
Abstract
Hepatic ischemia-reperfusion injury (IRI) is a common complication of hepatectomy and liver transplantation. As far as we know, mild hypothermia (MH) is an effective measure to alleviate liver IRI. However, the precise mechanisms have not been fully elucidated. This study investigated the regulatory role of FoxO1/PPARα-mediated energy metabolism in MH-alleviated IRI. Results showed a significant reduction in PPARα expression during liver IRI. Moreover, high PPARα level in post-IR liver specimens correlated with improved liver transplantation outcomes. MH increased blood glucose, elevated gluconeogenesis, and promoted fatty acid oxidation (FAO), indicating a high metabolic state, and also enhanced PPARα expression. Additionally, PPARα promoted FAO and gluconeogenesis, thereby enhancing MH’s protective effects on hepatocytes. Mechanistically, MH promoted FoxO1-PPARα interaction, enhancing PCK1 and CPT1a transcription and boosting hepatic metabolism. Selective FoxO1 inhibition by AS1842856 worsened IRI and apoptosis by reducing gluconeogenesis and FAO, MH partially mitigated this damage and promoted energy metabolism. Additionally, through the FoxO1/PPARα pathway, MH inhibits BAX/Cytochrome C/Caspase-3 cascade-initiated apoptosis. Our findings suggest that MH alleviates liver IRI by activating FoxO1/PPARα-mediated energy metabolism and inhibiting the mitochondria-dependent apoptosis pathway, potentially providing a basis for broader MH application.
Similar content being viewed by others


Introduction
Ischemia-reperfusion injury (IRI) is a major cause of liver damage during liver surgeries, transplantation, and treatment of hemorrhagic shock1. It is a significant risk factor for liver graft dysfunction and rejection following transplantation2. In the early stages of IRI, ROS causes hepatocyte damage through lipid peroxidation, protein oxidation, mitochondrial dysfunction, and DNA damage. Subsequently, inflammatory cells are activated in response to hepatocyte death and cause liver inflammation. Unfortunately, there is no effective prevention or treatment of liver IRI in clinical practice3. Currently, drug-free treatment to ameliorate hepatic IRI is receiving increasing attention4.
Mild hypothermia (MH) (32–35 °C) has emerged as a promising approach that balances metabolic suppression with a reduced risk of cold-induced injuries. Increasing research supports that MH demonstrates therapeutic potential in treating cardiac arrest, ischemic stroke, and liver injury5,6,7. Multiple mechanisms, including inhibition of apoptosis, necrosis, autophagy, and inflammatory response, are implicated in MH’s protective effects after IRI8,9. Notably, metabolic reprogramming may play a crucial role in mitigating IRI under MH7. Nonetheless, a more comprehensive understanding of the signaling pathways and downstream targets through which MH regulates liver metabolism and exerts protective effects is still lacking and warrants further investigation.
The liver is a high-energy-consuming organ, and energy substrate metabolism is crucial for maintaining overall metabolic function10. Alterations in energy substrate utilization, particularly glucose and fatty acid (FA) metabolism, are critical determinants of hepatocyte vulnerability to IRI11. In recent years, energy balance processes such as gluconeogenesis and fatty acid oxidation (FAO) have become increasingly important in the context of IRI2,12,13,14. During IRI, FAO is inhibited, leading to lipotoxicity and energy deprivation, characterized by intracellular lipid accumulation, decreased adenosine triphosphate (ATP) levels, and renal tubular cell injury and death. Conversely, upregulation of FAO has been shown to attenuate kidney injury15. Additionally, a recent study confirmed that overexpression of the gluconeogenic enzyme PCK1 can protect against hepatic IRI16.
Numerous nuclear receptors are involved in a wide range of metabolic processes, including lipid, bile acid, glucose and cholesterol metabolism17. Peroxisome proliferator-activated receptor-α (PPARα) is a ligand-activated transcription factor that belongs to the nuclear receptor superfamily18. PPARα plays a central role in energy metabolism by directly regulating fatty acid oxidation genes and influencing gluconeogenesis-related pathways19. PPARα directly regulates the transcription of key enzymes such as acyl-CoA oxidase (ACO) and the multifunctional enzyme hydratase/dehydrogenase (HD), enhancing the β-oxidation process of fatty acids20. This breaks down fatty acids into acetyl-CoA, which then enters the tricarboxylic acid (TCA) cycle or generates ketone bodies for energy production. Upon activation, PPARα promotes fatty acid oxidation, leading to the generation of acetyl-CoA. This acetyl-CoA can inhibit the activity of pyruvate carboxylase (PC), thereby reducing the production of oxaloacetate and decreasing the substrate supply for the gluconeogenesis pathway21. Studies have shown that PPARα regulates several genes involved in FAO. Upregulating FAO through overexpression of Ppargc1a or by using a PPARα agonist can attenuate acute organ injury22. Numerous studies have shown that PPARα is regulated by Forkhead box protein O1 (FoxO1) and participates in metabolic processes, such as FAO and gluconeogenesis, through the FoxO1 pathway.
FoxO1 is a transcription factor within the Forkhead box O subfamily, playing a pivotal role in regulating various cellular processes, including metabolism, oxidative stress response, cell cycle regulation, and apoptosis23,24. FoxO1 exerts its biological functions by binding to specific DNA sequences and regulating target gene expression25. One of FoxO1’s central roles in metabolism is managing cellular stress responses through regulation of gluconeogenesis and FAO, which are crucial for maintaining energy balance and mitigating oxidative damage26,27. Several studies indicate that FoxO1 primarily participates in IRI by regulating autophagy, apoptosis, inflammation, and maintaining mitochondrial function28,29,30. In recent years, energy balance processes such as gluconeogenesis and FAO have become increasingly important in the context of IRI2,31,13. Therefore, whether FoxO1 contributes to the alleviation of IRI under MH by regulating PPARα-mediated energy metabolism remains to be investigated.
Although both hypothermia and IRI significantly affect the expression of FoxO1, PPARα, and energy metabolism, the role of FoxO1/PPARα-mediated energy metabolism in reducing hepatic IRI during MH is not clear. Therefore, in this study, we investigated the effect of mitochondria-mediated energy metabolism and apoptosis on IRI and explored the role of FoxO1/PPARα in MH-mediated IRI tolerance.
Materials and methods
Animals
C57BL/6 male mice (6–8 weeks old, weight: 18–20 g) were purchased from Wuhan Wan Qian Jia Xing Bio-Technology Co. Ltd. (Wuhan, China). All mice had ad libitum access to food and water and were housed in a specific pathogen-free facility with a 12-hour light/dark cycle. All animal experiments were approved by the Animal Care Committee of Wuhan University Zhongnan Hospital (ZN2022214), and conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85 − 23, 1996, revised 2011) and also the ARRIVE guidelines (https://arriveguidelines.org) for the care and use of laboratory animals.
Animals experiment
The FoxO1 inhibitor AS1842856 (8 mg/kg) (Med Chem Express, New Jersey, USA) was dissolved in PBS containing 1% DMSO and administered intraperitoneally 1 h prior to warm ischemia. The mice in the MH and MH + IR groups were rapidly cooled to 32.0 ± 0.25 °C, maintained at this temperature for 2 h using an icebox and heating panel, and then gradually rewarmed to 36.2 ± 0.2 °C. For the liver IRI model, a 70% liver warm ischemia model was used7. Briefly, mice were anesthetized with an intraperitoneal injection of 1% sodium pentobarbital. After a median abdominal incision, an atraumatic clip was applied to obstruct the hepatic artery and portal venous blood supply to the left and middle liver lobes. After 1 h of ischemia, the clips were removed to initiate hepatic reperfusion. After 6 h of reperfusion, the mice were re-anesthetized and euthanized by cervical dislocation. After blood collection, 5 mL of cold heparinized Ringer’s solution was injected through the abdominal aorta to flush the liver, followed by immediate collection of liver samples.
On the day of the experiment, mice were randomly divided into 6 groups: (1) normal group (N), in which liver samples were obtained without IR; (2) MH group (MH), where mice were treated with MH for 2 h; (3) ischemia reperfusion (IR) group, in which mice were exposed to 70% hepatic ischemia for 1 h, followed by 6 h of reperfusion; (4) MH + IR group, where mice were treated with MH for 2 h and then exposed to IR; (5) IR + AS1842856 group, in which mice received AS1842856 for 1 h before IR; and (6) MH + IR + AS1842856 group, where mice received AS1842856 for 1 h before ischemia.
Cell culture
BRL-3 A cells (Bnbio, Beijing, China) were cultured in DMEM supplemented with 10% FBS and 1% (100x) penicillin-streptomycin solution (Biological Industries, Beit-Haemek, Israel) at 37 °C in a 5% CO2 atmosphere. Cells were used at passages 3–5 with a seeding density of 2 × 106 cells/well and achieved 80–90% confluency at the time of experiments. To simulate hypoxia/re-oxygenation (H/R) in vitro, oxygen and glucose supply to the cells were halted for 12 h, followed by culturing in 10% FBS medium under normal air conditions for 6 h, as previously described32. The MH + H/R group consisted of cells incubated with complete DMEM at 32 °C for 6 h prior to H/R. In the AS1842856 + H/R group, cells were pre-treated with the inhibitor AS1842856 (0.5 µM) at 37 °C for 12 h prior to H/R. In the AS1842856 + MH + H/R group, cells were pre-treated with AS1842856 for 12 h (6 h at 37 °C, 6 h at 32 °C), followed by exposure to H/R. Finally, cell samples were harvested.
Transfection
To investigate the role that PPARα plays in the protective effects of MH on IRI, a specific overexpressed plasmid (ov-PPARα) and siRNA (si-PPARα) were used. Cells were transfected with si-PPARα or ov-PPARα using Lipofectamine 3000 (Invitrogen, Shanghai, China), all processes were according to the manufacturers’ protocols. After transient transfection, the cells underwent H/R with MH pretreatment. The siRNA sequences used are listed in Supplementary Table 1.
Biochemical analysis
Plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), serum glucose content (GLU) and lactate were measured by automatic analysis (Beckman Coulter, Inc., California, USA) at Zhongnan Hospital of Wuhan University.
Liver histology and TUNEL staining
Ischemic lobes were perfused with cold (4 °C) Ringer’s solution via the abdominal aorta, fixed with 4% formalin, embedded in paraffin, cut into 5-µm sections, and stained with haematoxylin and eosin (H&E). The severity of tissue IRI was scored from 0 to 12 using Suzuki’s criteria. The proportion of apoptotic cells was determined using a terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) assay kit (Servicebio, Wuhan, China). Fluorescent images were selected in three random fields of view (100×) per section using a fluorescence microscope and analysed using Image-Pro Plus 6.0 image analysis software (Media Cybernetics, Rockville, MD, USA). The rate of apoptosis was calculated using the following formula: TUNEL-positive cells/total number of hepatocytes ×100%.
Western blot analysis
Western blotting was performed as described previously8. The following primary antibodies were used in the experiment: rabbit anti-FoxO1, rabbit anti-PPARα, rabbit anti-ACOX1, rabbit anti-CPT1a, rabbit anti-PCK1, rabbit anti-CS, rabbit anti-IDH2, rabbit anti-FASN, rabbit anti-SREBP1, rabbit anti-Cleaved caspase-3 (1:1000, Proteintech, Manchester, UK), mouse anti-β-Actin (1:5000, Proteintech, Manchester, UK), mouse anti-BCL-2, rabbit anti-BAX (1:600, Bioss Biotechnology, Beijing, China), rabbit anti-p-FoxO1 (phosphorylated at S256) (1:800, Cell Signaling, Danvers, USA). Immunoreactive bands were detected using enhanced chemiluminescence (Wuhan Biotechnology, Wuhan, China). Fluorescence intensity was measured using Image-Pro Plus 6.0 image analysis software.
qRT-PCR
Total RNA was extracted from animal tissues and cells using TRIzol reagent. RNA concentration and purity were determined using a NanoDrop 2000. Subsequently, 2 µg of RNA was reverse-transcribed into cDNA. Quantitative PCR (qPCR) analysis was performed using ChamQ SYBR qPCR Master Mix (Vazyme) according to the manufacturer’s instructions. β-Actin was used as the internal control. The primer sequences used are listed in Supplementary Table 2.
Apoptosis analysis
The BRL-3 A cells from each group were collected after experimental treatment. Thereafter, the cells were washed three times with PBS (0.01 M, pH 7.4), and suspended in Annexin V-FTIC (5 µl) and PI (10 µl). Next, the cells were incubated at 37 °C for 15 min in the dark. Finally, 400 µl of 1 × Binding Buffer was added, and the cells were analyzed by flow cytometry. The cell apoptosis rate was calculated.
Co-immunoprecipitation (Co-IP)
To determine whether FoxO1 directly interacts with PPARα in vitro, the ability of the two proteins to co-immunoprecipitate was detected using a Pierce Classic Magnetic IP/Co-IP Kit (Thermo Scientific, Massachusetts, USA). Anti-IgG (1:750, Proteintech, Manchester, UK) was used as a negative control. Aliquots of cell lysates were incubated with anti-FoxO1 (1:100, Proteintech, Manchester, UK) or anti-IgG (1:100, Proteintech, Manchester, UK) overnight at 4 °C. The antigen/antibody complex was bound to protein A/G magnetic beads for 1 h at room temperature. Then, the magnetic beads were washed three times with IP Lysis/Wash and once with purified water. Next, low-pH elution buffer was added and the supernatant containing the target antigen was saved. The immunoprecipitated protein complexes were separated by SDS-PAGE and analysed using an immunoblot assay using anti-FoxO1 or anti-PPARα antibody. Immunoreactive bands were detected using enhanced chemiluminescence (Wuhan Biotechnology, Wuhan, China). Fluorescence intensity was measured using Image-Pro Plus 6.0 image analysis software.
Immunofluorescence
BRL-3 A cells were seeded on cover glass and incubated at 37 °C overnight prior to treatment. The treated cells were fixed with 4% formalin for 20 min and processed for immunofluorescence staining using standard procedures. The cells were incubated with anti-FoxO1, anti-PPARα and anti-Cleaved caspase-3 at 4 °C in the dark overnight. Then, the samples were washed three times with PBS and incubated with secondary antibodies conjugated to Alexa Fluor 555. After extensive washing steps, the samples were stained with DAPI. Sections were imaged using a 3D Histech Panoramic digital slide scanner (3DHistech Ltd, Budapest, Hungary).
Human tissue specimens
Human liver tissues were obtained from donors at the Institute of Hepatobiliary Diseases of Wuhan University Zhongnan Hospital, Basic clinical information about the donor and recipient were provided in the Supplementary Table 3. The donation after circulatory death (DCD) liver were collected immediately after a period of warm and cold ischemia, and 1 h after opening the portal vein during orthotopic liver transplantation (OLT), and stored in a fixative or frozen at -80 °C. This study was conducted in accordance with the Declaration of Helsinki and approved by the Medical Ethics Committee of Wuhan University Zhongnan Hospital (Approval No: 2020122). Informed consent was obtained from all participants and their legal guardians.
RNA-sequencing
For the RNA-sequencing (RNA‐seq) analysis, total RNA was extracted from pre- and post-transplantation human DCD liver samples. Then, cDNA libraries were constructed and sequenced on an Illumina Novaseq 6000 platform with 2 × 150 bp read length. Expression levels of all mRNAs were evaluated using StringTie software (v1.3.44d), with the default parameters retained for other settings. DESeq2 software was used to calculate differentially expressed mRNAs according to the following two criteria: |log2FC| >= 0.585 and adjusted p value < 0.05.
Statistical analysis
All data were analyzed using the GraphPad Prism (version 9.3) software. The data are expressed as mean ± standard deviation (SD) of at least three experiments. Statistical analyses were performed with one-way ANOVA. Statistical significance was defined as P < 0.05.
Results
PPARα expression decreased after OLT and correlates with clinical outcomes
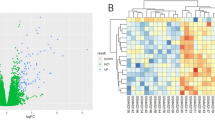
To investigate changes in gene expression following liver transplantation, we collected 3 pairs of clinical DCD liver specimens before and 1 h after reperfusion and conducted transcriptomic analysis. KEGG33,34 and GO analyses revealed significant regulation of energy metabolism pathways following liver transplantation (Fig. 1A). Hierarchical clustering of both upregulated and downregulated genes revealed distinctive patterns within these groups, and PPARα mRNA expression was significantly decreased after OLT (Fig. 1B,C), which was further confirmed by immunofluorescence, qRT-PCR and Western blot (Fig. 1D–F). Finally, logistic regression analysis was performed to investigate whether PPARα expression is associated with postoperative ALT and AST levels in clinical OLT patients. We found a significant negative correlation between PPARα expression levels and ALT and AST levels in patients following liver transplantation (Fig. 1G).

PPARα expression decreased after OLT and correlates with clinical outcomes. (A) GO and KEGG pathways altered in pre- vs. post-OLT liver tissues. (B) Heatmap analysis of pre-vs. post-OLT group. (C) Volcano plot of differentially regulated genes, P < 0.05 were considered significant. (D) Representative images of HE and PPARα immunofluorescence image in pre- vs. post-OLT liver tissues, scale bar = 100 μm and their quantification results. (E,F) Change of PPARα mRNA and protein expression in the paired liver tissues. (G) The Pearson correlation analysis of PPARα and serum ALT/AST levels.
MH attenuates hepatic IRI by regulating FAO and gluconeogenesis
We then established a mouse model of hepatic IRI to investigate whether MH could alleviate hepatic IRI. We found that MH significantly alleviated liver damage, reduced liver injury scores, and lowered serum ALT/AST levels (Fig. 2A,B). Next, we then investigated changes in energy metabolism following MH. We found that hepatic glucose and free fatty acids decreased following IR, while serum lactate levels increased. MH promotes triglyceride hydrolysis and enhances glucose levels, while hepatic glycogen levels remained unchanged (Fig. 2C–F). PAS staining revealed that glycogen levels were reduced following IRI and were not altered by MH (Fig. 2G). These data indicate that MH mediates its gluconeogenic effect by promoting fat mobilization and gluconeogenesis. Consequently, we measured the mRNA and protein expression of key enzymes involved in glucose and fatty acid metabolism. Accordingly, we found that the expression of CPT1α and PCK1 decreased following IR and was partly rescued by MH, while the expression of FASN showed the opposite trend (P < 0.05) (Fig. 2H–J).

MH attenuates hepatic IRI by regulating FAO and gluconeogenesis (A) Representative images of H&E stained liver tissues, 100x, and the Suzuki’s histological score of liver tissue. (B) Serum ALT levels and AST levels (n = 5). (C–F) Serum glucose (GLUC), free fatty acid (FFA), triglyceride (TRIG) and lactate levels (n = 5). (G) Representative images of PAS staining (100x) and the quantification data. (H–J) Representative blots of proteins that are related to FAO and gluconeogenesis and their quantification results. data are expressed as mean ± SD; *P < 0.05, **P < 0.01.
MH alleviates hepatic IRI by activating PPARα
As shown in Fig. 3A, PPARα expression was significantly reduced in the IR group compared to the control, while MH treatment enhanced PPARα expression both in normal and IR condition, suggesting a potential link between PPARα and the protective effects of MH. To further explore whether MH exerts its protective effects by activating PPARα, we manipulated its expression in BRL-3A and AML12 cells subjected to H/R. Western blot analysis confirmed the successful overexpression and knockdown of PPARα (Fig. 3B), validating the reliability of our experimental approach. We found that H/R increased the levels of ALT and AST in the culture medium, overexpression of PPARα reduced the levels of ALT and AST in the culture medium and decreased the cell apoptosis rate. In contrast, inhibition of PPARα increased the levels of ALT and AST in the cells and the cell apoptosis rate (Fig. 3C,D). Critically, PPARα knockdown in the MH + H/R (MH + H/R + si-PPARα) group abolished the protective effects of MH, with ALT/AST levels and apoptosis rates significantly elevated compared to the MH + H/R group. Conversely, PPARα overexpression (MH + H/R + ov-PPARα) synergized with MH, further reducing ALT/AST levels and apoptosis beyond the MH + H/R group alone (Fig. 3C,D).

Protective effects of PPARα on H/R of hepatocytes. (A) Representative blots of PPARα and their quantification results; Relative mRNA level of PPARα. (B) Western blotting verified the successful overexpression and knockdown of PPARα. (C) ALT and AST levels in culture medium of BRL-3 A cells. (D) Flow cytometry analysis of apoptosis in BRL-3 A cells and the quantification results. (E) Representative immunofluorescence image of cleaved-caspase-3 in BRL-3 A cells. (F) Representative blots of cleaved caspase-3, BCL2 and BAX and their quantification results. (G) Relative mRNA level of cytochrome C. n = 3 per group; data are expressed as mean ± SD; *P < 0.05, **P < 0.01.
Immunofluorescence and western blot analyses delineated PPARα’s regulatory role in apoptosis. Compared to the H/R group, MH treatment (MH + H/R group) significantly reduced caspase-3 cleavage and cytochrome C levels while elevating the BCL2/BAX ratio. Crucially, PPARα knockdown in the H/R (H/R + si-PPARα) group exacerbated H/R-induced apoptosis, with caspase-3 cleavage and cytochrome C levels surpassing those in the H/R group. In contrast, PPARα overexpression (H/R + ov-PPARα) significantly suppressed caspase-3 activation and cytochrome C release, recapitulating the anti-apoptotic effects of MH treatment (Fig. 3E–G).
To further elucidate the MH’s dependency on PPARα, we further compared MH-treated groups under PPARα-modulated conditions. In the MH + H/R group, PPARα knockdown (MH + H/R + si-PPARα) abolished MH’s protection, restoring caspase-3 activation and cytochrome C release to H/R levels. Conversely, PPARα overexpression (MH + H/R + ov-PPARα) synergized with MH, suppressing apoptosis markers beyond the MH + H/R group alone. These findings suggest a synergistic protective role of PPARα and MH against hepatic IRI by suppressing cytochrome C release and subsequent apoptotic signaling.
PPARα alleviates hepatic IRI under MH by regulating the expression of CPT1a and PEPCK1
During liver IRI, oxidative stress is a key mechanism contributing to tissue damage. Reactive oxygen species (ROS) and malondialdehyde (MDA) are well-established markers of oxidative damage. Excessive ROS production triggers lipid peroxidation, leading to the accumulation of MDA, which in turn causes protein denaturation, DNA damage, and apoptosis, thereby exacerbating inflammation and tissue injury35. Antioxidants, such as nitric oxide (NO) and GSH, play pivotal roles in regulating oxidative stress. NO exhibits vasodilatory, anti-inflammatory, and anti-apoptotic effects, while GSH serves as the primary intracellular antioxidant, effectively scavenging excess ROS and maintaining cellular redox balance36. To further investigate the role of PPARα in mitigating liver IRI under MH, we assessed changes in ROS and NO levels to explore its regulation of redox balance in BRL-3 A cells. Treatment of BRL-3 A cells with H/R significantly increased ROS and MDA production and reduced the NO level and GSH/GSSG ratio. This effect was suppressed in the MH + H/R group (Fig. 4A–C), indicating that MH can alleviate oxidative stress in H/R. Furthermore, we found that silencing PPARα induced cellular ROS and MDA accumulation and reduced the NO level and GSH/GSSG ratio, whereas PPARα overexpression exhibited the opposite trend (Fig. 4A–C). Crucially, PPARα knockdown in the MH + H/R group (MH + H/R + si-PPARα) significantly elevated ROS and MDA levels while suppressing NO production and the GSH/GSSG ratio compared to MH + H/R group. Conversely, PPARα overexpression (MH + H/R + ov-PPARα) further enhanced MH’s ability to suppress oxidative stress compared to the MH + H/R group (Fig. 4A–C).

MH promotes FAO and gluconeogenesis through PPARα on H/R. (A) Representative images of intracellular ROS and NO levels with various treatments. Experiments were performed three times with similar results. Scale bar: 100 μm. (B) MDA was measured in BRL-3 A cells as indicators of oxidative stress. (C) GSH/GSSG ratio was measured in BRL-3 A cells as indicators of antioxidant defense. (D–G) Representative blots of PPARα, CPT1a, PEPCK1 and their quantification results. n = 3 per group; data are expressed as mean ± SD; *P < 0.05, **P < 0.01.
PPARα is a known regulator of FAO and glucogenesis, we further tested the expression of CPT1A and PEPCK1 expression in PPARα silencing or overexpressing cells. We found that PPARα knockdown in the H/R (H/R + si-PPARα) group significantly suppressed CPT1a and PEPCK1 expression compared to H/R alone, whereas PPARα overexpression (H/R + ov-PPARα) partially restored CPT1a and PEPCK1 levels. Importantly, PPARα knockdown in the MH + H/R (MH + H/R + si-PPARα) group attenuated MH’s restorative effects on CPT1a and PEPCK1, while PPARα overexpression (MH + H/R + ov-PPARα) synergistically enhanced the expression of CPT1a and PEPCK1 beyond MH treatment alone (Fig. 4D–G). Consistent results were also obtained using AML12 cells, which are presented in the Supplementary Fig. 1 and 2. These findings align with those observed in BRL-3 A cells, confirming the consistency of our results across different cellular models.
MH promote FoxO1 and PPARα interaction
The FoxO1-PPARα interaction was initially predicted through multiscale structural modeling using AlphaFold 3 in rat (Fig. 5A). Cross-species validation confirmed conserved interaction patterns in human and mouse, as detailed in Supplementary Fig. 3A. The protein–protein interaction pairs were also identified using the STRING database (Supplementary Fig. 3B). The results predicted that the interaction between FoxO1 and PPARα (Fig. 5A). To explore the potential interaction between FoxO1 and PPARα, we used immunofluorescence staining to examine their co-localization after H/R. The results showed that the co-localization of PPARα and FoxO1 was reduced after H/R. Interestingly, MH enhanced the nuclear translocation of FoxO1 and PPARα and promoted their co-localization (Fig. 5B). We then performed immunoprecipitation to detect the potential interaction between PPARα and FoxO1. Initial co-immunoprecipitation (Co-IP) assays using FoxO1 as the bait protein demonstrated physical interaction between PPARα and FoxO1 under basal conditions, with attenuated binding after H/R that was restored by MH pretreatment (Fig. 5C). To exclude the possibility that reduced interaction resulted from decreased PPARα expression, we performed reciprocal Co-IP experiments using PPARα as the bait. Consistent with our previous findings, this reverse approach confirmed significant attenuation of PPARα-FoxO1 interaction post-H/R (p < 0.01 vs. control), which was remarkably rescued by MH treatment. Westernblot and qRT-PCR showed that, following IR injury, the levels of FoxO1 reduced. However, pre-treatment with MH increased the expression of FoxO1 and decreased the phosphorylated FoxO1 levels (Fig. 5D-E).

MH promote FoxO1 and PPARα interaction. (A) AlphaFold 3 websites predict protein interactions between PPARα and FoxO1. (B) Representative immunofluorescence image of FoxO1 and PPARα in BRL-3 A cells treated with IR or MH + H/R, scale bar = 10 μm. (C) The immunoprecipitates were blotted with the relative antibodies. (D) Representative blots of p-FoxO1, FoxO1 and their quantification results. (E) Relative mRNA level of FoxO1. n = 3 per group; data are expressed as mean ± SD; *P < 0.05, **P < 0.01.
AS1842856 reduced FoxO1 and PPARα interaction
To delineate the fundamental regulatory mechanism between FoxO1 and PPARα, we first investigated FoxO1-PPARα interplay without H/R or MH. Under basal physiological conditions, inhibition of FoxO1 using AS1842856 significantly reduced both FoxO1 and PPARα protein expression. Crucially, co-immunoprecipitation analysis demonstrated that the attenuation of FoxO1-PPARα physical interaction exceeded the observed protein reduction, establishing that FoxO1 principally regulates PPARα activity through direct molecular binding (Fig. 6A). The results showed that the co-localization of PPARα and FoxO1 was enhanced after H/R, and AS1842856 treatment inhibited FoxO1 expression and reduced the co-localization of FoxO1 and PPARα (Fig. 6B). We then performed immunoprecipitation to detect the potential interaction between PPARα and FoxO1. The results showed that overexpression of FoxO1 enhanced its interaction with PPARα, while inhibition of FoxO1 weakened its interaction with PPARα (Fig. 6C). Interestingly, MH enhanced the nuclear translocation of FoxO1 and PPARα and promoted their co-localization, while inhibition of FoxO1 suppressed the nuclear translocation of PPARα (Fig. 6D-E).

AS1842856 reduced FoxO1 and PPARα interaction. (A) Immunoprecipitation in BRL-3 A cells treated with AS1842856 followed by immunoblotting using anti-PPARα and anti-FoxO1 antibodies. (B) Representative immunofluorescence image of FoxO1 and PPARα in BRL-3 A cells treated with AS1842856 on H/R, scale bar = 10 μm. (C) Immunoprecipitation with anti-FoxO1 antibody in BRL-3 A cells followed by immunoblotting using anti-PPARα and anti-FoxO1 antibodies. (D) Representative immunofluorescence image of FoxO1 and PPARα in BRL-3 A cells treated with AS1842856 on MH + H/R, scale bar = 10 μm. (E) Immunoprecipitation with anti-FoxO1 antibody in BRL-3 A cells followed by immunoblotting using anti-PPARα and anti-FoxO1 antibodies.
FoxO1 Inhibition aggravated hepatic IRI
We further investigated whether FoxO1 is involved in hepatic IRI. The results shown that the inhibition effect was most significant at 8 mg/kg for 1 h in vivo (Supplementary Fig. 4B). We observed no effect on hepatic function and injury using 1% DMSO vehicle (Supplementary Fig. 4C-D). Compared with the IR group, AS1842856 treatment aggravated liver damage, promoted hepatocyte necrosis and apoptosis, elevated ALT and AST levels, whereas these increases were suppressed in the MH + IR + AS group (Fig. 7A–C). We then explored whether energy metabolism was changed after inhibiting FoxO1 expression. The results shown that serum TRIG and lactate were significantly increased in the IR + AS group compared with the IR group, whereas that of glucose and GSH/GSSG ratio showed the opposite result (Fig. 7D–H). However, these results were partially reversed by MH. Taken together, these data suggest that MH alleviates liver IRI by promoting the expression of FoxO1.

FoxO1 inhibition aggravated hepatic IRI. (A) Representative images of HE staining (100x) and the Suzuki’s score. (B) Representative images of TUNEL staining (100x) and cell apoptosis rate. (C) Serum ALT levels and AST levels (n = 5). (D–G) Serum GLUC, FFA, TRIG and lactate levels (n = 5). (H) Liver GSH/GSSG ratio (n = 5). (I) Representative blots of p-FoxO1, FoxO1, PCK1, CPT1a and PPARα and their quantification results. (J) Schematic illutration on the proposed mechanism of FoxO1/PPARα-mediated energy metabolism contributes to the alleviation of hepatic IRI in MH. Data are expressed as mean ± SD; *P < 0.05, **P < 0.01.
We next tested key FAO and gluconeogenesis enzymes, MH combined with AS1842856 increased CPT1a, PCK1, and PPARα expression compared with the IR + AS1842856 groups (Fig. 7I). These results strongly suggest that FoxO1 mediates fatty acid metabolism and gluconeogenesis and participates in the protective effect of MH.
Discussion
Liver IRI is a major complication closely associated with morbidity and mortality following liver transplantation37. Strategies to alleviate IRI can be roughly divided into pharmaceutical, chemical, genetic, and surgical interventions38. Unfortunately, there are currently no effective drugs available in clinical practice to alleviate hepatic IRI. This paper explores mild hypothermia as a potential drug-free therapeutic intervention that can significantly improve IRI and delves into its protective mechanisms. Recent studies demonstrated that MH is an effective neuroprotective therapy and plays a positive role in kidney transplantation39,40. These studies suggest that MH could be an effective strategy for overcoming hepatic IRI. Our previous studies indicated that MH alleviates hepatocyte IRI by regulating FAO7,41. However, the underlying mechanism remains elusive. Our findings indicate that the expression of PPARα, a key component in maintaining metabolic homeostasis, is upregulated in hepatic IRI during MH. Upregulating PPARα further promotes the protective effect of MH on hepatocyte IRI. Furthermore, MH, by activating FoxO1 expression, promotes the interaction with PPARα, which in turn enhances gluconeogenesis and FAO to improve metabolic efficiency. Simultaneously, FoxO1/PPARα also upregulates the expression of the mitochondrial anti-apoptotic gene BCL2 and the antioxidant GSH. Together with the oxidative benefits from FAO, this helps maintain the redox balance within mitochondria and prevents mitochondrial dysfunction. Ultimately, this protective mechanism shields cells from apoptosis mediated by the BAX /Cytochrome C/Caspase-3 pathway (Fig. 7J).
The liver is the largest central organ in energy homeostasis, relies on the tightly regulated balance of glucose and lipid metabolism to meet its energy demands, particularly during and after IRI42. Disruptions in these metabolic processes can exacerbate cellular damage and compromise mitochondrial function, leading to further injury. The metabolic shift from oxidative phosphorylation to glycolysis during ischemia increased glucose consumption and accelerated lactate production2while initially protective, can result in acidosis and exacerbate cellular injury. High PCK1 (the rate-limiting enzyme of gluconeogenesis) level in human IR liver specimens was closely correlated with better outcomes of liver transplantation2. One study showed that during ischemia-reperfusion, hepatic glycogen and glucose are significantly depleted through anaerobic glycolysis, leading to lactate accumulation. MH pretreatment promotes gluconeogenesis by activating PCK1 expression, resulting in a decrease in lactate levels and an increase in blood glucose levels. This provides sufficient energy supply to the injured liver tissue. Similarly, FAO a vital mitochondrial process for generating ATP, is impaired during IRI43leading to reduced energy availability and increased susceptibility to oxidative stress. In mitochondrial FAO, CPT1a (mitochondrial FAO rate-limiting enzyme, liver isoform) facilitates the entry of fatty acyl-CoAs into the mitochondria, where dehydrogenation, hydration, dehydrogenation, and thiolysis produce a large amount of energy and acetyl-CoA44. Here, we found that MH promoted lipid metabolism, as evidenced by an upward trend in the levels of free fatty acids, which serve as substrates for FAO. Additionally, the key enzyme for FAO, CPT1a, showed a significant increase, indicating that MH enhances FAO. Acetyl-CoA produced during FAO can enter the tricarboxylic acid cycle to produce ATP or be used as a substrate for cellular energy production via gluconeogenesis45. In our study, MH promoted the expression of citrate synthase (CS), a key enzyme in the tricarboxylic acid (TCA) cycle during IRI. This result suggests that MH ultimately enhances ATP production. Although FAO is crucial for energy production, it is also accompanied by potential oxidative stress risks46. Cells utilize antioxidant mechanisms, such as GSH, to remove excess ROS, thereby maintaining a balance between FAO and oxidative stress47. Therefore, we measured the GSH/GSSG ratio to assess the mitochondria’s ability to eliminate ROS. As expected, MH greatly enhanced the production of GSH during IRI. In addition, the expression of ACOX1, the key enzyme in peroxisomal β-oxidation48was also altered, but not as significantly as CPT1a expression. The effective progression of FAO and the protection of the antioxidant system rely on functional and efficient mitochondria. Thus, it is reasonable to speculate that MH significantly ameliorated IR-induced mitochondrial injury.
FAO regulation may be mediated through the PPARα pathway15AMPK pathway49and JAK2/STAT3 pathway50. Our previous studies showed that MH alleviates hepatic IRI by activating the JAK2/STAT3 signaling pathway. However, the role of PPARα signaling pathways has not been fully elucidated. PPARα, a nuclear receptor and essential transcriptional regulator of FAO, primarily regulates the canonical target genes CPT1a and ACOX151. PPARα protein expression is reduced in murine fatty liver following IRI52. In this study, our transcriptomic analysis of clinical DCD liver specimens revealed a significant reduction in PPARα expression after liver transplantation, which correlated with poorer postoperative clinical outcomes, as indicated by higher levels of ALT and AST. RNA sequencing analysis revealed significant changes in metabolic and PPAR signaling pathways during liver IRI under MH. Given PPARα’s role in maintaining hepatic energy homeostasis, its reduction after transplantation likely compromises the liver’s metabolic resilience, contributing to exacerbated injury. This aligns with studies indicating that low PPARα expression impairs FAO, leading to lipotoxicity and oxidative stress in hepatocytestrategies aimed at maintaining or enhancing PPARα expression, such as MH, might offer a protective advantage by promoting FAO and preventing hepatic lipid accumulation under IRI conditions.
FoxO1, a transcription factor known for its role in regulating cellular stress response and metabolic53was found to be significantly activated during MH treatment. Tomoyuki et al. reported that inhibiting FoxO1 significantly exacerbates liver IRI during a 12-h fast54. Interestingly, inhibition of FoxO1 significantly inhibited the expression of PPARα. More importantly, our immunoprecipitation and immunofluorescence results confirmed that MH promotes the nuclear translocation and co-localization of FoxO1 and PPARα. The upregulation of FoxO1 under MH conditions suggests that it plays a important role in shifting the metabolic state towards energy production and storage, which is essential for protecting liver from IRI. Our results demonstrated that the inhibition of FoxO1 using AS1842856 aggravated hepatic damage, reduced glucose levels and FAO, further underscoring the importance of FoxO1 in energy metabolism during IRI. Our findings further suggest that the FoxO1/PPARα interaction promotes metabolic reprogramming under MH, which is beneficial for reducing hepatic IRI. Specifically, the interaction between FoxO1 and PPARα under MH conditions may help maintain a balance in energy metabolism by enhancing FAO and gluconeogenesis, thereby sustaining energy levels and mitigating oxidative stress.
During IRI, mitochondrial dysfunction often triggers the release of cytochrome C, which, in turn, activates the caspase-3-mediated apoptotic pathway55. This study provides compelling evidence that MH mitigates mitochondrial damage and reduces apoptosis by modulating the FoxO1/PPARα axis. We observed that MH decreased the the production of ROS and BAX, a pro-apoptotic protein, and inhibited the release of cytochrome C and activation of cleaved caspase-3. These findings indicate that MH effectively interrupts the mitochondria-dependent apoptotic cascade, thereby preserving mitochondrial function during IRI. This is consistent with the observed reduction in hepatocyte apoptosis and liver damage in MH-treated groups, both in vivo and in vitro. Further, the protective role of PPARα in mitochondrial function was further validated by our experiments involving the overexpression and silencing of PPARα. We verified that overexpression of PPARα enhanced mitochondrial function, reduced oxidative stress, and decreased apoptosis, as evidenced by the increased BCL2/BAX ratio. Conversely, silencing PPARα exacerbated mitochondrial dysfunction and increased cell apoptosis, highlighting the pivotal role of PPARα in maintaining mitochondrial homeostasis during IRI. Finally, the restrained expression of PPARα was confirmed in human liver specimens subjected to IRI in DCD liver transplantation and its correlation with poor postoperative outcomes suggest that enhancing PPARα activity could be a promising therapeutic strategy for alleviating hepatic IRI.
However, this study also has some limitations. First, while we have demonstrated the critical role of FoxO1 and PPARα in MH-mediated protection, which may promote the stability of PPARα protein, thereby maintaining its expression and enhancing the transcriptional activation of downstream targets such as CPT1a and PEPCK1. However, the precise molecular mechanisms underlying the FoxO1-PPARα interaction and its functional consequences remain incompletely understood. While our data suggest a potential role for PPARα stabilization, rigorous validation of this mechanism is lacking, which limits the persuasiveness of our conclusions. This represents a notable limitation of our study, further research is needed to identify these signals and to understand how they are modulated by MH. Interestingly, CPT1a mRNA expression is significantly induced in the MH group, while its protein expression remains unchanged. This discrepancy between mRNA and protein expression trends requires further investigation. Future studies should investigate the underlying mechanisms, including potential microRNA interactions or ubiquitin-mediated proteasomal degradation.Additionally, while our study focused on the role of FoxO1 and PPARα in energy metabolism and apoptosis, other pathways and factors may also contribute to the protective effects of MH. Future studies should explore these potential mechanisms to provide a more comprehensive understanding of MH’s protective role in hepatic IRI.
Conclusion
In conclusion, our results revealed the potential protective effects of MH via activating the FoxO1/PPARα pathway, which enhances energy metabolism and inhibits mitochondria-dependent apoptosis. These findings not only provide new insights into the mechanisms underlying the protective effects of MH but also suggest potential therapeutic targets for improving outcomes in patients undergoing liver surgeries. Furthermore, our findings provide a foundation for a wider application of MH therapy in clinics.
Data availability
RNA-Seq data used in this study are deposited in the GEO database under the accession number GSE283907. The datasets obtained in this study are availableare accessible at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE283907.
References
Hirao, H., Nakamura, K. & Kupiec-Weglinski, J. W. Liver ischaemia-reperfusion injury: a new Understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 19(4), 239–256. https://doi.org/10.1038/s41575-021-00549-8 (2022).
Yu, S. et al. M 6 A-mediated gluconeogenic enzyme PCK1 upregulation protects against hepatic ischemia-reperfusion injury. Hepatology 12 https://doi.org/10.1097/HEP.0000000000000716 (2023).
Ding, M. J. et al. E3 ubiquitin ligase ring finger protein 5 protects against hepatic ischemia reperfusion injury by mediating phosphoglycerate mutase family member 5 ubiquitination. Hepatology 76(1), 94–111. https://doi.org/10.1002/hep.32226 (2022).
Polderman, K. H. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet 371(9628), 1955–1969. https://doi.org/10.1016/S0140-6736(08)60837-5 (2008).
Choi, H. A., Badjatia, N. & Mayer, S. A. Hypothermia for acute brain injury–mechanisms and practical aspects. Nat. Rev. Neurol. 8(4), 214–222. https://doi.org/10.1038/nrneurol.2012.21 (2012).
Wei, S. et al. Acute and delayed protective effects of Pharmacologically induced hypothermia in an intracerebral hemorrhage stroke model of mice. Neuroscience 252, 489–500 (2013).
Wang, W. et al. Mild hypothermia attenuates hepatic Ischemia-Reperfusion injury through regulating the JAK2/STAT3-CPT1a-Dependent fatty acid β-Oxidation. Oxid. Med. Cell. Longev. 2020, 5849794. https://doi.org/10.1155/2020/5849794 (2020).
Xiao, Q. et al. Mild hypothermia ameliorates hepatic ischemia reperfusion injury by inducing RBM3 expression. Apoptosis 27(11–12), 899–912. https://doi.org/10.1007/s10495-022-01757-6 (2022).
Wang, L. et al. Mild hypothermia alleviates complement C5a-Induced neuronal autophagy during brain Ischemia-Reperfusion injury after cardiac arrest. Cell. Mol. Neurobiol. 43(5), 1957–1974. https://doi.org/10.1007/s10571-022-01275-8 (2023).
Rui, L. Energy metabolism in the liver. Compr. Physiol. 4(1), 177–197. https://doi.org/10.1002/cphy (2014).
Ren, J. et al. Alteration in gut microbiota caused by time-restricted feeding alleviate hepatic ischaemia reperfusion injury in mice. J. Cell. Mol. Med. 23(3), 1714–1722. https://doi.org/10.1111/jcmm.14069 (2019).
Li, F., Gao, J., Kohls, W., Geng, X. & Ding, Y. Perspectives on benefit of early and prereperfusion hypothermia by Pharmacological approach in stroke. Brain Circ. 8(2), 69–75. https://doi.org/10.4103/bc.bc_27_22 (2022). Published 2022 Jun 30.
Xu, S. et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 101(5), 987–1002. https://doi.org/10.1016/j.kint.2022.01.029 (2022).
Piret, S. E. et al. Loss of proximal tubular transcription factor Krüppel-like factor 15 exacerbates kidney injury through loss of fatty acid oxidation. Kidney Int. 100(6), 1250–1267. https://doi.org/10.1016/j.kint.2021.08.031 (2021).
Liu, L. et al. Twist1 downregulation of PGC-1α decreases fatty acid oxidation in tubular epithelial cells, leading to kidney fibrosis. Theranostics 12(8), 3758–3775. https://doi.org/10.7150/thno.71722 (2022).
Yu, S. et al. m 6 A-mediated gluconeogenic enzyme PCK1 upregulation protects against hepatic ischemia-reperfusion injury. Hepatology https://doi.org/10.1097/HEP.0000000000000716 (2023).
Christofides, A., Konstantinidou, E., Jani, C. & Boussiotis, V. A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 114, 154338. https://doi.org/10.1016/j.metabol.2020.154338 (2021).
Montaigne, D., Butruille, L. & Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 18(12), 809–823. https://doi.org/10.1038/s41569-021-00569-6 (2021).
Nakamura, M. T., Yudell, B. E. & Loor, J. J. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 53, 124–144. https://doi.org/10.1016/j.plipres.2013.12.001 (2014).
Banke, N. H. et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 107(2), 233–241. https://doi.org/10.1161/CIRCRESAHA.110.221713 (2010).
Ahmad, A. et al. Adiponectin homolog novel osmotin protects obesity/diabetes-induced NAFLD by upregulating AdipoRs/PPARα signaling in ob/ob and db/db Transgenic mouse models. Metabolism 90, 31–43. https://doi.org/10.1016/j.metabol.2018.10.004 (2019).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21(1), 37–46. https://doi.org/10.1038/nm.3762 (2015).
O-Sullivan, I. et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization [published correction appears in Nat commun. 24(6), 7861. https://doi.org/10.1038/ncomms8861 (2015).
Kamo, N., Ke, B., Busuttil, R. W. & Kupiec-Weglinski, J. W. PTEN-mediated Akt/beta-catenin/FoxO1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology 57(1), 289–298. https://doi.org/10.1002/hep.25958 (2013).
Sanchez, A. M., Candau, R. B. & Bernardi, H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell. Mol. Life Sci. 71(9), 1657–1671. https://doi.org/10.1007/s00018-013-1513-z (2014).
Pan, A. et al. The mitochondrial β-oxidation enzyme HADHA restrains hepatic glucagon response by promoting β-hydroxybutyrate production. Nat. Commun. 13(1), 386. https://doi.org/10.1038/s41467-022-28044-x (2022).
Chen, S. et al. Hepatocyte-specific Mas activation enhances lipophagy and fatty acid oxidation to protect against acetaminophen-induced hepatotoxicity in mice. J. Hepatol. 78(3), 543–557. https://doi.org/10.1016/j.jhep.2022.10.028 (2023).
Wang, D. et al. FoxO1 Inhibition prevents renal ischemia-reperfusion injury via cAMP-response element binding protein/PPAR-γ coactivator-1α-mediated mitochondrial biogenesis. Br. J. Pharmacol. 177(2), 432–448. https://doi.org/10.1111/bph.14878 (2020).
Shao, D. et al. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 5, 3315. https://doi.org/10.1038/ncomms4315 (2014).
Puthanveetil, P., Wan, A. & Rodrigues, B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 97(3), 393–403. https://doi.org/10.1093/cvr/cvs426 (2013).
Li, F., Gao, J., Kohls, W., Geng, X. & Ding, Y. Perspectives on benefit of early and prereperfusion hypothermia by Pharmacological approach in stroke. Brain Circ. 8(2), 69–75. https://doi.org/10.4103/bc.bc_27_22 (2022).
Tu, Q. et al. Peroxiredoxin 6 attenuates ischemia and hypoxia induced liver damage of braindead donors. Mol. Med. Rep. 13, 753e761. https://doi.org/10.3892/mmr.2015.4587 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Lamb, R. J. et al. ALDH2 polymorphism and myocardial infarction: from alcohol metabolism to redox regulation. Pharmacol. Ther. 259, 108666. https://doi.org/10.1016/j.pharmthera.2024.108666 (2024).
Hao, T. et al. An injectable Dual-Function hydrogel protects against myocardial ischemia/reperfusion injury by modulating ROS/NO disequilibrium. Adv. Sci. 9(15), e2105408. https://doi.org/10.1002/advs.202105408 (2022).
Zhai, Y., Petrowsky, H., Hong, J. C., Busuttil, R. W. & Kupiec-Weglinski, J. W. Ischaemia-reperfusion injury in liver transplantation–from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 10(2), 79–89. https://doi.org/10.1038/nrgastro.2012.225 (2013).
Ito, T. et al. Ischemia-reperfusion injury and its relationship with early allograft dysfunction in liver transplant patients. Am. J. Transpl. 21(2), 614–625. https://doi.org/10.1111/ajt.16219 (2021).
Kurisu, K. & Yenari, M. A. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology 134, 302–309. https://doi.org/10.1016/j.neuropharm.2017.08.025 (2018).
Niemann, C. U. et al. Therapeutic hypothermia in deceased organ donors and Kidney-Graft function. N. Engl. J. Med. 373, 405–414. https://doi.org/10.1056/NEJMoa1501969 (2015).
Wang, W. et al. Mild hypothermia pretreatment attenuates liver ischemia reperfusion injury through inhibiting c-Jun NH2-terminal kinase phosphorylation in rats. Transpl. Proc. 50, 259–266. https://doi.org/10.1016/j.transproceed.2017.12.019 (2018).
He, L. Metformin and systemic metabolism. Trends Pharmacol. Sci. 41(11), 868–881. https://doi.org/10.1016/j.tips.2020.09.001 (2020).
Yue, T. L. et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation 108(19), 2393–2399. https://doi.org/10.1161/01.CIR.0000093187.42015.6C (2003).
Ploton, M. et al. The nuclear bile acid receptor FXR is a PKA- and FOXA2-sensitive activator of fasting hepatic gluconeogenesis. J. Hepatol. 69, 1099–1109. https://doi.org/10.1016/j.jhep.2018.06.022 (2018).
Perry, R. J. et al. Leptin mediates a glucose-Fatty acid cycle to maintain glucose homeostasis in starvation. Cell 172, 234–248e17. https://doi.org/10.1016/j.cell.2017.12.001 (2018).
Chen, C. L. et al. NANOG metabolically reprograms Tumor-Initiating Stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell. Metab. 23(1), 206–219. https://doi.org/10.1016/j.cmet.2015.12.004 (2016).
Chalifoux, O., Faerman, B. & Mailloux, R. J. Mitochondrial hydrogen peroxide production by pyruvate dehydrogenase and α-ketoglutarate dehydrogenase in oxidative eustress and oxidative distress. J. Biol. Chem. 299(12), 105399. https://doi.org/10.1016/j.jbc.2023.105399 (2023).
He, A. et al. Acetyl-CoA derived from hepatic peroxisomal β-Oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol. Cell. 79, 30–42e4. https://doi.org/10.1016/j.molcel.2020.05.007 (2020).
Gao, Y. et al. Self-amplified ROS production from fatty acid oxidation enhanced tumor immunotherapy by atorvastatin/PD-L1 SiRNA lipopeptide nanoplexes. Biomaterials 291, 121902. https://doi.org/10.1016/j.biomaterials.2022.121902 (2022).
Wang, T. et al. JAK/STAT3-Regulated fatty acid β-Oxidation is critical for breast Cancer stem cell Self-Renewal and chemoresistance. Cell. Metab. 27(6), 1357. https://doi.org/10.1016/j.cmet (2018).
Zhu, L. H. et al. Mindin/Spondin 2 inhibits hepatic steatosis, insulin resistance, and obesity via interaction with peroxisome proliferator-activated receptor α in mice. J. Hepatol. 60(5), 1046–1054. https://doi.org/10.1016/j.jhep.2014.01.011 (2014).
Lam, V. H. et al. Activating PPARα prevents post-ischemic contractile dysfunction in hypertrophied neonatal hearts. Circ. Res. 117(1), 41–51. https://doi.org/10.1161/CIRCRESAHA.117.306585 (2015).
Chu, Y. et al. Liver Med23 ablation improves glucose and lipid metabolism through modulating FoxO1 activity. Cell. Res. 24(10), 1250–1265. https://doi.org/10.1038/cr.2014.120 (2014).
Fontes, P. et al. Liver preservation with machine perfusion and a newly developed cell-free oxygen carrier solution under subnormothermic conditions. Am. J. Transplant. 15(2), 381–394. https://doi.org/10.1111/ajt.12991 (2015).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10(1), 1689. https://doi.org/10.1038/s41467-019-09397-2 (2019).
Acknowledgements
This research was supported by the the National Natural Science Foundation of China (No. 81970548).
Author information
Authors and Affiliations
Contributions
Z.S.L carried out the main experiments. H.C supervised the work and wrote the manuscript. A.X.L and S.F. N collected the human liver tissue specimens. W.W, Z.B.Z and Q.F.Y gave advice and assisted in the experiments. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Z., Chen, H., Liu, A. et al. Mild hypothermia attenuates hepatic ischemia-reperfusion injury by regulating FoxO1/PPARα pathway. Sci Rep 15, 28800 (2025). https://doi.org/10.1038/s41598-025-09725-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09725-1
