
Abstract
Colorectal cancer (CRC) is a prevalent type of cancer affecting the digestive system, characterized by a complex and not fully understood development process. Inflammation and metabolic processes are thought to play significant roles in both the initiation and progression of this disease. This study aimed to clarify the causal relationships between CRC and specific inflammatory proteins, immune cell types, and metabolites, while also exploring potential bidirectional associations and identifying possible therapeutic targets. We employed Mendelian Randomization (MR) analysis to investigate a wide array of biological factors, including 4,907 plasma proteins, 91 inflammatory proteins, 731 immune cell characteristics, and 1,400 metabolites. Additionally, we utilized bioinformatics techniques such as constructing Protein-Protein Interaction (PPI) networks, performing Gene Ontology (GO) enrichment, and conducting Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Our results indicated significant associations between CRC and 104 circulating proteins, 5 inflammatory proteins, 24 immune cell traits, and 28 metabolites. Through subnetwork analysis, we identified 15 central proteins, among which Tissue Inhibitor of Metalloproteinase 1 (TIMP1) showed notable variability in expression. This finding was computationally validated through survival analysis data from the Human Protein Atlas (HPA) and Gene Expression Profiling Interactive Analysis (GEPIA2) databases. Furthermore, molecular docking studies confirmed TIMP1 as a promising drug target, demonstrating stable interactions with compounds such as meclizine and megestrol. Overall, this research enhances our understanding of CRC pathogenesis by emphasizing the roles of inflammation, immune response, and metabolic pathways, and it suggests TIMP1 as a particularly promising target for future therapeutic development.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is one of the most common cancers of the digestive system. According to the latest cancer statistics, it is projected that by 2024, over 150,000 new cases of CRC will be diagnosed in the United States, with an estimated 53,010 deaths attributed to the disease. This accounts for approximately 10% of the total cancer incidence and mortality1. Globally, CRC has risen from the fifth to the second most common cancer between 2018 and 20202,3. The exact causes and development of CRC are still not fully understood, though they likely involve a complex interaction of genetic and environmental factors. Recent research has highlighted the crucial role of inflammatory and metabolic pathways in CRC development4,5.
Recent studies indicate that dysregulated inflammation and metabolic pathway dysfunction are linked to various cancers, including CRC. CRC risk is influenced by metabolic byproducts and immune factors, with high-fat diets increasing CRC risk through bile acid secretion and harmful secondary bile acid production by gut microbes. While excessive ω-6 fatty acids promote CRC, ω-3 polyunsaturated fatty acids (PUFAs) have anti-inflammatory properties. Vitamin D, via the nuclear vitamin D receptor (VDR), modulates genes related to cell proliferation and angiogenesis, inversely correlating with CRC risk6. The CRC tumor microenvironment (TME) has high inflammation and cytokine levels. Toll-like receptors (TLRs), especially TLR2 and TLR4, regulate cytokine production (IL-1, IL-6, IL-17 A, STAT3), promoting CRC progression7,8. Interferons from NK and Th1 cells help restrict tumor growth, while Th1 polarization markers are linked to reduced recurrence9,10,11. However, TGF-β suppresses immune responses, aiding CRC development12,13. Tissue inhibitor of metalloproteinase 1 (TIMP1), which regulates extracellular matrix remodeling, is paradoxically associated with CRC progression. Elevated TIMP1 correlates with advanced tumor stage, lymphatic metastasis, and poor survival. TIMP1 activates PI3K/AKT and FAK pathways to promote epithelial-mesenchymal transition (EMT) and chemoresistance. TIMP1 (61%) showed significantly higher sensitivity than CEA (39%) and CA19-9 (11%), especially in distinguishing colorectal adenomas from cancer (AUC = 0.73). When combined with MMP-7 or CEA, TIMP1’s sensitivity increased to 75%, enhancing early detection of adenoma-cancer progression14.
Previous research has largely relied on observational studies and simple experimental models. Observational studies, while useful for identifying associations, are fundamentally limited by confounding bias (e.g., unmeasured lifestyle factors) and reverse causality (e.g., disease progression altering exposure measurements). Similarly, conventional experiments often lack external validity, as controlled laboratory conditions poorly reflect human environmental complexity15.
These limitations necessitate an alternative approach. Mendelian Randomization (MR) overcomes these issues by utilizing genetic variants as instrumental variables (IVs). Specifically, MR exploits the random allocation of genetic variants (e.g., SNPs) at conception, which mimics the randomization process in RCTs16. Recent methodological advances, such as multivariable MR and pleiotropy-robust methods, further enhance its ability to disentangle complex causal pathways17. Crucially, genetic variants are fixed prior to disease onset and are unaffected by environmental confounders (e.g., diet, socioeconomic status), thereby satisfying the IV independence assumption18. Furthermore, because SNPs influence outcomes solely through their biological effects on the exposure (exclusion restriction), MR avoids reverse causality a critical advantage when studying chronic diseases with long latency periods19. For instance, a 2021 MR study of body mass index (BMI) and COVID-19 severity overturned earlier observational claims of obesity’s protective effects, demonstrating MR’s capacity to correct survival bias in pandemic research20. By integrating genetic epidemiology with causal inference principles, MR provides a robust framework for inferring causality that is both biologically grounded and methodologically rigorous16.
In this study, we employed a bidirectional, two-sample MR approach, utilizing large-scale datasets covering a range of biological factors, including inflammatory proteins, immune cell traits, and metabolites. Through bioinformatics analysis, we aimed to uncover the functional roles of key proteins in cellular processes, thereby identifying potential therapeutic targets. Additionally, potential therapeutic strategies were explored through drug target screening and molecular docking validation.
Materials and methods
Study design
We conducted a bidirectional two-sample MR analysis, combined with bioinformatics approaches, to explore how inflammation and metabolism contribute to the development of CRC and to identify potential pharmacological targets and biomarkers. This study used data exclusively from publicly available genome-wide association studies (GWAS) datasets. The design of this study was inspired to some extent by the work of Ming-jie He and colleagues21. MR analysis relies on three core assumptions: (1) the genetic variants used as IVs are strongly associated with the exposure of interest; (2) these genetic variants are not associated with any confounders that influence both the exposure and the outcome; and (3) the genetic variants affect the outcome only through the exposure and not through any other pathways (exclusion restriction).
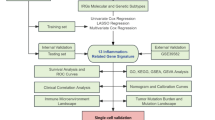
In this study, we first selected the most comprehensive and well-researched GWAS data for circulating proteins, inflammatory proteins, immune cell traits, and metabolites as exposure variables, and used the CRC GWAS data from the Finnish database as the outcome variable. By selecting IVs (P < 5 × 10− 8, r² < 0.001, kb > 10,000, F-statistic > 10) and performing MR analysis, we identified immune cells, metabolites, inflammatory proteins, and circulating proteins that have causal relationships with CRC. To further investigate the role of inflammatory proteins in CRC and identify potential therapeutic targets, we combined the Gene Set Enrichment Analysis (GSEA) database22 to select related inflammatory genes. Subsequently, key proteins were identified through Protein-Protein Interaction (PPI) network analysis, and their expression and biological functions were further validated using the Gene Expression Profiling Interactive Analysis (GEPIA2)23 and Human Protein Atlas (HPA) databases24. Finally, drug prediction and molecular docking analyses were performed to identify potential therapeutic drugs. For detailed research procedures, please refer to Fig. 1.

Flowchart describing the research process. GO: gene ontology, IVs: instrumental variables, KEGG: kyoto encyclopedia of genes and genomes, MR: mendelian randomization, PPI: protein–protein interaction, GEPIA2:Gene Expression Profiling Interactive Analysis.
Data sources
The data used for the MR study were divided into two main categories: exposure data and outcome data. Outcome data related to CRC were obtained from the Finnish R11 database25(https://www.finngen.fi/en/access_results) on September 15, 2024 .The latest release of the large-scale FinnGen study, reseased in July 2024, includes analyses of over 500,000 Finnish biobank samples, correlating genetic variation with health data to understand disease mechanisms and predispositions.
Exposure data concentrated on two main areas: inflammation and metabolism. For metabolic analysis, 1,091 blood metabolites and 309 metabolite ratios were included, with data sourced from the GWAS catalog26(https://www.ebi.ac.uk/gwas/studies/GCST90199621-902010209)27. The inflammation analysis consisted of three components: 4,907 plasma proteins2891 inflammatory proteins29and 731 immune cell types30. All GWAS data used in the MR analysis were restricted to individuals of European ancestry. The exposure data and outcome data in this study come from different databases, so there is no need to consider the issue of sample overlap, and the overlap rate does not need to be calculated.Supplementary Table S1 provides comprehensive details on the datasets used in this study, including information about the sources, sample sizes, and specific variables included in each dataset.
The bioinformatics data came from the GEPIA2 database(http://gepia2.cancer-pku.cn/#index), primarily focusing on colon adenocarcinoma in CRC. These data were sourced from The Cancer Genome Atlas (TCGA)and Genotype-Tissue Expression (GTEx) databases, which included 275 cases of CRC and 349 normal controls.
Screening of 4,907 Circulating plasma proteins
To identify protein quantitative trait loci (pQTL), we utilized data from the circulating protein expression GWAS study conducted by deCODE Genetics, which measured 4,907 proteins in 35,559 Icelandic individuals. Because this study focuses on the immune and inflammatory aspects of CRC, we conducted an additional screening to select proteins related to immunity and inflammation.
Gene sets related to humans (H, C1–C8) were retrieved from the Molecular Signatures Database via the GSEA platform (https://www.gsea-msigdb.org/gsea). We screened these gene sets using keywords like “inflammation” and “immunity” resulting in the identification of 5,886 genes associated with these processes. Additional details are provided in Supplementary Table S2.
Subsequently, a MR analysis was performed on the 4,907 plasma proteins in relation to CRC. We cross-referenced plasma proteins that exhibit a causal relationship with CRC against the 5,886 genes associated with immunity and inflammation from the GSEA database to identify the proteins involved in these biological processes.
Screening of genetic IVs
Selection of SNPs significantly associated with exposure phenotypes
To identify SNPs significantly associated with the exposure phenotypes, we applied a stringent threshold (P < 5 × 10⁻⁸). The details of the selected SNPs, including their corresponding p-values, effect sizes, and other relevant statistics, are provided in Supplementary Table S3-S6, which contain information on SNPs related to circulating plasma proteins, inflammatory proteins, immune cell traits, and metabolites associated with CRC.
Removal of SNPs in linkage disequilibrium
To eliminate linkage disequilibrium among SNPs, we applied the criteria of r² < 0.001 and a minimum distance of 10,000 kb31. This approach ensures the selection of SNPs that are adequately spaced and minimally correlated, thereby reducing the potential impact of linkage disequilibrium on the results.
Integration, harmonization, and palindromic SNP correction
We integrated and assessed the consistency between the exposure and outcome datasets. Additionally, palindromic SNPs with ambiguous strand alignments were corrected using allele frequency information to ensure accurate alignment and interpretation. This process utilized the “harmonise_data” function from the “TwoSampleMR” R package. This function automatically excludes SNPs with palindromic sequences during the final MR analysis.
Evaluation of IVs strength
We assessed the strength of IVs by calculating the F-statistic, applying a threshold of F > 10 to exclude weak IVs. This approach enhances the stability and reliability of our results.The F-statistic in this study is defined by the formula: F = R² × (N − K − 1) / (1 − R²) where R² represents the variance in the exposure factor explained by each IVs, K stands for the number of IVs, and N is the sample size of the GWAS. R² was calculated using the following formula: R² = 2 × (1-MAF) ×MAF×b2 where b denotes the effect size of the allele, MAF is the minor allele frequency32,33. Further details on the SNPs, including their associated p-values and effect sizes, are provided in Supplementary Table S3-S6.
MR analysis and sensitivity analyses
In this study, analyses were conducted primarily using R version 4.4.1, with “Two-Sample MR” and “MRPRESSO” as the main packages34.The primary method used was the Inverse Variance Weighted (IVW) approach35.This method calculated the odds ratio (OR) and its 95% confidence interval (CI) to evaluate the causal relationships between the exposure and the outcome.Supplementary analyses also included the MR-Egger method36,weighted median37and weighted mode38.Sensitivity analyses were performed to ensure robustness and validity. Heterogeneity was assessed using Cochran’s Q test39.A P-value of less than 0.05 indicated heterogeneity, leading to the application of a random-effects model. If P ≥ 0.05, a fixed-effects model was used.
To detect pleiotropy, we employed the MR-Egger and MRPRESSO (MR Pleiotropy Residual and Outlier) methods34excluding exposure data with horizontal pleiotropy to ensure reliability.The Benjamini-Hochberg method, incorporating the false discovery rate (FDR)40was used to address multiple testing issues. A significance threshold of P < 0.05 was set. If both the original and FDR-adjusted P-values(P_fdr) were below 0.05, the exposure factor was considered to have a significant causal relationship with CRC; if only the original p-value was below 0.05, the exposure factor was regarded as having a potential causal relationship.
Bioinformatics analysis
Following the MR analysis, we identified 56 inflammation- and immunity-related proteins associated with CRC which were further screened and analyzed to gain insights into their roles in CRC development.
Enrichment analysis
To elucidate the functional characteristics and biological relevance of the identified proteins, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses using the R packages clusterProfiler and Pathview41. GO analysis covered three categories: biological processes (BP), molecular functions (MF), and cellular components (CC), while KEGG analysis provided insights into the metabolic pathways associated with these proteins.
Protein interaction network construction
We constructed a PPI network to understand interactions among the proteins. The STRING database42was used with the minimum interaction score set to 0.4, corresponding to medium confidence, and default parameter settings.We performed visualization of the PPI network using Cytoscape(V3.10.2)43. To identify functional modules and key regulatory proteins, we used the Molecular Complex Detection (MCODE) plugin in Cytoscape, with parameters optimized (degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max depth = 100).
TCGA and GTEx database screening
Differential expression and survival analyses of the identified proteins were performed using the GEPIA2 tool. Parameters for differential expression analysis included |Log2FC| Cutoff = 1, p-value Cutoff = 0.01, Jitter Size = 0.4, and TCGA-GTEx matched data. Overall Survival was used for survival analysis with the Group Cutoff set to the median. GEPIA2 is an enhanced version of GEPIA, analyzing RNA sequencing data from 9,736 tumor and 8,587 normal samples from TCGA and GTEx with uniformly processed data.
HPA database screening
To further understand protein localization within tissues and cells, we searched the identified molecules in the HPA database (v23.0). Immunohistochemistry images of target gene expression in CRC tissues were retrieved and validated. Since 2003, the HPA project has aimed to map human protein distribution across cells, tissues, and organs using various omics technologies. This open-access resource provides valuable data for researchers in the field of oncology research.
Drug enrichment analysis
Evaluating protein-drug interactions is essential to assess the druggability of target genes. We conducted this analysis using data from the Drug Signatures Database (DSigDB, http://dsigdb.tanlab.org/DSigDBv1.0/)44, which contains 22,527 gene sets and 17,389 compounds. The drug-gene data from DSigDB were cross-referenced with the identified proteins, and drug enrichment analysis was conducted using R.We also screened drugs approved by the Food and Drug Administration (FDA)(https://www.fda.gov/) to identify those that may be effective in treating CRC.
Molecular docking
Molecular docking simulations were performed at the atomic level to evaluate binding energy and interaction patterns between candidate drugs and target proteins. These simulations allow analysis of binding affinity, aiding in the prioritization of drug targets and optimization of candidate drug designs. CB-Dock244,46was used for molecular docking, integrating cavity detection and template fitting to predict binding sites and affinities.
We obtained drug structure data, including compound IDs, from the PubChem Compound Database (https://pubchem.ncbi.nlm.nih.gov/)47. Protein structure data were downloaded from the Protein Data Bank (PDB) (http://www.rcsb.org/)48, with relevant PDB IDs provided.
Results
MR and sensitivity analysis results
Through MR analysis of 4,907 circulating plasma proteins, 91 inflammatory proteins, 731 immune cell traits, and 1,400 metabolites, we identified a series of biomarkers with causal relationships to CRC. Detailed results can be found in Fig. 2 and Supplementary Table S7-S10, with sensitivity analysis results presented in Supplementary Table S11-S14.

MR analysis illustrating causal relationships between circulating proteins, inflammatory proteins, immune cells, and metabolites with CRC. (A) Volcano plot showing causal relationships between 104 circulating proteins and CRC. (B) Volcano plot displaying causal relationships between 5 inflammatory proteins and CRC. (C) Forest plot presenting causal relationships between 24 immune cell types and CRC. (D) Forest plot illustrating causal relationships between 28 metabolites and CRC.
Circulating plasma proteins
Following the designed steps, 109 of the 4,907 circulating plasma proteins demonstrated a causal relationship with CRC (P < 0.05). Five proteins (GREM1, DEFA5, RSPO1, BMP6, PRSS8) were excluded due to outliers detected by MRPRESSO.Consequently, 104 circulating plasma proteins were identified as having a causal relationship with CRC. The detailed results are shown in Fig. 2A (volcano plot), where green dots represent proteins associated with a reduced risk of CRC, and red dots represent proteins associated with an increased risk of CRC. The specific MR results are provided in Supplementary Table S7, while the sensitivity analysis results can be found in Supplementary Table S11.After FDR correction, two proteins remained significant (P_fdr < 0.05): PDE5A and PRRG4.
Inflammatory proteins
After stringent IV selection and MR analysis, 5 of the 91 inflammatory proteins exhibited a causal relationship with CRC (P < 0.05).These proteins include eukaryotic translation initiation factor 4E-binding protein 1(4EBP-1), C-C motif chemokine 19 (CCL19), Delta and Notch-like epidermal growth factor-related receptor(DNER), monocyte chemoattractant protein 2(MCP-2), and vascular endothelial growth factor A(VEGFA). The detailed results are shown in Fig. 2 (volcano plot), where CCL19 is represented by a green dot, indicating its association with a reduced risk of CRC. The other inflammatory proteins are shown as red dots, indicating their association with an increased risk of CRC.After FDR correction, two proteins, CCL19 and MCP-2, still showed a causal relationship (P_fdr < 0.05). The specific MR results are provided in Supplementary Table S8, while the sensitivity analysis results can be found in Supplementary Table S12.
Screening of immune and inflammatory related proteins
By intersecting the 104 plasma proteins identified in Sect. 3.1.1 with the immune and inflammation-related proteins downloaded from GSEA, We identified 52 plasma proteins that were associated with both immune and inflammatory processes, as well as CRC. Combining these with the five inflammatory proteins identified in Sect. 3.1.2, a total of 56 immune and inflammation-related proteins were selected (with CCL19 present in both datasets), The list of these specifically identified immune and inflammation-related proteins is provided in Supplementary Table S15.
Immune cells
The MR analysis of 731 immune cell traits revealed 24 with causal relationships to CRC, and no pleiotropy was detected. However, after FDR correction, all 24 immune cell traits had P_fdr values greater than 0.05, indicating a potential causal relationship with CRC. Figure 2C shows the forest plot for the 24 immune cell traits with causal relationships to CRC identified through MR analysis. The detailed results are provided in Supplementary Table S9, while the sensitivity analysis results can be found in Supplementary Table S13.
Metabolites
After MR analysis of the 1,400 metabolites and metabolite ratios, 28 were found to have a causal relationship with CRC with no pleiotropy detected. The MR results are shown in the forest plot in Fig. 2D and detailed in Supplementary Table S10, while the sensitivity analysis results can be found in Supplementary Table S14.After FDR correction, six metabolites and metabolite ratios remained significantly associated with CRC (P_fdr < 0.05): 1-palmitoyl-2-linoleoyl-GPC (16:0/18:2), 1-stearoyl-2-linoleoyl-GPC (18:0/18:2), 1-palmitoyl-2-arachidonoyl-GPC (16:0/20:4n6), 1-stearoyl-2-arachidonoyl-GPC (18:0/20:4), 1-arachidonoyl-GPC (20:4n6), and aspartate to mannose ratio .
Reverse MR analysis
In the reverse MR analysis, CRC data were used as the exposure, and the positive exposures identified earlier (52 plasma proteins, 5 inflammatory proteins, 24 immune cell traits, and 28 metabolites and ratios) were treated as outcomes. The results showed no causal relationship, indicating that CRC does not influence the immune and metabolic exposures we studied, as shown in Supplementary Table S16. Sensitivity analyses demonstrated that the results were stable, with no evidence of heterogeneity or pleiotropy, as shown in Supplementary Table S17.
Bioinformatics analysis
GO and KEGG enrichment analysis
GO enrichment analysis revealed interactions between genes and functional terms, while KEGG enrichment analysis highlighted the relationships between genes and functional pathways. As shown in Fig. 3A, the most significant pathways in the BP category were related to cell migration and chemotaxis, such as neutrophil chemotaxis, monocyte migration, and lymphocyte migration. In the CC category, genes were enriched in immune-related structures, including secretory granule lumen, cytoplasmic vesicle lumen, and lateral plasma membrane. For MF, these genes were involved in immune functions, including cytokine activity, cytokine receptor binding, and chemokine activity.

Enrichment analysis results.(A) Results of GO enrichment analysis sorted by gene count, displaying the top 10 terms; (B) Results of KEGG pathway analysis. BP: biological processes; CC: cellular components; MF: molecular functions.
KEGG analysis indicated that the most significant pathways were related to cytokine signaling and cancer, including cytokine-cytokine receptor interaction (hsa04060, P < 0.05), viral protein interaction with cytokines and cytokine receptors (hsa04061, P < 0.05), and the HIF-1 signaling pathway (hsa04066, P < 0.05). The enrichment of these pathways emphasizes the critical role of the related genes in immune responses, viral infections, and cancer, as shown in Fig. 3B.
PPI network analysis
We performed PPI analysis on the 56 identified proteins using the STRING database, with the minimum interaction score set to medium confidence (0.400). Under this threshold, we identified interactions among 24 proteins, as detailed in Supplementary Figure S1A. Notably, TNF and CXCL10 had the most connections with other proteins. The top 10 proteins with the highest connectivity included IL2, CXCL10, TIMP1, CXCL11, MPO, CDH1, CCL8, CCL7, CCL19, and PLAU.
Using the MCODE plugin in Cytoscape, we identified three subnetworks containing 15 core proteins: CXCL11, IL2, CCL8, CCL19, CXCL10, CCL7, TGFB3, TIMP1, PLAU, CDH1, HP, MPO, PRKG1, PDE4A, and PDE5A. These proteins may play crucial regulatory roles, as detailed in Supplementary Figure S1B-S1D.
Differential expression and survival analysis
We queried the 15 core proteins identified in Sect. 3.3.2 using the GEPIA2 platform and obtained differential expression data for these proteins in CRC. Among them, CDH1, CXCL10, CXCL11, PLAU, and TIMP1 were upregulated in CRC tissues (Figs. 4 A–4E), while HP, PDE5A, PRKG1, and TGFB3 were downregulated (Fig. 4F and I). Six proteins, including CCL19, CCL7, CCL8, IL2, MPO, and PDE4A, showed no significant difference in expression between CRC tissues and normal tissues (Fig. 5).

Differential expression analysis of selected genes in CRC using the GEPIA2 database. (A) CDH1; (B) CXCL10; (C) CXCL11; (D) PLAU; (E) TIMP1; (F) HP; (G) PDE5A; (H) PRKCB; (I) TGFB3.

Differential expression analysis of selected genes in CRC using the GEPIA2 database. (A) CCL7; (B) MPO; (C) PDE4B; (D) IL2; (E) CCL19; (F) CCL28.
Survival analysis was performed on the nine proteins with differential expression. Only the high and low expression groups for TIMP1 showed a significant difference in overall survival (HR = 1.7, P < 0.05)(Fig. 6A), while the other proteins did not display significant survival differences between the expression groups (Fig. 6B-F).

Overall survival analysis of selected genes in CRC using Kaplan-Meier plots.
(A)TIMP1; (B) CDH1; (C) TGFB3; (D) PRKCB; (E) PLAU; (F) PDE5A; (G) HP; (H) CXCL11; (I) CXCL10.
Validation with the HPA database
TIMP1 was further validated using the HPA database, which confirmed that TIMP1 is prognostic, and high expression is unfavorable in CRC. This finding aligns with the differential expression analysis. As shown in Fig. 7, immunohistochemical staining revealed strong positive staining for TIMP1 in tumor cells, though the percentage of positive cells was relatively low (< 25%). The staining was predominantly cytoplasmic and membranous, suggesting a specific functional distribution of this protein in some tumor cells. The overall staining intensity was moderate, indicating localized protein activity.

Immunohistochemical staining of TIMP1 in CRC tissue versus normal tissue, based on data from the HPA database. HPA: Human Protein Atlas, CRC: colorectal cancer.
Drug enrichment analysis
Using the DSigDB database, we predicted potential effective intervention drugs. Based on parameters such as Rich Factor, Fold Enrichment, Z-Score, and P-value, the top ten potential chemical compounds were listed in Table 1. Among them, meclizine and megestrol are FDA-approved drugs that may be relevant for CRC treatment.
Molecular docking
To evaluate the binding affinity of candidate drugs to their targets and assess druggability, molecular docking was performed. The molecular structures of meclizine (Compound CID:4034) and megestrol (Compound CID:19090) were retrieved from the PubChem Compound Database. The three-dimensional coordinates of the protein TIMP1 (PDB code: 3V96; resolution: 1.7 A) were downloaded from the PDB .
Molecular docking results showed that both meclizine and megestrol could bind to TIMP1, with binding energies of -9.8 kcal/mol and − 10.8 kcal/mol, respectively, indicating highly stable binding interactions. The detailed results are depicted in Fig. 8, which shows the cartoon representation of the overlay of the crystal structures of TIMP1 protein with the small molecule compounds meclizine and megestrol.

Cartoon representation: overlay of the crystal structures of TIMP1 protein with small molecule compounds meclizine and megestrol.(A) Cartoon representation of molecular docking and interaction of TIMP1 with meclizine; (B)Cartoon representation of molecular docking and interaction of TIMP1 with megestrol; (C)The 3D structure of TIMP1 protein; (D)The 3D structure of meclizine; (E)The 3D structure of megestrol.
Discussion
This study used MR analysis to validate causal relationships between 56 inflammation and immunity related proteins and CRC. GO enrichment analysis revealed significant pathways in the BP category, such as cell migration and chemotaxis, including neutrophil chemotaxis, monocyte migration, and lymphocyte migration. For CC, genes were enriched in immune-related structures like the secretory granule lumen, cytoplasmic vesicle lumen, and lateral plasma membrane. In terms of MF, these genes were primarily involved in immune activities, including cytokine activity, cytokine receptor binding, and chemokine activity. KEGG pathway analysis highlighted pathways associated with cytokine signaling and cancer, such as cytokine-cytokine receptor interaction, viral protein interaction with cytokines and receptors, and the HIF-1 signaling pathway. These enriched pathways underscore the critical roles these genes play in immune responses, viral infections, and cancer development. PPI analysis indicated that TNF and CXCL10 had the most interactions. By utilizing Cytoscape’s MCODE plugin, we identified three core subnetworks involving 15 proteins, including CXCL11, IL2, CCL8, CCL19, CXCL10, CCL7, TGFB3, TIMP1, PLAU, CDH1, HP, MPO, PRKG1, PDE4A, and PDE5A, which may serve as key regulatory hubs.Among these, TIMP1 was further validated as being associated with CRC through differential expression and survival analyses in the GEPIA2 database using TCGA and GTEx data. HPA analysis further confirmed that TIMP1 predominantly localizes to the cytoplasm and cell membrane in CRC tissues, reinforcing its potential as a therapeutic target. Drug enrichment analysis identified meclizine and megestrol as potential inhibitors of TIMP1, with molecular docking results confirming their stable affinity for this target. However, further studies are needed to assess whether these drugs could effectively inhibit TIMP1 in CRC therapy.Our MR analysis of 1,400 metabolites and their ratios identified 28 with causal relationships to CRC five of which are associated with arachidonic acid metabolism. Arachidonic acid, an ω-6 fatty acid, plays a key role in inflammatory pathways and serves as a precursor for pro-inflammatory molecules like prostaglandins and leukotrienes, which contribute to CRC progression, particularly in inflammation-associated tumorigenesis49. Additionally, we identified causal relationships between 731 immune cell traits and CRC, with 24 traits showing significant associations. Notably, CD8 on central memory CD8 + T cells showed the highest significance (P < 0.05). CD8 on naive CD8 + T cells also showed relevance (P < 0.05), supporting existing findings that CD8 + T cell infiltration correlates with better prognosis in CRC patients, despite functional exhaustion of these cells in some tumor microenvironments, leading to reduced anti-tumor activity50. Collectively, our findings indicate that CD8 + T cell infiltration may offer protective effects against CRC, while TIMP1 protein and arachidonic acid metabolism may contribute to CRC risk.
The enrichment analysis results provide significant insights into the molecular mechanisms driving CRC progression. The biological processes identified, such as cell migration and chemotaxis, are crucial for tumor cell invasion and metastasis, which are key features of CRC progression. These processes enable the tumor cells to spread to distant organs, contributing to CRC’s aggressive nature. Moreover, the enrichment of genes involved in immune functions, such as cytokine activity and receptor binding, suggests that immune cells play a critical role in the tumor microenvironment. Cytokines and chemokines can promote immune cell infiltration into the tumor, but also facilitate immune evasion, which is a hallmark of CRC development. Furthermore, the HIF-1 signaling pathway, which is enriched in our analysis, supports tumor survival in hypoxic conditions, promoting angiogenesis and metastasis, and is well-established as a critical pathway in CRC pathophysiology50,51.These findings reinforce the direct link between the enriched pathways and CRC’s biological and clinical behaviors, highlighting their potential as therapeutic targets.
CD8 + T cells are essential in tumor immunity, particularly for immune surveillance and evasion processes. These cells can directly kill tumor cells expressing abnormal antigens by releasing perforin and granzymes, and they further stimulate the anti-tumor immune response by secreting cytokines like IFN-γ. CD8 + T cell infiltration is generally linked to a favorable prognosis in CRC[49],, as these cells effectively recognize and eradicate cancer cells. However, tumors often evade immune destruction by expressing PD-L1, which binds to PD-1 receptors on CD8 + T cells, resulting in T cell exhaustion and impaired functionality52. Exhausted T cells remain in a low-activity state in the tumor microenvironment, hindering effective tumor clearance.In addition to T cell exhaustion, CRC is influenced by other immunosuppressive factors, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which secrete factors like IL-10 and TGF-β, further diminishing CD8 + T cell activity53. Consequently, immune checkpoint inhibitors targeting PD-1/PD-L1 may hold promise in CRC treatment by reactivating T cells and restoring their anti-tumor capabilities. Arachidonic acid, a crucial fatty acid, generates bioactive molecules like prostaglandins (PGE2) and leukotrienes (LTs) through its metabolic pathways, which are integral to inflammation, cell proliferation, and tumor progression. Elevated PGE2 levels in CRC patients are closely linked to tumor growth, immune suppression, and angiogenesis. Arachidonic acid metabolites, particularly PGE2, have been associated with macrophage infiltration in CRC, which in turn promotes tumor progression51,54. Aberrant arachidonic acid metabolism via COX and LOX pathways may drive tumorigenesis and progression, making COX-2 inhibitors promising therapeutic options in this context[53]. However, further research is required to clarify the specific mechanisms of arachidonic acid metabolism in different cancers.
Previous studies have explored the expression and mechanisms of TIMP1 in CRC. These studies found that TIMP1 expression is significantly elevated in CRC tissues compared to normal tissues and is associated with regional lymph node metastasis, distant metastasis, vascular invasion, and AJCC staging. The Cox proportional hazard model identifies TIMP1 as an independent prognostic marker for disease-free and overall survival in CRC patients55,56. TIMP1 plays a dual role in CRC, On one hand, as a key inhibitor of matrix metalloproteinases, TIMP1 restricts tumor invasion by blocking MMP-9-mediated extracellular matrix degradation, thus inhibiting tumor cell invasion and metastasis57. On the other hand, TIMP1 accelerates CRC progression by activating the PI3K/AKT signaling pathway, promoting the proliferation of cancer-associated fibroblasts, which in turn supports CRC growth58. Our study further supports the role of TIMP1 as an oncogene, particularly in CRC, where its upregulation may contribute to tumorigenesis and cancer progression. Therefore, TIMP1 could serve as a promising therapeutic target for CRC.
Meclizine and megestrol are two FDA approved drugs with clinical applications. meclizine, an antihistamine, is commonly used to prevent and treat nausea, vomiting, and dizziness associated with motion sickness, while megestrol, a synthetic progestin, serves as an appetite stimulant for cancer or HIV/AIDS patients with anorexia and cachexia. megestrol is also used to treat advanced breast and endometrial cancers. However, neither drug has been reported in CRC treatment. In this study, we identified meclizine and megestrol as potential TIMP1 inhibitors, suggesting they may have therapeutic relevance in CRC. Molecular docking confirmed their binding affinity for TIMP1, but further studies are necessary to evaluate their clinical potential.
This study has several limitations. First, the GWAS data predominantly represent European populations, which may restrict the generalizability of our findings to other racial and ethnic groups. Second, in the selection of IVs, although we applied stringent criteria and performed sensitivity analyses to assess the robustness of the results, unknown confounding factors and pleiotropy may still influence the findings. Therefore, caution is needed when interpreting the causal relationships between the exposure factors and CRC. Third, our analysis relied on summary-level GWAS data without access to individual-level data (e.g., age and gender), limiting our ability to perform detailed stratified analyses. Lastly, due to study design and funding constraints, our research focused on data analysis and did not include clinical specimen validation or cell-based experimental verification.
This study identified causal relationships between CRC and 56 proteins, 24 immune cell traits, and 28 metabolites. Our findings address several controversies in traditional observational studies and provide in-depth insights into CRC pathology. Notably, TIMP1 emerges as a potential therapeutic target for CRC. These findings offer valuable directions for understanding CRC mechanisms and developing future therapeutic strategies.
Data availability
All original data is derived from publicly available databases. All results are presented within the article or supplementary files. For more detailed data, please contact the corresponding author.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA-CANCER J. CLIN. 74, 12–49 (2024).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-CANCER J. CLIN. 68, 394–424 (2018).
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-CANCER J. CLIN. 71, 209–249 (2021).
Li, Y. et al. BMP suppresses Wnt signaling via the Bcl11b-regulated NuRD complex to maintain intestinal stem cells. EMBO J. 43, 6032–6051 (2024).
Zhang, J., Chen, C., Yan, W. & Fu, Y. New sights of immunometabolism and agent progress in colitis associated colorectal cancer. FRONT. PHARMACOL. 14, 1303913 (2023).
Zhang, Y. et al. Insights of expression profile of chemokine family in inflammatory bowel diseases and carcinogenesis. INT J. MOL. SCI. 25, 10857 (2024).
Tang, J. et al. CXC chemokine receptor 4 - mediated immune modulation and tumor microenvironment heterogeneity in gastric cancer: utilizing multi-omics approaches to identify potential therapeutic targets. BIOFACTORS. 51, e2130 (2024).
Zhou, Y. et al. IL-1 receptor-associated kinase 3 (IRAK3) in lung adenocarcinoma predicts prognosis and immunotherapy resistance: involvement of multiple inflammation-related pathways. TRANSL LUNG CANCER R. 13, 2139–2161 (2024).
Rattray, N. et al. Environmental influences in the etiology of colorectal cancer: the premise of metabolomics. Curr. Pharmacol. Rep. 3, 114–125 (2017).
Mukherjee, S. et al. Toll-like receptor-guided therapeutic intervention of human cancers: molecular and immunological perspectives. FRONT. IMMUNOL. 14, 1244345 (2023).
Behzadi, P. et al. The Interleukin-1 (IL-1) Superfamily Cytokines and Their Single Nucleotide Polymorphisms (SNPs). J IMMUNOL RES 2054431 (2022). (2022).
Tannenbaum, C. S. & Hamilton, T. A. Immune-inflammatory mechanisms in IFNgamma-mediated anti-tumor activity. SEMIN CANCER BIOL. 10, 113–123 (2000).
Maccalli, C., Scaramuzza, S. & Parmiani, G. TNK cells (NKG2D + CD8 + or CD4 + T lymphocytes) in the control of human tumors. CANCER IMMUNOL. IMMUN. 58, 801–808 (2009).
Sconocchia, G. et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. ONCOIMMUNOLOGY. 3, e952197 (2014).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. SCIENCE. 313, 1960–1964 (2006).
Patra, R., Das, N. C. & Mukherjee, S. Exploring the differential expression and prognostic significance of the COL11A1 gene in human colorectal carcinoma: an integrated bioinformatics approach. FRONT. GENET. 12, 608313 (2021).
Patra, R., Dey, A. K. & Mukherjee, S. Identification of genes critical for inducing ulcerative colitis and exploring their tumorigenic potential in human colorectal carcinoma. PLOS ONE. 18, e289064 (2023).
Smith, G. D. et al. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLOS MED. 4, e352 (2007).
Davies, N. M., Holmes, M. V. & Davey, S. G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ-BRIT MED. J. 362, k601 (2018).
He, M. J. et al. Exploring the roles and potential therapeutic strategies of inflammation and metabolism in the pathogenesis of vitiligo: a Mendelian randomization and bioinformatics-based investigation. FRONT. GENET. 15, 1385339 (2024).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. NATURE. 613, 508–518 (2023).
Buniello, A. et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. NUCLEIC ACIDS RES. 47, D1005–D1012 (2019).
Chen, Y. et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. NAT. GENET. 55, 44–53 (2023).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. NAT. GENET. 53, 1712–1721 (2021).
Zhao, J. H. et al. Genetics of Circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. NAT. IMMUNOL. 24, 1540–1551 (2023).
Orrù, V. et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. NAT. GENET. 52, 1036–1045 (2020).
Li, C., Tang, Z., Zhang, W., Ye, Z. & Liu, F. GEPIA2021: integrating multiple deconvolution-based analysis into GEPIA. NUCLEIC ACIDS RES. 49, W242–W246 (2021).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. P NATL. ACAD. SCI. USA. 102, 15545–15550 (2005).
Pierce, B. L., Ahsan, H. & Vanderweele, T. J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. INT. J. EPIDEMIOL. 40, 740–752 (2011).
Burgess, S. & Thompson, S. G. Avoiding bias from weak instruments in Mendelian randomization studies. INT. J. EPIDEMIOL. 40, 755–764 (2011).
Bowden, J. et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. INT. J. EPIDEMIOL. 48, 728–742 (2019).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal Pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. NAT. GENET. 50, 693–698 (2018).
Burgess, S., Scott, R. A., Timpson, N. J., Davey, S. G. & Thompson, S. G. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. EUR. J. EPIDEMIOL. 30, 543–552 (2015).
Bowden, J., Davey, S. G. & Burgess, S. Mendelian randomization with invalid instruments: effect Estimation and bias detection through Egger regression. INT. J. EPIDEMIOL. 44, 512–525 (2015).
Bowden, J., Davey, S. G., Haycock, P. C. & Burgess, S. Consistent Estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. GENET. EPIDEMIOL. 40, 304–314 (2016).
Hartwig, F. P., Davey, S. G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal Pleiotropy assumption. INT. J. EPIDEMIOL. 46, 1985–1998 (2017).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. GENET. EPIDEMIOL. 37, 658–665 (2013).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc.: Ser. B (Methodol.). 57, 289–300 (1995).
Chen, L., Zhang, Y. H., Lu, G., Huang, T. & Cai, Y. D. Analysis of cancer-related LncRNAs using gene ontology and KEGG pathways. ARTIF. INTELL. MED. 76, 27–36 (2017).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. NUCLEIC ACIDS RES. 51, D638–D646 (2023).
Otasek, D., Morris, J. H., Bou as, J., Pico, A. R. & Demchak, B. Cytoscape automation: empowering workflow-based network analysis. GENOME BIOL. 20, 185 (2019).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. SCIENCE 347, 1260419 (2015).
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. BIOINFORMATICS 31, 3069–3071 (2015).
Liu, Y. et al. CB-Dock2: improved protein-ligand blind Docking by integrating cavity detection, Docking and homologous template fitting. NUCLEIC ACIDS RES. 50, W159–W164 (2022).
Yang, X., Liu, Y., Gan, J., Xiao, Z. X. & Cao, Y. FitDock: protein-ligand Docking by template fitting. BRIEF BIOINFORM. 23, bbac087 (2022).
Kim, S. et al. PubChem 2023 update. NUCLEIC ACIDS RES. 51, D1373–D1380 (2023).
Berman, H. M. et al. The protein data bank. NUCLEIC ACIDS RES. 28, 235–242 (2000).
Liu, W., Kuang, T., Liu, L. & Deng, W. The role of innate immune cells in the colorectal cancer tumor microenvironment and advances in anti-tumor therapy research. FRONT. IMMUNOL. 15, 1407449 (2024).
Burgos-Molina, A. M., Téllez, S. T., Redondo, M. & Bravo, R. M. The crucial role of inflammation and the immune system in colorectal Cancer carcinogenesis: A comprehensive perspective. INT J. MOL. SCI. 25, 6188 (2024).
Guo, L. et al. Regulation of IFN-γ-mediated PD-L1 expression by MYC in colorectal cancer with wild-type KRAS and TP53 and its clinical implications. FRONT. PHARMACOL. 13, 1022129 (2022).
Jorgovanovic, D., Song, M., Wang, L. & Zhang, Y. Roles of IFN-γ in tumor progression and regression: a review. BIOMARK. RES. 8, 49 (2020).
Zhang, C. et al. The arachidonic acid metabolome reveals elevation of prostaglandin E2 biosynthesis in colorectal cancer. ANALYST 149, 1907–1920 (2024).
Fan, Y. Y. et al. A new model to study the role of arachidonic acid in Colon cancer pathophysiology. CANCER PREV. RES. 9, 750–757 (2016).
Qiu, X. et al. Unraveling TIMP1: a multifaceted biomarker in colorectal cancer. FRONT. GENET. 14, 1265137 (2023).
Meng, C. et al. TIMP-1 is a novel serum biomarker for the diagnosis of colorectal cancer: A meta-analysis. PLOS ONE. 13, e207039 (2018).
Tarpgaard, L. S. et al. TIMP-1 is under regulation of the EGF signaling axis and promotes an aggressive phenotype in KRAS-mutated colorectal cancer cells: a potential novel approach to the treatment of metastatic colorectal cancer. Oncotarget 7, 59441–59457 (2016).
Acknowledgements
We acknowledge that all genetic summary data utilized in this study were obtained from the FinnGen, GWAS Catalog, and deCODE consortium. We extend our sincere thanks to all participants and investigators who contributed to the GWAS data.
Funding
This research was supported by the Yongchuan District Natural Science Foundation of Chongqing, China (Project No. 2023yc-jckx20060) in 2023.
Author information
Authors and Affiliations
Contributions
L.Z. wrote the main manuscript text and performed statistical analysis. Y.L. and J.Z. supervised the study, designed the research framework, and revised the manuscript. M.J.L., L.L.Z., T.T.P., C.Y.H., Q.Z., X.R.H., A.Y.D., Z.Y.G., J.L., J.T., and J.C. contributed to data collection, interpretation of results, and manuscript review. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, L., Liao, M., Zhang, L. et al. Exploring the role of inflammation, immunity, and metabolism in colorectal cancer via mendelian randomization approach. Sci Rep 15, 24466 (2025). https://doi.org/10.1038/s41598-025-10318-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-10318-1
