electronic transitions of the remaining aromatic C = C bonds49. This is essentially a characteristic peak which appears at ~ 270 nm for pure graphene and blueshifts for graphene oxide as the degree of oxidation increases50. A shoulder is visible at 300 nm, which is also characteristic of GO and appears due to n→ π* transitions of the oxygen functional groups51. The sharp peak at 224 nm in the GO-PEDOT: PSS spectrum is a characteristic PEDOT: PSS absorption peak52.
The GO flake sizes were measured by Dynamic Light Scattering (DLS) to ensure that they are within the acceptable range for the inkjet-printer nozzles, with results presented in Fig. 4c. The graphs are the result of the averaging of four measurements for each sample. Pure GO has a narrower distribution peak and flake sizes that range from ~ 30 nm to ~ 950 nm, with the maximum peak corresponding to an average hydrodynamic diameter of 220 nm. For the GO-PEDOT: PSS sample, the distribution curve is broader, with flake sizes varying from ~ 40 to ~ 1700 nm and an average hydrodynamic diameter of 396 nm.

(a) Chemical structures of GO and PEDOT: PSS. (b) UV-Vis absorption spectra of GO and GO-PEDOT: PSS dispersions (c) Flake size distribution of GO flakes in pure GO ink and GO-PEDOT: PSS ink, as measured by DLS.
The interaction between the PEDOT: PSS molecules and the exfoliated GO flakes was investigated using Raman and ATR-IR spectroscopy. The recorded ATR-IR transmittance spectra of GO and GO-PEDOT: PSS are presented in Fig. 5a. The transmittance spectrum of the pure GO flakes is comparable to the previously recorded spectrum for the synthesised bulk graphite oxide, exhibiting multiple oxygen-containing group peaks. Specifically, the GO spectrum features peaks at 1724.3 cm−1 and 1643.3 cm−1, which correspond to the stretching vibrations of the C = O and C = C groups, respectively53,54. The peaks at 756.0 cm−1, 875.6 cm−1, 987.5 cm−1, and 1421.5 cm−1 are attributed to the C-H benzene, C = C, C-H and O-H bending vibrations, respectively55. Finally, the broad shoulder centred around 3155.4 cm−1 corresponds to the O-H stretching vibrations of GO’s hydroxyl groups56. The GO-PEDOT: PSS blend exhibits ATR-IR peaks from both components. GO peaks are observed at 2912.4 cm−1, 1627.8 cm−1, 1515.7 cm−1, 1292.2 cm−1 and 1122.5 cm−1 which correlate to the C-H, C = C, C-H benzene and C-O epoxy stretching modes, respectively57,58. The peak at 997.1 cm−1 is also attributed to the C-O stretching of the epoxide groups, while the bending vibration of alcohol O-H appears at 1407.9 cm−127. Peaks related to the sulfur-containing groups of PEDOT: PSS are also detected. Specifically, the bands appearing at 671.0 cm−1, 941.2 cm−1 and 864.1 cm−1 arise from the C-S stretching mode of the PEDOT thiophene groups59,60,61. The peak at 1161.1 cm−1 is attributed to the SO3H groups of PSS62. This peak appears red-shifted to that of pristine PEDOT: PSS, which is an indication of interaction between the GO flakes and the PEDOT: PSS polymer63.
The Raman results are presented in Fig. 5b and summarised in Table 2. The GO flakes exhibit the characteristic D and G bands at 1345.7 cm−1 and 1589.9 cm−1, respectively. Also visible are the 2D and D + D’ peaks at 2675.9 cm−1 and 2931.0 cm−1, respectively64. The ID/IG ratio of GO is 0.98, which is virtually the same as that calculated for the bulk graphite oxide precursor (ID/IG=0.99). The GO-PEDOT: PSS Raman spectrum exhibits multiple features that are attributed to both the GO and the PEDOT: PSS components. Apart from the characteristic GO Raman modes, additional overlapping low-intensity peaks are detected between ~ 400–1600 cm−1 that are attributed to the presence of PEDOT: PSS65. These Raman features are more clearly indicated in Fig. 5c. The most characteristic PEDOT: PSS Raman peak appears at 1437.4 cm−1 due to the benzenoid units of PEDOT66. More specifically, the peaks at 1437.4 cm−1 and 1507.8 cm−1 arise from the symmetric and asymmetric stretching modes of the aromatic Cα=Cβ bands within the PEDOT thiophene rings, respectively67,68. The peaks at 1240.1 cm−1 and 1353.7 cm−1, on the other hand, arise from the Cα-Cα and Cβ-Cβ single bond stretching modes of the PEDOT chains, respectively16,69. The peak at 1385.3 cm−1 is attributed to the Cβ-Cβ stretching deformation70. Two weak signals are detected at 985.2 cm−1 and 1076.0 cm−1, and are associated with the oxyethylene ring and C-O-C deformations of the PSS units, respectively71. The peak at 712.2 cm−1 is assigned to the symmetric C-S-C deformation, while the 440.8 cm−1 peak appears due to the SO3– ions of the PSS units72,73. The most prominent peaks, however, are the D and G bands of the GO component, which appear red-shifted compared to the pristine GO sample. Specifically, the D band shifts from 1345.7 cm−1 to 1334.6 cm−1, while the G band shifts from 1589.9 cm−1 to 1577.6 cm−1. The intensity of the D band also decreases upon incorporation of the PEDOT: PSS polymer74. The 2D and D + D’ peaks both red-shift to 2674.5 cm−1 and 2917.8 9 cm−1, respectively, and become broader. The ID/IG ratio decreases from 0.99 for pristine GO to 0.88 for GO-PEDOT: PSS, which, along with the shifting of the D and G bands, suggests strong π-π* interactions between the PEDOT chains and the GO lattice and the formation of a composite75,76.

(a) ATR-IR and (b) Raman spectra of exfoliated GO flakes and GO-PEDOT: PSS blend (c) Detailed Raman spectrum of GO-PEDOT: PSS wherein the peaks that arise from the PEDOT: PSS component are indicated.
To ensure the prepared materials meet the criteria for correct printability, we characterised the rheological properties, i.e. the viscosity (η), and surface tension (γ), of the two prepared inks. The viscosity was measured using a viscosimeter at different sheer rates to obtain the flow models for the inks. Both systems behave like classic Newtonian fluids, observed by a classic linear regression intercepting the y-axis to 0 (Figure S1). The presence of PEDOT: PSS in the blend greatly increases the overall viscosity of the system due to the large number of Van der Waals forces between the polymer chains. For this reason, no additive was necessary to improve the printability. For the GO ink, 20% v/v isopropanol (IPA) and 10% v/v propylene glycol (PG) were added to increase the viscosity and reduce surface tension. The average viscosities resulting from the extrapolation of the slope from the flow models were 4.36 cP for GO-PEDOT: PSS ink and 2.45 cP for GO ink (Figure S1).
Surface tension was calculated using the pending drop method (Figure S3). The presence of IPA in the GO ink, decreases the surface tension to 42.54 ± 1.37 mN/m, from the starting 72.36 ± 0.05 mN/m of the GO dispersion in water. For the GO-PEDOT: PSS ink, the measured surface tension is 80.83 ± 0.06 mN/m. Although the addition of IPA to GO-PEDOT: PSS would have lowered the surface tension, it would have also increased the already high viscosity. Consumer printer’s inks typically have viscosities, which range between 2 and 6 cP, and in our formulation, we tried to remain in this range, to avoid possible damaging or clogging of the printhead, without any additive to the blend. For this reason, neither IPA nor PG was added to the GO-PEDOT: PSS aqueous blend, which was printed as is.
Finally, the contact angle (CA) of the two inks with the semi-absorbent plastic substrate was determined by the sessile drop method (Figure S2). The CA is directly correlated with the homogeneity of the printed film and its regularity. Due to the lower surface tension GO ink exhibited better adhesion to the substrate, with an average contact angle of 45.85º, compared to that of GO-PEDOT: PSS which was 83.69º. The rheological profiles of the inks are summarised in Table 3.
The formulated GO and GO-PEDOT: PSS inks were then inkjet-printed along with AgNP ink on PET substrates to fabricate flexible, lightweight MEGs with interdigitated Ag contacts. Digital photographs of the resulting devices are available in the Supplementary Information (Figure S4). The dimensions of each device were 22 × 14 mm, as seen in the optical microscope image (Fig. 6a). The Ag electrodes form a comb-like structure in the centre, with each contact being 2.850 × 0.4 mm2. The GO or GO-PEDOT: PSS moisture-adsorbing layer is printed in between the Ag contacts with an area of 2.825 × 0.4 mm2 or 19.2 mm2. There are 17 of these segments of active material, making the total area A = 19.2 mm2 or A = 0.192 cm2. The flexibility of the substrate used is demonstrated in Fig. 6(b) and (c). Atomic Force Microscopy (AFM) topography and Scanning Electron Microscopy (SEM) images of the inkjet-printed GO and GO-PEDOT: PSS films are available in Figures S5 and S6 of the Supplementary Information, respectively. The surface roughness of the films expressed in Root Mean Square (RMS) was calculated and compared. GO-PEDOT: PSS yielded a smoother film (RMS = 5.14 nm), compared to pure GO (RMS = 21.77 nm).

(a) Optical microscope image of printed MEG layout with dimensions given in mm. (b, c) Demonstration of the flexibility of the developed MEGs. Voltage output as a response to applied relative humidity (RH) variation of (d) GO-based MEG and (e) GO-PEDOT: PSS.
The moisture-induced Vo results are presented in Fig. 6(d) and (e). In each humidity response cycle, the relative humidity (RH) was increased from 40 to 95%, then back to 40%. Both devices exhibited a response to the RH variations, with the Vo increasing and then decreasing with the RH, as the chamber is filled with moisture and subsequently purged. Both MEG types require about 10 min to reach their maximum Vo as the RH increases from 40 to 95%, while they exhibit a rapid response to the RH decrease. The GO device yielded an average voltage output of Vo=183 (± 1.76) mV, while for GO-PEDOT: PSS this value increases to Vo=194 (± 0.97) mV. Dividing the average output voltage by the area A calculated earlier, yields an output of 950 mV·cm−2 for and 1010 mV·cm−2. This increase in Vo can be ascribed to the fact that GO, on one hand, facilitates moisture adsorption, while PEDOT: PSS, on the other hand, acts as an efficient charge transport material77,78. Even though GO promotes ion transport upon moisture absorption, it exhibits intrinsically insulating behaviour, which would reduce the efficiency of charge collection79. The inclusion of PEDOT: PSS likely results in the enhancement of electronic conductivity due to the intrinsic π-conjugated polymer structure of the PEDOT component. Therefore, the addition of PEDOT: PSS may offer better a balance of ionic and electronic conduction, allowing for more efficient charge separation and collection, leading to higher voltage generation from the GO-PEDOT: PSS MEG. It is also worth considering that since GO-PEDOT: PSS has a lower surface roughness (Figure S5) it is likely that the generated surface charges meet less structural resistance during their diffusion, thus reaching the respective electrodes with higher efficiency.
The Sheet Resistance (RSH) and volume resistivity (ρ) of GO and GO-PEDOT: PSS solid films were measured using the Van der Pauw method. For GO, the RSH ranged from 1.00 to 1.51 GΩ−1, while ρ ranged from 14.2 to 21.8 KΩ·cm, results which agree with previous reports80,81,82,83. For GO-PEDOT: PSS the RSH decreases to the range of 3.3–8.1 MΩ−1, while the ρ decreases to 1.1–2.3 KΩ·cm. The decrease in both RSH and ρ can be attributed to the presence of PEDOT: PSS which, as a semiconducting polymer, typically exhibits ρ values between 0.5 and 5 KΩ·cm and RSH values in the range of 103−106 Ω−184,85. This decrease in surface and volume resistance may facilitate enhanced movement of the generated H+ and therefore result in a higher voltage output for the GO-PEDOT: PSS active layer.
A final round of experiments was performed to gain insight into the stability and durability of the devices (a) under prolonged exposure to high humidity conditions (b) over a longer operation period and (c) after undergoing mechanical stress. Firstly, the long-term exposure to high humidity conditions was explored by measuring the output voltage under continuously applied RH = 95% for 3 h (Figure S7). Results show that both devices continue to stably generate voltage under continuous exposure to high humidity, without exhibiting signs of degradation. In the second stability test, each device underwent multiple humidity response cycles in the range of RH = 40–95%, over a prolonged period of 12 h and the output voltage was recorded (Figure S8). Both devices continuously generated voltage for up to 40 cycles. Lastly, the effect of mechanical stress on the devices’ performance was investigated by measuring the humidity-induced Vo before and after 100 bending cycles (Figure S9). The stress caused by bending appears to have a more significant effect on the GO-PEDOT: PSS device with the repeatability of Vo being disrupted. The GO device, on the other hand, exhibits repeatable response cycles and no evident output voltage losses after bending. For comparative purposes, the results were normalised by dividing the voltage output by the maximum value of each measurement during all three stability tests.
A noteworthy topic to be discussed based on this work is the upscalability of the demonstrated MEG devices. An appropriate interconnection of such devices in series or parallel has been reported to increase the overall voltage or current output, respectively, in many dedicated research works on energy harvesting devices86. For example, a voltage output of 2.0 V has been achieved by connecting GO-based MEGs in series, while the same method has been used to power an LCD screen with carbon material-based MEGs15,87.
This course of action appears promising for achieving higher overall generated power from GO- and GO-PEDOT: PSS-based MEGs and working toward powering electronics. The matter of upscalability and large-area manufacturing of energy harvesters can be successfully addressed with techniques described in this manuscript, i.e. inkjet printing and printable inks comprised of liquid-processed functional materials. This research has aimed to yield a proof of concept that can pave the way for viable manufacturing of MEGs in large quantities since they are based on low-capex converting techniques such as inkjet printing and eco-friendly, robust and upscalable materials. Maximizing generated power so far has not been this work’s intention but constitutes a subsequent research objective towards a future publication. Another avenue of research may be the effect of the amount of printed active material on the performance of the MEG device. Our first experiments involved devices fabricated using the same GO and GO-PEDOT: PSS inks described above, but with an inkjet-printed active layer comprised of 10 layers. These devices were found to be non-operational when measuring the humidity response, possibly due to non-compact film formation. By increasing the number of printed layers to 20, the operational devices reported herein were achieved.
Conclusions
In this work, we fabricated and evaluated thin, light, flexible and fully printed GO- and GO-PEDOT: PSS-based MEGs, that convert ambient moisture variation into voltage output through a moisture-induced H+ generation and diffusion. The water-based GO and GO-PEDOT: PSS printable inks were formulated following simple, user-friendly and scalable methods from inexpensive materials. The moisture-induced responses of both devices were tested using a custom-made automated humidity chamber, with results revealing an average voltage output of Vo=183 (± 1.76) mV for pure GO and Vo=194 (± 0.97) mV for GO-PEDOT: PSS for an RH range of 45–95%. We trust that this study may act as proof of concept for the development of low-cost energy harvesters for self-powered portable and wearable technologies that will minimise the dependence on batteries and, by extension, reduce electronic waste.
Materials and methods
Materials
For graphite oxide synthesis, pure graphite powder (< 20 μm) and sodium nitrate were purchased from Sigma Aldrich, potassium permanganate was purchased from Fisher Scientific, while sulfuric acid (95–97%) and hydrogen peroxide (30%) were purchased from Honeywell. Ultrapure water purchased from Honeywell was used to prepare aqueous GO dispersions. PEDOT: PSS AI4083 was purchased from Ossila. Silver Nano™ AgNP ink (NBSIJ-MU01) and semi-absorbent plastic substrates (NB-TP-3GU100) were purchased from Mitsubishi Paper Mills (Tokyo, Japan).
Equipment
LPE of graphite oxide was carried out using a Hielscher (Teltow, Germany) UP200Ht (200 W, 26 kHz) ultrasonic probe. Centrifugations were performed in a Hettich (Tuttlingen, Germany) UNIVERSAL 320 centrifuge. ATR-IR transmission spectra were obtained with a Bruker Vertex 70v FT-IR vacuum spectrometer equipped with a A225/Q Platinum ATR unit with a single reflection diamond crystal. Raman spectra were obtained using a modified HORIBA Scientific LabRAM HR Raman Spectrometer (Kyoto, Japan) using a 532 nm laser UV–Vis absorption spectra were obtained with a Shimadzu UV-2401 PC (Kyoto, Japan) Recording Spectrophotometer. The XPS surface analysis measurements were performed in a UHV chamber (P ~ 5 × 10−10 mbar) equipped with a SPECS Phoibos 100-1D-DLD hemispherical electron analyser and a non-monochromatized dual-anode Mg/Al X-ray source. SpecsLab Prodigy (Specs GmbH, Berlin, Germany) software was used for spectra collection and processing. XRD patterns were obtained with a RIGAKU (Tokyo, Japan) D/MAX-2500 powder diffractometer equipped with a monochromated Cu Kα radiation (λ = 1.5418 Å). DLS was performed with a Nano-ZS Zetasizer from Malvern Panalytical (Worcestershire, United Kingdom) using a Folded Capillary Zeta Cell (DTS1070), and automatic focus and calibration mode. Viscosity measurements were done with an AMETEK Brookfield Viscometer (Massachusetts, USA) for low torque ranges (CZ-40 spindle). Surface tension and contact angle measurements were executed with Drop Shape Analyzer DSA25S (KRÜSS GmbH, Hamburg, Germany). Inkjet printing was carried out with a commercial EPSON XP15000 using refillable cartridges purchased from InkOwl. AFM images were captured using a Park XE7 AFM system from Park Systems (Suwon, South Korea) and subsequently processed and analysed using Gwyddion software. SEM images were obtained using a XHRSEM Magellan 400 L Thermo Fisher Scientific microscope. The humidity-response performance of the devices was carried out using a custom-made automated humidity chamber that utilises electronic solenoids controlled by an Arduino Uno (Monza, Italy) microcontroller board. The Arduino Uno allows for automatic adjustment of the chamber’s relative humidity (RH %), the number of cycles and the wait time of each cycle. The humidity chamber is filled with water vapours and then purged with N2 gas, with the gas pressure standardised at 2 bar. The Arduino Uno is equipped with an AM2302 (DHT22) sensor for real-time monitoring of humidity and temperature levels. Data acquisition was achieved with a Keithley 2700 multimeter. The Van der Pauw Sheet Resistance (RSH) and resistivity (ρ) were measured using a KEITHLEY 4200 A-SCS Parameter Analyzer measurement system and a Cascade Microtech® EPS150TRIAX probe station. Finally, to test the bending stability of the devices, MEGs were attached with a double-sided adhesive tape to a PET substrate and mounted onto the bending apparatus. In this custom-made setup, an Arduino-driven bipolar stepper motor (SM15DD) with a L298N dual H-bridge motor driver was programmed to reach bending values between 0o (flat) and 90o.
Experimental procedures
GO and GO-PEDOT: PSS dispersion preparation and characterisation
To formulate the moisture-active layer, graphite oxide powder was synthesised from pure graphite powder following a modified Hummers’ method. The chemical composition of the as-prepared graphite oxide was investigated using ATR-IR and Raman spectroscopy, as well as XRD and XPS. The synthesised graphite oxide was dispersed in ultrapure water (2 mg/ml, 40 ml) via LPE using an ultrasonic homogeniser (26 kHz, 100% A, Pmax = 200 W, 1 h). The mixture was subsequently centrifugated (9000 rpm, 5 min) to remove any unexfoliated particles, yielding an aqueous GO dispersion. 20 ml of the GO dispersion were used as-is, while the other 20 ml were mixed with PEDOT: PSS at a ratio of 1:1 v/v and stirred at room temperature overnight to create a GO-PEDOT: PSS blend. The as-prepared GO and GO-PEDOT: PSS dispersions were diluted 100 times and their UV-Vis absorption spectra were recorded in the 200–800 nm region.
GO and GO-PEDOT: PSS printable ink formulation
Before formulating printable inks, both dispersions were filtered using a 0.45 μm PVDF filter to remove the larger flakes (> 500 nm) that would clog the inkjet-printer nozzles. To determine the size of the GO flakes in each dispersion and ensure that they are within the acceptable range for the inkjet-printer nozzles, both samples were diluted to 20 µg/ml and DLS was performed to obtain an estimation of the flake size distribution.
The final printable GO ink was formulated by mixing 70% GO aqueous dispersion with 20% IPA and 10% PG to increase the viscosity and reduce surface tension. This resulted in a final concentration of ~ 1 mg ml−1. The viscosities were then measured using a rotational rheometer performing a full scan at different shear speeds and fitting a linear curve extracting the viscosity from the slope of the curve.
The contact angles of the inks on the flexible PET substrate were determined using the sessile drop method, wherein drops of 2 µl were cast onto the surfaces with an automatically software-controlled syringe and 0.5 mm diameter steel needles.
Surface tension values were obtained with the pendant drop method, and the pendant drops were generated by an automatically software-controlled dosser in the tip of a 1.8 mm diameter steel needle.
Consumer inkjet printing of MEGs
MEGs were fabricated using an Epson XP15000 consumer printer loaded with commercial AgNPs ink (15% wt.) for the contacts, and either GO or GO-PEDOT: PSS inks for the active layer. 6 ml of the prepared ink (final concentration ~ 1 mg mL−1) were loaded into an empty cartridge (InkOwl) and installed in the printer in the magenta and cyan colour slot, respectively. The printed patterns are designed using Autodesk AutoCAD 2025, coding each part of the MEGs, with the corresponding colour slot of the printer (magenta for the contacts and cyan for the active layer), and exported as a PDF file. The substrate used (NB-TP-3GU100 from Mitsubishi Paper Mills) is a semi-absorbent plastic (PET) substrate which allows rapid drying of the ink upon contact, inducing room-temperature sintering of the AgNPs made by the same producer. The optimised settings to ensure the maximum possible printing resolution have been described in a previous publication14.
The printing follows the normal consumer procedure, inserting the substrate into the rear feed cassette of the printer and initiating the printing process from the created PDF file. A first layer of the two materials AgNPs and GO or GO-PEDOT: PSS, was printed simultaneously onto the substrate. Additionally, another 19 layers of only GO or GO-PEDOT: PSS were printed on top of the previously deposited one, to ensure sufficient coverage of the substrate and a uniform surface. After the last layer was deposited, the devices were left to dry in a fume hood overnight, completing the fabrication process.
MEG performance evaluation
The performance of the printed MEGs was investigated using a custom humidity test chamber and data acquisition system, described in the “Equipment” section. The Vo of the devices was measured against the applied RH for five response cycles. The wait time of each cycle was set at 10 min, during which the RH varied from 45 to 95% (high RH state) and then back to 45% (low RH state). The temperature was monitored consistently at 26 oC.
Bending stability evaluation
To further validate the flexibility of resulting MEGs, a GO- and a GO: PEDOT: PSS-based MEG were attached atop flexible PET substrates that were subjected to bending via an Arduino-driven stepper motor. Bending started from the flat state (0o bending angle) and ended at almost 90o bending angle. The upper (outer), coated surface of the attached bent sample undergoes greater tensile stresses. No compressive stress has been exerted to the GO- and PEDOT: PSS coating. More relevant information regarding the bending setup can be found in the SI. Before bending, each MEG was subjected to 5 humidity cycles ranging from RH = 40% to RH = 95% and the output voltage was measured. 100 bending cycles were then performed on the devices which were subsequently subjected to an additional 5 humidity cycles ranging from 40 to 95% while having their output voltage measured. During both humidity-induced output voltage measurements, the MEGs were kept in a flat position. The bending equipment is demonstrated in Figure S10, while Figure S11 shows a bending snapshot.
Preparation of GO and GO-PEDOT: PSS films for resistance measurements
To measure the resistance of the developed materials, solid films of GO and GO-PEDOT: PSS were prepared on glass substrates by spray-coating the respective inks88,89. The glass substrates were treated with O2-plasma (50 W, 5 min), first to increase the surface hydrophilicity and ensure better film formation. The spray coating parameters were as follows: nozzle-to-substrate distance: 10 cm, length: 6 cm, substrate temperature: 70 oC, spray speed: 0.025 ml s−1, deposition rate: 0.15 ml cm−2.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Medhekar, N. V., Ramasubramaniam, A., Ruoff, R. S. & Shenoy, V. B. ‘Hydrogen Bond Networks in Graphene Oxide Composite Paper: Structure and Mechanical Properties’, ACS Nano, vol. 4, no. 4, pp. 2300–2306, Apr. (2010). https://doi.org/10.1021/nn901934u
Dinh Trung, V., Chen, S., Xia, H., Natsuki, T. & Ni, Q. Q. ‘A Moisture-Induced Electric Generator with High Output Voltage for Self-Powered Wearable Electronics’, ChemNanoMat, vol. 8, no. 12, p. e202200395, (2022). https://doi.org/10.1002/cnma.202200395
Ali, A. et al. Recent progress in energy harvesting systems for wearable technology. Energy Strategy Reviews. 49, 101124. https://doi.org/10.1016/j.esr.2023.101124 (Sep. 2023).
Liu, G. et al. High-output moisture-enabled electricity generator for fully self-powered wearable physical and biochemical monitoring. Nano Energy. 119, 109098. https://doi.org/10.1016/j.nanoen.2023.109098 (Jan. 2024).
Chen, Y., Gao, Z., Zhang, F., Wen, Z. & Sun, X. ‘Recent progress in self-powered multifunctional e-skin for advanced applications’, Exploration, vol. 2, no. 1, p. 20210112, (2022). https://doi.org/10.1002/EXP.20210112
Anagnostou, K. et al. ‘Geometric Tuning for Enhanced Moisture-Driven Electricity Generation Enabled by Graphene-Oxide Flakes’, Coatings, vol. 12, no. 12, Art. no. 12, Dec. (2022). https://doi.org/10.3390/coatings12121970
Chen, F. et al. ‘High-Performance Flexible Graphene Oxide-Based Moisture-Enabled Nanogenerator via Multilayer Heterojunction Engineering and Power Management System’, Small, vol. 20, no. 39, p. 2304572, (2024). https://doi.org/10.1002/smll.202304572
Huang, Y. et al. Graphene oxide assemblies for sustainable Clean-Water harvesting and Green-Electricity generation. Acc. Mater. Res. 2 (2), 97–107. https://doi.org/10.1021/accountsmr.0c00073 (Feb. 2021).
Qi, X. et al. Ultralight pedot:pss/graphene oxide composite aerogel sponges for electric power harvesting from thermal fluctuations and moist environment. Nano Energy. 77, 105096. https://doi.org/10.1016/j.nanoen.2020.105096 (Nov. 2020).
Chen, Q., Zhao, J. & Cheng, H. Graphene-Based assemblies for Moist-Electric generation. Front. Energy Res. 9 https://doi.org/10.3389/fenrg.2021.738142 (Dec. 2021).
Huang, Y. et al. ‘All-region-applicable, continuous power supply of graphene oxide composite’, Energy Environ. Sci., vol. 12, no. 6, pp. 1848–1856, Jun. (2019). https://doi.org/10.1039/C9EE00838A
Cheng, H. et al. Flexible in-plane graphene oxide moisture-electric converter for touchless interactive panel. Nano Energy. 45, 37–43. https://doi.org/10.1016/j.nanoen.2017.12.033 (Mar. 2018).
Yang, C., Huang, Y., Cheng, H., Jiang, L. & Qu, L. Rollable, stretchable, and reconfigurable graphene hygroelectric generators. Adv. Mater. 31 (2), 1805705. https://doi.org/10.1002/adma.201805705 (2019).
Rosati, G. et al. A plug, print & play inkjet printing and impedance-based biosensing technology operating through a smartphone for clinical diagnostics. Biosens. Bioelectron. 196, 113737. https://doi.org/10.1016/j.bios.2021.113737 (Jan. 2022).
Liang, Y. et al. Jul., ‘Electric power generation via asymmetric moisturizing of graphene oxide for flexible, printable and portable electronics’, Energy Environ. Sci., vol. 11, no. 7, pp. 1730–1735, (2018). https://doi.org/10.1039/C8EE00671G
Yoo, D., Kim, J. & Kim, J. H. Direct synthesis of highly conductive poly(3,4-ethylenedioxythiophene):poly(4-styrenesulfonate) (PEDOT:PSS)/graphene composites and their applications in energy harvesting systems. Nano Res. 7 (5), 717–730. https://doi.org/10.1007/s12274-014-0433-z (May 2014).
Hassan, G., Sajid, M. & Choi, C. Highly sensitive and full range detectable humidity sensor using PEDOT:PSS, Methyl red and graphene oxide materials. Sci. Rep. 9 (1), 15227. https://doi.org/10.1038/s41598-019-51712-w (Oct. 2019).
Popov, V. I., Kotin, I. A., Nebogatikova, N. A., Smagulova, S. A. & Antonova, I. V. ‘Graphene-PEDOT: PSS Humidity Sensors for High Sensitive, Low-Cost, Highly-Reliable, Flexible, and Printed Electronics’, Materials, vol. 12, no. 21, Art. no. 21, Jan. (2019). https://doi.org/10.3390/ma12213477
Sarkar, B., Jaiswal, M. & Satapathy, D. K. Swelling kinetics and electrical charge transport in PEDOT:PSS thin films exposed to water vapor. J. Phys. : Condens. Matter. 30 (22), 225101. https://doi.org/10.1088/1361-648X/aabe51 (May 2018).
Zhanshayeva, L., Favaron, V. & Lubineau, G. ‘Macroscopic Modeling of Water Uptake Behavior of PEDOT:PSS Films’, ACS Omega, vol. 4, no. 26, pp. 21883–21890, Dec. (2019). https://doi.org/10.1021/acsomega.9b02866
Yen, C. T. et al. ‘Charge transfer highways in polymer solar cells embedded with imprinted PEDOT:PSS gratings’, RSC Adv., vol. 4, no. 102, pp. 58342–58348, Nov. (2014). https://doi.org/10.1039/C4RA10268A
‘FTIR Functional Group Database Table with Search - InstaNANO’. Accessed: Dec. 20, 2024. [Online]. Available: https://instanano.com/all/characterization/ftir/ftir-functional-group-search/
Si, Y. & Samulski, E. T. ‘Synthesis of Water Soluble Graphene’, Nano Lett., vol. 8, no. 6, pp. 1679–1682, Jun. (2008). https://doi.org/10.1021/nl080604h
Guo, H. L., Wang, X. F., Qian, Q. Y., Wang, F. B. & Xia, X. H. ‘A Green Approach to the Synthesis of Graphene Nanosheets’, ACS Nano, vol. 3, no. 9, pp. 2653–2659, Sep. (2009). https://doi.org/10.1021/nn900227d
He, J. & Fang, L. Controllable synthesis of reduced graphene oxide. Curr. Appl. Phys. 16 (9), 1152–1158. https://doi.org/10.1016/j.cap.2016.06.011 (Sep. 2016).
Sudesh, N., Kumar, S., Das, C., Bernhard & Varma, G. D. Effect of graphene oxide doping on superconducting properties of bulk MgB2. Supercond Sci. Technol. 26 (9), 095008. https://doi.org/10.1088/0953-2048/26/9/095008 (Jul. 2013).
He, D. et al. Mechanism of a green graphene oxide reduction with reusable potassium carbonate. RSC Adv. 5 (16), 11966–11972. https://doi.org/10.1039/C4RA14511A (Jan. 2015).
Malard, L. M., Pimenta, M. A., Dresselhaus, G. & Dresselhaus, M. S. ‘Raman spectroscopy in graphene’, Physics Reports, vol. 473, no. 5, pp. 51–87, Apr. (2009). https://doi.org/10.1016/j.physrep.2009.02.003
Zólyomi, V., Koltai, J. & Kürti, J. ‘Resonance Raman spectroscopy of graphite and graphene’, physica status solidi (b), vol. 248, no. 11, pp. 2435–2444, (2011). https://doi.org/10.1002/pssb.201100295
Ferrari, A. C. ‘Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects’, Solid State Communications, vol. 143, no. 1, pp. 47–57, Jul. (2007). https://doi.org/10.1016/j.ssc.2007.03.052
Ferrari, A. C. et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 97, 187401. https://doi.org/10.1103/PhysRevLett.97.187401 (Oct. 2006).
Ferrari, A., Robertson, J., Reich, S. & Thomsen, C. ‘Raman spectroscopy of graphite’, Philosophical Transactions of the Royal Society of London. Series A: Mathematical, Physical and Engineering Sciences, vol. 362, no. 1824, pp. 2271–2288, Sep. (2004). https://doi.org/10.1098/rsta.2004.1454
Childres, I., Jauregui, L. A., Park, W., Cao, H. & Chen, Y. P. ‘RAMAN SPECTROSCOPY OF GRAPHENE AND RELATED MATERIALS’, p. 20 .
Kudin, K. N. et al. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 8 (1), 36–41. https://doi.org/10.1021/nl071822y (Jan. 2008).
Trzaska, M. & Bartoszek, W. ‘Hybrid nanocomposite layers Ni/Al2O3/C graphite produced by electrocrystallization method’, Archives of Metallurgy and Materials; ; vol. 64; No 1; 167–173, 2019, Accessed: Dec. 11, 2024. [Online]. (2019). Available: https://journals.pan.pl/dlibra/publication/126233/edition/110146
Lucchese, M. M. et al. Apr., ‘Quantifying ion-induced defects and Raman relaxation length in graphene’, Carbon, vol. 48, no. 5, pp. 1592–1597, (2010). https://doi.org/10.1016/j.carbon.2009.12.057
Hong, J. et al. Origin of new broad Raman D and G peaks in annealed graphene. Sci. Rep. 3 (1), 2700. https://doi.org/10.1038/srep02700 (Sep. 2013).
Claramunt, S. et al. The importance of interbands on the interpretation of the Raman spectrum of graphene oxide. J. Phys. Chem. C. 119 (18), 10123–10129. https://doi.org/10.1021/acs.jpcc.5b01590 (May 2015).
Ban, F. Y., Majid, S. R., Huang, N. M. & Lim, H. N. Graphene oxide and its electrochemical performance. Int. J. Electrochem. Sci. 7 (5), 4345–4351. https://doi.org/10.1016/S1452-3981(23)19543-5 (Jan. 2012).
Siburian, R., Sihotang, H., Raja, S. L., Supeno, M. & Simanjuntak, C. New route to synthesize of graphene nano sheets. Orient. J. Chem. 34 (1), 182–187 (Feb. 2018).
Stobinski, L. et al. Graphene oxide and reduced graphene oxide studied by the XRD, TEM and electron spectroscopy methods. J. Electron Spectrosc. Relat. Phenom. 195, 145–154. https://doi.org/10.1016/j.elspec.2014.07.003 (Aug. 2014).
Sheng, Y., Tang, X., Peng, E. & Xue, J. ‘Graphene oxide based fluorescent nanocomposites for cellular imaging’, J. Mater. Chem. B, vol. 1, no. 4, pp. 512–521, Dec. (2012). https://doi.org/10.1039/C2TB00123C
Powell, C. X-ray photoelectron spectroscopy database xps,version 4.1, NIST standard reference database 20. Natl. Inst. Stand. Technol. https://doi.org/10.18434/T4T88K (1989).
Almalki, M. et al. Improving the operational stability of perovskite solar cells with cesium-doped graphene oxide interlayer. J. Energy Chem. 96, 483–490. https://doi.org/10.1016/j.jechem.2024.04.037 (Sep. 2024).
Rozada, R. et al. From graphene oxide to pristine graphene: revealing the inner workings of the full structural restoration. Nanoscale 7 (6), 2374–2390. https://doi.org/10.1039/C4NR05816J (Jan. 2015).
Al-Gaashani, R., Najjar, A., Zakaria, Y., Mansour, S. & Atieh, M. A. XPS and structural studies of high quality graphene oxide and reduced graphene oxide prepared by different chemical oxidation methods. Ceram. Int. 45 (11), 14439–14448. https://doi.org/10.1016/j.ceramint.2019.04.165 (Aug. 2019).
Stankovich, S. et al. Jun., ‘Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide’, Carbon, vol. 45, no. 7, pp. 1558–1565, (2007). https://doi.org/10.1016/j.carbon.2007.02.034
Johra, F. T., Lee, J. W. & Jung, W. G. ‘Facile and safe graphene preparation on solution based platform’, Journal of Industrial and Engineering Chemistry, vol. 20, no. 5, pp. 2883–2887, Sep. (2014). https://doi.org/10.1016/j.jiec.2013.11.022
Luo, Z., Lu, Y., Somers, L. A. & Johnson, A. T. C. High yield Preparation of macroscopic graphene oxide membranes. J. Am. Chem. Soc. 131 (3), 898–899. https://doi.org/10.1021/ja807934n (Jan. 2009).
Emiru, T. F. & Ayele, D. W. Controlled synthesis, characterization and reduction of graphene oxide: A convenient method for large scale production. Egypt. J. Basic. Appl. Sci. 4 (1), 74–79. https://doi.org/10.1016/j.ejbas.2016.11.002 (Mar. 2017).
Yang, Y. & Liu, T. ‘Fabrication and characterization of graphene oxide/zinc oxide nanorods hybrid’, Applied Surface Science, vol. 257, no. 21, pp. 8950–8954, Aug. (2011). https://doi.org/10.1016/j.apsusc.2011.05.070
Ouyang, J. & ‘Solution-Processed PEDOT:PSS Films with Conductivities as Indium Tin Oxide through a Treatment with Mild and Weak Organic Acids’, ACS Appl. Mater. Interfaces, vol. 5, no. 24, pp. 13082–13088, Dec. (2013). https://doi.org/10.1021/am404113n
Romero, A., Lavin-Lopez, M. P., Sanchez-Silva, L., Valverde, J. L. & Paton-Carrero, A. Comparative study of different scalable routes to synthesize graphene oxide and reduced graphene oxide. Mater. Chem. Phys. 203, 284–292. https://doi.org/10.1016/j.matchemphys.2017.10.013 (Jan. 2018).
Mafamadi, M., Etale, A. & Daramola, M. O. ‘Synthesis and characterization of graphene oxide functionalized with dicarboxylic acids’, Materials Today: Proceedings, vol. 105, pp. 201–208, Jan. (2024). https://doi.org/10.1016/j.matpr.2023.08.291
Pu, L. et al. Feb., ‘Simple method for the fluorinated functionalization of graphene oxide’, RSC Adv., vol. 3, no. 12, pp. 3881–3884, (2013). https://doi.org/10.1039/C2RA20585H
Drewniak, S. et al. ‘Studies of Reduced Graphene Oxide and Graphite Oxide in the Aspect of Their Possible Application in Gas Sensors’, Sensors, vol. 16, no. 1, Art. no. 1, Jan. (2016). https://doi.org/10.3390/s16010103
Khalili, D. ‘Graphene oxide: a promising carbocatalyst for the regioselective thiocyanation of aromatic amines, phenols, anisols and enolizable ketones by hydrogen peroxide/KSCN in water’, New J. Chem., vol. 40, no. 3, pp. 2547–2553, Mar. (2016). https://doi.org/10.1039/C5NJ02314A
Khan, S. A. et al. ‘Fourier transform infrared spectroscopy: fundamentals and application in functional groups and nanomaterials characterization.’ In Handbook of Materials Characterization (eds Sharma, S. K. et al.) 317–344 (Springer International Publishing, 2018). https://doi.org/10.1007/978-3-319-92955-2_9.
Xu, S. et al. Cooperative effect of carbon black and dimethyl sulfoxide on PEDOT:PSS hole transport layer for inverted planar perovskite solar cells. Sol. Energy. 157, 125–132. https://doi.org/10.1016/j.solener.2017.08.009 (Nov. 2017).
Xiao, Y. et al. ‘Pulse electropolymerization of high performance PEDOT/MWCNT counter electrodes for Pt-free dye-sensitized solar cells’, J. Mater. Chem., vol. 22, no. 37, pp. 19919–19925, Aug. (2012). https://doi.org/10.1039/C2JM34425D
Yoo, D. et al. Mar., ‘Effects of one- and two-dimensional carbon hybridization of PEDOT:PSS on the power factor of polymer thermoelectric energy conversion devices’, J. Mater. Chem. A, vol. 3, no. 12, pp. 6526–6533, (2015). https://doi.org/10.1039/C4TA06710J
Jiang, Q. et al. Free-standing PEDOT:PSS film as electrode for the electrodeposition of bismuth telluride and its thermoelectric performance. Int. J. Electrochem. Sci. 9 (12), 7540–7551. https://doi.org/10.1016/S1452-3981(23)10986-2 (Dec. 2014).
Song, H., Liu, C., Xu, J., Jiang, Q. & Shi, H. ‘Fabrication of a layered nanostructure PEDOT:PSS/SWCNTs composite and its thermoelectric performance’, RSC Adv., vol. 3, no. 44, pp. 22065–22071, Oct. (2013). https://doi.org/10.1039/C3RA42414F
López-Díaz, D. et al. ‘Towards Understanding the Raman Spectrum of Graphene Oxide: The Effect of the Chemical Composition’, Coatings, vol. 10, no. 6, Art. no. 6, Jun. (2020). https://doi.org/10.3390/coatings10060524
Garreau, S., Louarn, G., Buisson, J. P., Froyer, G. & Lefrant, S. ‘In Situ Spectroelectrochemical Raman Studies of Poly(3,4-ethylenedioxythiophene) (PEDT)’, Macromolecules, vol. 32, no. 20, pp. 6807–6812, Oct. (1999). https://doi.org/10.1021/ma9905674
Tung, V. C., Kim, J., Cote, L. J. & Huang, J. ‘Sticky Interconnect for Solution-Processed Tandem Solar Cells’, J. Am. Chem. Soc., vol. 133, no. 24, pp. 9262–9265, Jun. (2011). https://doi.org/10.1021/ja203464n
Xu, B. et al. Mar., ‘Functional solid additive modified PEDOT:PSS as an anode buffer layer for enhanced photovoltaic performance and stability in polymer solar cells’, Sci Rep, vol. 7, no. 1, p. 45079, (2017). https://doi.org/10.1038/srep45079
Sharma, R. K. et al. Graphene oxide modified PEDOT:PSS as an efficient hole transport layer for enhanced performance of hybrid silicon solar cells. Surf. Interfaces. 36, 102577. https://doi.org/10.1016/j.surfin.2022.102577 (Feb. 2023).
Kim, Y., Kim, Y. & Kim, J. H. ‘Highly Conductive PEDOT:PSS Thin Films with Two-Dimensional Lamellar Stacked Multi-Layers’, Nanomaterials, vol. 10, no. 11, Art. no. 11, Nov. (2020). https://doi.org/10.3390/nano10112211
Singh, V., Arora, S., Arora, M., Sharma, V. & Tandon, R. P. Characterization of doped PEDOT: PSS and its influence on the performance and degradation of organic solar cells. Semicond. Sci. Technol. 29 (4), 045020. https://doi.org/10.1088/0268-1242/29/4/045020 (Mar. 2014).
Chiu, W. W., Travaš-Sejdić, J., Cooney, R. P. & Bowmaker, G. A. Studies of Dopant effects in poly(3,4-ethylenedi-oxythiophene) using Raman spectroscopy. J. Raman Spectrosc. 37 (12), 1354–1361. https://doi.org/10.1002/jrs.1545 (2006).
Kumar, S. R. S., Kurra, N. & Alshareef, H. N. ‘Enhanced high temperature thermoelectric response of sulphuric acid treated conducting polymer thin films’, J. Mater. Chem. C, vol. 4, no. 1, pp. 215–221, Dec. (2015). https://doi.org/10.1039/C5TC03145A
Jucius, D. et al. Dec., ‘Structure and Properties of Dual-doped PEDOT:PSS Multilayer Films’, Mat. Res., vol. 22, p. e20190134, (2019). https://doi.org/10.1590/1980-5373-MR-2019-0134
Borges, B. G. A. L. et al. Molecular orientation and femtosecond charge transfer dynamics in transparent and conductive electrodes based on graphene oxide and PEDOT:PSS composites. Phys. Chem. Chem. Phys. 21 (2), 736–743. https://doi.org/10.1039/C8CP05382K (2019).
Zhang, J. & Zhao, X. S. ‘Conducting Polymers Directly Coated on Reduced Graphene Oxide Sheets as High-Performance Supercapacitor Electrodes’, J. Phys. Chem. C, vol. 116, no. 9, pp. 5420–5426, Mar. (2012). https://doi.org/10.1021/jp211474e
Anindya, W., Wahyuni, W. T., Rafi, M. & Putra, B. R. ‘Electrochemical sensor based on graphene oxide/PEDOT:PSS composite modified glassy carbon electrode for environmental nitrite detection’, International Journal of Electrochemical Science, vol. 18, no. 3, p. 100034, Mar. (2023). https://doi.org/10.1016/j.ijoes.2023.100034
Yu, J. C. et al. Highly efficient and stable inverted perovskite solar cell employing PEDOT:GO composite layer as a hole transport layer. Sci. Rep. 8 (1), 1070. https://doi.org/10.1038/s41598-018-19612-7 (Jan. 2018).
Reza, K. M. et al. Tailored PEDOT:PSS hole transport layer for higher performance in perovskite solar cells: enhancement of electrical and optical properties with improved morphology. J. Energy Chem. 44, 41–50. https://doi.org/10.1016/j.jechem.2019.09.014 (May 2020).
Sharma, N., Tomar, S., Shkir, M., Kant Choubey, R. & Singh, A. ‘Study of Optical and Electrical Properties of Graphene Oxide’, Materials Today: Proceedings, vol. 36, pp. 730–735, Jan. (2021). https://doi.org/10.1016/j.matpr.2020.04.861
Kumar, R. et al. Investigation of sheet resistance variation with annealing temperature and development of highly sensitive and selective room temperature ammonia gas sensor using functionalized graphene oxide. J. Mater. Sci: Mater. Electron. 32 (2), 1716–1728. https://doi.org/10.1007/s10854-020-04940-0 (Jan. 2021).
Foller, T. et al. Enhanced graphitic domains of unreduced graphene oxide and the interplay of hydration behaviour and catalytic activity. Mater. Today. 50, 44–54. https://doi.org/10.1016/j.mattod.2021.08.003 (Nov. 2021).
Morant-Miñana, M. C. et al. ‘Spatially resolved solid-state reduction of graphene oxide thin films’, Materials Horizons, vol. 5, no. 6, pp. 1176–1184, (2018). https://doi.org/10.1039/C8MH00895G
Wu, Z. Yang,Haolei, Xie, Weibin, and D. and Lu, ‘Resistivity of graphene oxide/alumina-mullite-cordierite ceramic conductive composites’, Fullerenes, Nanotubes and Carbon Nanostructures, pp. 1–8. https://doi.org/10.1080/1536383X.2025.2483358
‘AI 4083 PEDOT:PSS’, Ossila & Accessed May 23, 2025. [Online]. Available: https://www.ossila.com/products/pedot-pss-ai-4083
Huang, Y., Tang, L. & Jiang, Y. ‘Chemical Strategies of Tailoring PEDOT:PSS for Bioelectronic Applications: Synthesis, Processing and Device Fabrication’, CCS Chemistry, vol. 6, no. 8, pp. 1844–1867, Apr. (2024). https://doi.org/10.31635/ccschem.024.202403858
Rogdakis, K., Karakostas, N. & Kymakis, E. ‘Up-scalable emerging energy conversion technologies enabled by 2D materials: from miniature power harvesters towards grid-connected energy systems’, Energy Environ. Sci., vol. 14, no. 6, pp. 3352–3392, Jun. (2021). https://doi.org/10.1039/D0EE04013D
Xue, G. et al. Water-evaporation-induced electricity with nanostructured carbon materials. Nat. Nanotech. 12 (4), 317–321. https://doi.org/10.1038/nnano.2016.300 (Apr. 2017).
Tountas, M., Anagnostou, K., Sotiropoulos, E., Polyzoidis, C. & Kymakis, E. ‘Spray-Coated Transition Metal Dichalcogenides as Hole Transport Layers in Inverted NFA-Based Organic Photovoltaics with Enhanced Stability under Solar and Artificial Light’, Nanoenergy Advances, vol. 4, no. 3, Art. no. 3, Sep. (2024). https://doi.org/10.3390/nanoenergyadv4030014
Anagnostou, K. et al. ‘Enhancing the Dispersibility and Stability of Graphene in Water Using Porphyrin-Based Compounds’, Small Methods, vol. n/a, no. n/a, p. 2401431. https://doi.org/10.1002/smtd.202401431
Acknowledgements
The authors would like to acknowledge the following contributors: For the ATR-IR and Raman spectroscopy measurements, Klytaimnistra Katsara (Department of Agriculture, Hellenic Mediterranean University, Institute of Electronic Structure and Laser, Foundation for Research and Technology—Hellas, (FORTH), GreeceFor the XPS measurements, Emmanuel I. Spanakis (Department of Materials Science and Engineering, University of Crete, Greece)For the XPS analysis, Dimitris Tsikritzis (Department of Electrical & Computer Engineering, Hellenic Mediterranean University, Greece)For the realisation of the bending and angle measurement setup, Angelos Mastrokostas, Lazaros Anthymidis, Antonios Arvanitis (Department of Electrical & Computer Engineering, Hellenic Mediterranean University, Greece)The work has been supported by the European Union’s Horizon 2020 research and Innovation program under project EMERGE. The EMERGE project has received funding under Grant Agreement No. 101008701. The authors acknowledge the Departament de Recerca i Universitats of Generalitat de Catalunya for the grant 2021 SGR 01464.
Author information
Authors and Affiliations
Contributions
K.A. contributed to the research conceptualisation and methodology, the execution of all experiments, the analysis of obtained data, the interpretation of the results, and the writing & visualisation of the original manuscript draft. K.A. synthesised and characterised the graphite oxide powder, prepared and characterised the GO and GO-PEDOT: PSS dispersions, recorded and analysed all UV-Vis absorption spectra and XRD patterns, and analysed all ATR-IR, and Raman results. M.U. performed viscosity, DLS, CA and surface tension measurements on the printable inks. M.U. obtained SEM images of the printed GO and GO-PEDOT: PSS films. M.U. planned and executed the inkjet printing of the MEG devices. M.U. contributed to the writing of the original draft, data analysis and interpretation of results. E.S. performed the AFM measurements and the humidity response measurements of the MEG devices. K.K. performed the stability measurements for evaluation of MEG performance over time and after stress under bending. K.M. optimised the electrical characterisation setup and developed a dedicated LabView-based software for data acquisition. C.P., K.R., K.M. and G.R. contributed to data analysis, interpretation of results, mechanism discussion, visualisation and writing and editing of the final draft. K.R., A.M. and E.K. contributed to the research conceptualisation, while also providing resources and supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Anagnostou, K., Urban, M., Sotiropoulos, E. et al. Water-based graphene oxide inks for inkjet-printed flexible moisture energy generators. Sci Rep 15, 24685 (2025). https://doi.org/10.1038/s41598-025-09628-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09628-1
Keywords
This article is cited by
-
Printing technologies for monitoring crop health
Nature Communications (2026)
