
Abstract
New Delhi metallo-beta-lactamase-1 (NDM-1) is a protein produced by bacteria carrying the blaNDM-1 gene, leading to antibiotic resistance, a major global health issue. NDM-1 hydrolyzes nearly all beta-lactam antibiotics, including last-resort carbapenems. Developing effective NDM-1 inhibitors is crucial, as none are currently available for clinical use. This study investigates the potential anti-NDM-1 effects of reported marine fungi-derived metabolites. Two hundred metabolites of fungal origin with antibacterial activity were collected and virtually screened targeting key active site residues of NDM-1. We examined the drug-likeness characteristics, interaction profile, and pharmacophoric features of the shortlisted metabolites followed by molecular dynamic (MD) simulation: RMSF, RMSD, Rg, SASA, hydrogen bond analysis, MMGBSA, PCA, and FEL. Fifty-eight metabolites exhibited greater binding affinity than the reference drug (meropenem) with the highest score of -6.59 kcal/mol. The top 7 metabolites were selected for interaction analysis based on drug-likeness criteria. The diverse interactions of our top metabolites with key residues (HIS-122, GLN-123, GLU-152, and ASN-220) suggest potential NDM-1 inhibition and disruption of bacterial resistance. Pharmacophoric features highlighted the effective structural characteristics of the top 3 metabolites. Three metabolites namely, 8-O-4-dehydrodiferulic acid, Emerixanthone B, and Trichaspside B demonstrated favorable dynamic behavior and binding stability, with 8-O-4-dehydrodiferulic acid showing the most promising potential. Furthermore, the top metabolites are non-toxic, compatible with β-lactam antibiotics, and show no predicted interactions with human proteins. These selected compounds exhibit favorable physiological compatibility and molecular characteristics that enhance their potential as therapeutic candidates. Given the urgent need for novel treatments targeting NDM-1, the investigated metabolites warrant further experimental validation.
Similar content being viewed by others

Introduction
New Delhi metallo-beta-lactamase (NDM-1) is a novel type of metallo-beta-lactamase called after the place of origin. In 2009, NDM-1 was reported for the first time in Klebsiella pneumoniae from a Swedish patient who had undergone treatment in New Delhi, India1. A multinational team reported the emergence of antibiotic resistance by reporting 180 cases of patients infected by NDM-1 in the August issue of The Lancet: Infectious Diseases2. The cases included 37 cases in the United Kingdom and 143 cases in various locations in Pakistan and India, indicating a widespread dissemination2. Gram-negative bacteria like Escherichia coli and Klebsiella pneumoniae are the most common ones that produce NDM-1, which renders the bacterium resistant to beta-lactam antibiotics. Numerous defense mechanisms have evolved in bacteria to fend off antibiotics3. The synthesis of the enzyme beta-lactamases, which hydrolyzes beta-lactam antibiotics like cephalosporins and carbapenems, is one of the mechanisms. B-lactamases are categorized by the Ambler classification into four classes (A, B, C, and D) that comprise two main families: metallo-beta-lactamases (MBLs) and serine-beta-lactamases (SBLs). Serine hydrolases comprise Ambler classes A, C, and D, whereas metalloenzymes, or MBLs (class B), carry zinc ions in the active site4. Sulbactam, tazobactam, clavulanic acid, and the most recent drugs, avibactam and vaborbactam, are examples of SBL inhibitors that have been effectively developed and are used in clinical settings5. Distinct from SBLs in terms of mechanism, MBLs depend on zinc ions in the active site to stimulate a water molecule, which subsequently breaks down the beta-lactam ring6. Because of this mechanistic difference, SBL inhibitors used in clinical settings hardly affect MBLs at all.
BlaNDM-1 genes are primarily found in bacterial plasmids, which can be obtained by transposition and can render neighboring bacteria resistant. Through conjugation transfer, it spreads horizontally with ease and rapidly disseminates all over the world7,8,9. Research on the NDM-1 target has been the main focus and as a result numerous potent NDM-1 inhibitors, including captopril, thiol compounds10aspergillomarasmine A11boric acid derivatives, sulfonamides12succinic acid derivatives, and several natural products, have been discovered to inhibit NDM-1 by interacting with zinc ions in the active core and significant catalytic amino acid residues. Nonetheless, no NDM-1 inhibitor has received approval because of its physicochemical characteristics, safety, and selectivity toward the human body. This suggests that the need to create novel, potent metallo-beta-lactamase inhibitors is critical and urgent.
In ancient times, humans relied primarily on microorganisms for various purposes, and the majority of antibiotics currently used to treat various infectious diseases are derived from microbial natural products. In 1928, Alexander Fleming used microorganisms to retrieve secondary metabolites. He discovered penicillin, which is a secondary metabolite produced by the fungus Penicillium notatum used to treat bacterial infection13. Some secondary metabolites have been identified as potential inhibitors of NDM-1, such as Aspergillomarasmine A a compound derived from the fungus Aspergillus versicolor which inhibits NDM-1 by chelating zinc ions from its active site14. However, such compounds also present certain limitations. Therefore, the development of new therapeutic agents is essential for the effective and targeted treatment of metallo-β-lactamase-producing pathogens.
Bacterial infections can become life-threatening if not treated promptly, especially given the growing concern of antibiotic resistance. Therefore, inhibiting β-lactamases is crucial for overcoming resistance and restoring the efficacy of β-lactam antibiotics. In-silico approaches such as these are particularly valuable in the early stages of drug discovery, as they accelerate the identification of promising candidates while significantly reducing time and cost15. In this study, we leveraged the rich diversity of marine-derived fungal secondary metabolites to target and inhibit the active site of the potent NDM-1 enzyme. This study employed computer-aided drug design techniques to virtually screen these metabolites against the β-lactamase target protein (PDB ID: 4EYL). The fungal-derived secondary metabolites show useful biological properties, i.e., anti-inflammatory, antioxidant, antimicrobial, and anticancer16. Our methodology comprises molecular docking of 200 metabolites of fungal origin, evaluations of drug-likeness and pharmacokinetic properties, toxicity analysis, target fishing analysis, medicinal chemistry analysis, molecular dynamic simulations, PASS prediction, and pharmacophore analyses. These comprehensive evaluations aimed to identify potential drug candidates that could overcome the limitations of current NDM-1 therapies and warrant further validation through wet-lab experiments.
Materials and methods
Collection and Preparation of metabolites
A total of 200 marine fungal metabolites were selected for virtual screening based on an extensive literature survey of bioactive compounds reported for their antibacterial activity (Table S1). The compounds were sourced from both original research and review papers, and the search was iteratively expanded until no additional unique metabolites were found. Meropenem (PubChem CID: 441130), a carbapenem antibiotic, was taken as a reference drug. It is co-crystalized with metallo beta-lactamase (PDB: 4EYL), allowing us to define the binding site of the enzyme. It provides valuable insights into the specificity of NDM-1 binding to its substrate and the enzyme inhibition mechanisms17. A chemical database, i.e., PubChem, was used to retrieve the SMILES (Simplified Molecular Input Line Entry System), which were pasted in the ChemDraw 12.0 software to accurately draw the structures of fungal-derived metabolites and subsequently save them in MDL mol format. Within the Molecular Operating Environment (MOE), these files were imported as an in-house database and proceeded with energy minimization for compound stability employing default parameters as performed previously18.
Preparation of protein
The crystal structure of NDM-1 (PDB ID: 4EYL) was obtained from the RCSB Protein Data Bank. The 4EYL was selected for the presence of meropenem in its catalytic domain and its crystallization with X-ray diffraction. The single unique chain of NDM-1 and its high resolution of 1.9 Å offer a coherent framework for accurate docking simulations. Before molecular docking, the target protein was prepared via MOE software. This involved eliminating the second chain, allocating bond ordering using default settings, and eliminating all attached ligands and water molecules (apart from the catalytic ones). Moreover, much attention was paid to preserving the integrity of the zinc ions inside the protein structure. It was saved as MOE molecule file after being energy minimized and protonated using default settings19.
Selection of active site
The NDM-1 active site was defined by using the MOE Site Finder program, which predicted pockets of variable sizes. Among them, the optimum binding pocket was chosen based on the position of the co-crystallized meropenem, which is further corroborated by the previously reported active sites of NDM-120,21,22. Dummy atoms were constructed to make the binding pockets accessible to the ligand atoms.
Molecular Docking
Molecular docking is a computational technique that predicts how a ligand binds to target to form a stable complex. It’s a key tool in computer-assisted drug design and structural molecular biology. As performed previously23,24 docking was performed through MOE (Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, MOE2022.v11.18.1). Dummy atoms were chosen from the site module; they are used as protein-active sites. The DOCK command of MOE was computed, and the induced-fit model of the receptor was employed as a multi-step protocol. The Triangle Matcher algorithm was used for initial ligand placement, followed by a London dG scoring function with a retention of 10 poses per ligand. These initial poses were refined using MMFF94x forcefield-based minimization, and a second rescoring was conducted using GBVI/WSA dG, with the top five poses retained. Duplicates were removed at each stage and the highest-scoring position of each ligand was analyzed based on the binding energy represented as docking score (S score). The S scores of the ligands were compared with the S scores of the reference inhibitor, meropenem. Ligands that exhibited a greater docking score compared to the reference inhibitor were shortlisted for further analyses.
Validation of the Docking protocol
Docking validation by superimposition is a process that involves comparing a docked ligand to its native ligand in a co-crystal structure25,26. We used the X-ray structure of NDM-1 (PDB ID: 4EYL) with co-crystallized Meropenem (PubChem CID: 441130) as a reference to compare our docked NDM-1-meropenem complex. Using PyMOL, the co-crystallized protein structure was superimposed with the docked protein-ligand complex, and the output was configured to compute the RMSD. An effective docking procedure was defined by an RMSD value of ≤ 2.0 A27.
Molecular dynamic simulation analysis
We performed molecular dynamics (MD) simulations using the AMBER force field (ff14SB) and the AMBER18 software suite28. For solvation, we used the TIP3P water model, and the systems were contained in a 12 Å × 12 Å × 12 Å orthorhombic box28. Each system was subjected to energy minimization, both with and without restrictions, before the MD simulations to guarantee complete enzyme-ligand complex convergence. The system was heated to 300 K before being incrementally equilibrated every 5 ns in the simulations, which were carried out under the NVT ensemble. Eventually, a 200 ns production run was completed. The remaining protocols followed the steps described in our earlier papers29,30. The CPPTRAJ module of AmberTools18 was used for trajectory analysis, and VMD was used for visualization31,32. PyMOL was used to generate the images, and XMGrace was used for all data processing in accordance with established procedures from earlier research33. Different calculations including Root Mean Squared Deviation (RMSD), Radius of gyration (Rg), Solvent Accessible Surface Area (SASA), and Hydrogen bonds calculation, and Root Mean Square Fluctuation (RMSF).
Binding free energy calculations
The binding free energy of three selected compounds 8-o-4-dehydrodiferulic acid (Compound 1), Emerixanthone B (Compound 2), and Trichaspside B (Compound 3)—with the 4ELY receptor was evaluated using the widely recognized molecular mechanics–generalized Born surface area (MMGBSA) approach34. To ensure accurate and statistically robust calculations, 2000 snapshots were extracted from the 200 ns MD simulation trajectories.
In this method, the binding free energy (ΔGbinding) is determined by subtracting the free energies of the unbound receptor and ligand from the free energy of the receptor-ligand complex. The binding free energy is calculated using the following equation:
Where: ΔGsolvation, complex, ΔGsolvation, receptor, and ΔGsolvation, ligand are the solvation free energies of the complex, receptor, and ligand, respectively. ΔGvacuum, binding is the vacuum free energy of binding, calculated as the average interaction energy between the receptor and ligand in the absence of solvent. The vacuum free energy (ΔGvacuum) includes contributions from van der Waals, electrostatic, and internal energies, reflecting the direct interactions between the receptor and the ligand. Solvation free energy components incorporate polar and nonpolar solvation effects, comprehensively representing the binding energetics. This approach decomposes the binding free energy into distinct contributing factors, enabling a detailed assessment of the forces driving ligand-receptor interactions. Molecular mechanics calculations provided van der Waals, electrostatic, and internal energy components, while solvation effects were calculated to account for the environment’s influence on binding. The combined contributions facilitated a thorough evaluation of ligand-binding energetics. These methods effectively capture the physical and solvation effects governing ligand-receptor binding, offering valuable insights into the stability and affinity of the complexes35,36.
Drug-likeness analysis
The drug-ability analysis was performed for the top metabolites with the docking scores superior to the standard compound. The software used for this analysis was the SwissADME online server37. The input was given as the mol files of the structures of metabolites. In drug discovery, drug-likeness is a crucial concept, assessing properties and structures to find promising candidates and exclude problematic ones early, saving time and resources. In this analysis, several filters were applied, such as Lipinski, Ghose, Veber, Egan, and Muegge, and the Bioavailability Score.
Pharmacokinetic analysis
Pharmacokinetics is a fundamental aspect of drug development that plays an important role in evaluating the effectiveness and safety of pharmaceutical compounds. It comprises the study of how the body interacts with a drug, including its absorption, distribution, metabolism, and excretion38. The pharmacokinetic properties of the top-selected metabolites were determined using the SwissADME server37. The pharmacokinetic analysis provides information about the pharmacokinetic properties of compounds, including GI (gastrointestinal) absorption, BBB (blood-brain barrier) permeability, P-glycoprotein (P-gp) substrate activity, and its interaction with five important enzymes of human cytochromes P450 (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4).
Toxicity analysis
ProTox-II server was used for toxicity analysis39. Here, the aim was to find the LD50 value (mg/kg), cytotoxicity probability, and hepatoxicity of the top selected metabolites.
Medicinal chemistry
Medicinal chemistry analysis plays an important role in drug design. It helps medicinal chemists in their daily drug discovery activities37. These analyses were accomplished by using the Swiss ADME server. The SMILES representations of the ligands were entered into the online interface of the server. In particular, the ligands were examined in the medicinal chemistry portion for possible Pan-Assay Interference Structures (PAINS), Brenk fragments, lead-likeness, and synthetic accessibility scores of the server.
Pharmacophore modeling
MOE has been established as an effective tool for creating a pharmacophoric model40. The mol files of the top hit ligands were opened in MOE software, and then a potent pharmacophore model was developed by exploiting the Pharmacophore Query Editor. The model was improved by adding certain pharmacophore properties, like bonding strength, spatial organization, and chemical functionality, specifically designed to represent the key molecular interactions.
Target fishing analysis
The metabolites that looked the most promising were subjected to target-fishing tests to assess their potential for interacting with human targets. The Swiss Target Prediction server was used for this purpose37. For this analysis, the SMILES codes of all selected metabolites were prepared and used as input files. The analysis results were predicted in the form of scores ranging from 0 to 1, and here the value 1 suggests that the target is considered to have the most likelihood of interaction with specific compounds.
PASS prediction
PASS (Prediction of Activity Spectra for Substances) analysis was performed on the top hits to identify possible biological activities in addition to their estimated antibacterial activity for NDM-123. Based on the structure-activity connections of known chemical compounds, the PASS server predicts the pharmacological effects, action mechanisms, and biological activities of compounds. It expresses its predictions as probabilities, representing the likelihood that a compound will have a particular biological activity (Pa). Properties with a Pa value greater than 0.7 were the ones we concentrated on in our study since there was an excellent likelihood that the hits we found would show these kinds of actions.
Result and discussion
Selected potential active site
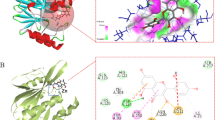
The catalytically significant residues of NDM-1 include HIS-120, HIS-122, ASP-124, HIS-189, CYS-208, HIS-250, 2(ZN-302 ZN-303)20LEU-65, PHE-70, VAL-73, TRP-93, LYS-211, ASN-22021, ALA-121, GLN-123, GLU-152, MET-154, LEU-218, GLY-219, and ASP-22322 (Fig. 1A). According to the crystal structure analysis, the two positively charged zinc ions that hydrolyze beta-lactam rings are found in NDM-1 active sites, forming a broad, shallow substrate-binding capsule cavity. Therefore, different types of beta-lactam antibiotics can be readily accepted. Zinc that is firmly bonded is known as Zn1 (ZN-302). Zinc 1 coordination with three identical amino acids (His120, His122, and His189) and with water molecules, or more probably hydroxyl groups, creates a tetrahedral geometry referred to as the histidine site; Zn2 (ZN-303), a loosely bound zinc, surrounds three different amino acids (ASP-124, CYS-208, and HIS-250) to form a triangular conical structure known as the cysteine site. It is also applicable as ZN-302 ligands41. Zn1 and Zn2 are joined by side chains of ASP-12443. In addition to the active site, some other important residues like LEU-65, PHE-70, VAL-73, TRP-93, LYS-211, ASN-220, ALA-121, GLN-123, GLU-152, MET-154, LEU-218, GLY-219, and ASP-223 have been proposed to play a crucial role in the stability of metallo beta-lactamase and hydrolysis of the beta-lactam ring44,45. Like, ASN-220 was a crucial residue for the substrate binding and integrity of the intermediate. The hydrophilic portion of the molecule forms a hydrogen bond with Asn220. At the same time, the hydrophobic part interacts with the L3 ring to indirectly decrease NDM-1 action9. Furthermore, GLN-123, GLU-152, HIS-122, LYS-211, ASN-220, and ASP-124 are important NDM-1 residues that help to establish H-bonds with antibiotics, and a few NDM-1 residues, including HIS-122, TRP-93, VAL-73, ILE-35, MET-154, and HIS- 250, also contributed to the formation of hydrophobic contacts with antibiotics22.

(A) Important active site residues of MBLs. Residues in dark magenta (hot pink)—GLN-123, ASN-220, ASP-223, and GLU-152—form hydrogen bonds with our top hits, while residues in white/grey—His120, His122, His189, Asp124, Cys208, and His250—coordinate with two zinc ions to stabilize the active site of NDM-1, which facilitates its catalytic activity and enables it to hydrolyze antibiotics, while an active water molecule (HOH 420) plays a crucial role in the hydrolysis reaction. Other residues in green color also play a role in the stability of NDM-1. (B) Graphical representation of the docking score of our top metabolites, namely 8-O-4-dehydrodiferulic acid, Emerixanthone B, and Trichaspside B, in comparison to standard metallobeta-lactamase inhibitors (TPEN (N, N,N’,N’-Tetrakis(2-pyridylmethyl) ethylene diamine), EDTA (Ethylenediaminetetraacetic acid), Captopril, and Dipicolinic acid) and muted antibiotic meropenem.
Molecular Docking analysis
Molecular docking was performed using 200 fungal-derived secondary metabolites with known antibacterial activity against the target protein NDM-1. The binding energies of the metabolites were noted and rated to determine the best hits (Table S1). The reference ligand displayed a −5.43 kcal/mol docking score. Here, it was discovered that 58 metabolites outperformed the reference in terms of docking score. Their docking scores were in the range of −6.76 kcal/mol to −5.44 kcal/mol, whereas the top selected metabolites were in the range of −6.59 to −5.99 kcal/mol (Table S1). Based on molecular docking results, it was found that the ten metabolites, namely, Auxarthrol D, Phomopsichalasin, Helvolic acid, Lapatin B, Equisetin, 7,10-epoxycycloneran-3,11,12-triol, 11-hydroxy-9-harzien-3-one, Austalide M, and GKK1032B, were inactive and failed to exhibit any kind of interaction with the target protein NDM-1 (Table S1), This could be as a result of the structural flexibility or complexity of metabolites, which can make it more difficult to generate an appropriate conformation for docking. Furthermore, it is possible that these chemicals were not compatible with the selected binding site, which led to an unsatisfactory docking process for these metabolites. Since these 10 metabolites are unable to interact with the target protein effectively, it is evident that they are not effective as prospective NDM-1 inhibitors. This suggests that to improve the binding affinity of these specific chemicals, more structural alteration would be required.
We also performed a comparative study involving five standard compounds alongside our three top hits, which were selected based on additional screening criteria described in the subsequent sections. We assessed the docking scores of each metabolite against a target protein. The top metabolites, namely, 8-O-4-dehydrodiferulic acid, Emerixanthone B, and Trichaspside B, showed greater docking scores as compared to five standard compounds (Fig. 1B). In standard compounds, meropenem is a modified antibiotic, while TPEN (N, N,N’,N’-Tetrakis(2-pyridylmethyl) ethylene diamine)44EDTA (Ethylenediaminetetraacetic acid), Captopril45and Dipicolinic acid46 are previously reported metallo beta-lactamase inhibitors. This comparison highlights the promising therapeutic potential of our top hits as they showcased more negative docking scores than these reported inhibitors.
Validation of Docking protocol
The co-crystallized Meropenem-NDM-1 complex (PDB ID: 4EYL) and the re-docked Meropenem-NDM-1 complex were used to validate the docking approach. A comparison of 231 residues from both structures was done in this process. We achieved a match align score of 1205.000 during this alignment, showing that the two structures are congruent. Notably, a tiny group of atoms was excluded throughout the rigorous refinement, obtaining a more accurate alignment. Particularly, in the first cycle, eight atoms were rejected; in the second, four atoms; and in the third, one atom. Finally, the ultimate alignment resulted in the low RMSD value of 0.697 Å, calculated across 218 atoms. This value not only highlights the excellent quality of our alignment but also the accuracy and dependability of our docking process in structural comparisons. Similar binding pockets were shown by superimposing these structures, indicating that the ligands interact with these targets in a manner that is similar to one another (Fig. 2A, B, C, and D).

Superimposition of the reference complex with co-crystallized ligand and the docked Meropenem_4EYL complex. (A) The binding of the standard compound, meropenem (red), in the 3D structure of NDM-1 (4EYL). (B) Binding of the docked meropenem (blue) in NDM-1. (C) Superimposition of A and B complexes shows binding of meropenem at the same location. (D) Superimposition of native and docked meropenem with the NDM-1 complex.
Drug-likeness analysis
After molecular docking, the drug-likeness analysis of the top 20 metabolites based on their binding energies was conducted by determining their scores for Lipinski’s (Pfizer, New York, NY, USA), Ghose, Veber (GSK, London, UK), Egan (Pharmacia, Uppsala, Sweden), and Muegge (Bayer, Leverkusen, Germany), along with the bioavailability scores. According to Lipinski’s rule, a druglike molecule should have a molecular mass of less than 500 Daltons, a maximum of 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, and a lipophilicity value (MlogP) that should be less than 550. Similarly, the Ghose filter indicates that a drug-like molecule should have a molecular weight (MW) in the range of 160–480 Daltons, its lipophilicity (WlogP) should be in the range of −0.4 to 5.6, it should have a molar refractivity of approximately − 130, and the number of atom counts should be in the range of 20–7051. In the same way, Veber’s filter suggests that a molecule should have no more than 10 rotatable bonds and a total polar surface area of 140 or less49. Further, Egan’s guideline specifies that lipophilicity (WlogP) should not exceed 5.88, and the total polar surface area should be 131 or less50. Furthermore, Muegge’s states that a drug-like molecule should have a MW between 200 and 600 Daltons, a lipophilicity (XlogP3) ranging from − 2 to 5, a total polar surface area of 150 or less, no more than 7 rings, more than 4 carbon atoms, more than 1 heteroatom, no more than 15 rotatable bonds, no more than 10 hydrogen bond acceptors, and no more than 5 hydrogen bonds51.
After analysis, 13 metabolites followed some rules while violating others, and only 7 metabolites, as shown in (Table 1), followed all rules for drug-likeness, which indicates that these metabolites have potential as drug candidates. Concerning the bioavailability score, all top-selected metabolites show an optimal score of 0.55, except for 8-O-4-dehydrodiferulic acid, which shows 0.56. In this analysis, all the top 7 selected metabolites show zero violation with the Lipinski, Ghose, Veber, Egan, and Muegge filters. Following the drug-likeness analysis, additional analyses were carried out to comprehensively evaluate various aspects of a potential drug candidate.
Protein-ligand interactions
Seven ligands were chosen to examine their interactions with the receptor depending on the drug-likeness criterion. These metabolites include 8-O-4-dehydrodiferulic acid, Emerixanthone B, Trichaspside B, Emerixanthone A, Tajixanthone hydrate, Trichaspside A, and Aspergixanthone–K (Table 2). After performing ligand-receptor molecular docking, MOE software was used to examine the 2D interactions. Other interactions were found by employing PyMOL software to create 3D representations of ligand-protein interactions. This variation in binding pattern highlights how crucial it is to use a variety of software tools to conduct a thorough analysis of ligand-protein interactions. However, because PyMOL has restrictions when it comes to displaying bond energies, the binding energies of these extra interactions are not mentioned52. A hydrogen bond was observed between the ASN-220 residue of the receptor and the oxygen atom of meropenem at 2.90 Å distance and − 2.2 kcal/mol bond energy. H-pi interactions were also formed between the 5-ring ligand and HIS-250 residue at 3.80 Å distance and − 0.9 kcal/mol bond energy. In addition, meropenem, with the help of its oxygen atom, also forms metal interactions with the ZN-303 atom at a 2.77 Å distance and − 1.2 kcal/mol bond energy, respectively (Fig. 3A, B). Here, also some metal and ionic interactions were seen between zinc atoms and receptor residues. ZN-303 forms metal interactions with HIS-120 and HIS-189 at 2.34 and 2.25 Å distances with − 3.8 and − 4.5 bond energies, respectively. Moreover, ZN-303 also forms ionic interactions with ASP-124 and HIS-189 residues at 3.34 and 2.25 Å distances with − 2.6 and − 12.1 kcal/mol bond energies, respectively. Moreover, ZN-302 also shows metal and ionic interactions. ZN-302 shows metal interactions with ASP-124 and CYS-208 residues at 2.16 and 2.66 Å distances and − 4.4 and − 6.3 kcal/mol bond energies, respectively. ZN-302 also shows ionic interactions with ASP-124 and CYS-208 residues at 2.16 and 2.66 Å distances with − 13.4 and − 0.9 kcal/mol bond energies, respectively (Table 2).
8-O-4-dehydrodiferulic acid with a remarkable docking score of −6.59 kcal/mol forms a hydrogen bond with NDM-1 (Fig. 3C, D). In this interaction, the oxygen atom of the ligand at position 21 forms a hydrogen bond (H-donor) with the GLU-152 residue of 4EYL, at a distance of 2.88 Å and − 7.4 kcal/mol bond energy. Interestingly, GLU-152 is an important NDM-1 residue that helps in the formation of hydrogen bonds between NDM-1 and antibiotics, leading to the hydrolysis of the antibiotics and muting them22. By inhibiting this residue, our compound is most likely to inhibit the hydrolyzing potential of NDM-1, and it can help combat the antibiotic resistance. Some other interactions between the Zn atom and receptor 4EYL were also seen, except for ligand-receptor interaction. These interactions are naturally present and help stabilize the active site of NDM-1. ZN-303 exhibits metal interactions with HIS-120 and HIS-189 residues at 2.34 and 2.25 Å distances with − 3.8 kcal/mol and − 4.0 kcal/mol, respectively. In addition, ZN-303 shows ionic interactions with ASP-124 and HIS-189 residues at 3.29 and 2.25 Å distances with − 2.8 and − 12.1 kcal/mol bond energies, respectively (Table 2).
In metallo beta-lactamase, two Zn ions show interactions with the ligand and receptor residues43. Our analysis revealed that zinc ions exclusively interacted with protein residues and not with ligands. This is a significant accomplishment, as research indicates that certain inhibitors exhibit strong inhibitory function without interacting with metal ions. While an inhibitor doesn’t need to demonstrate that it can also chelate with metal ions, those that do so are preferable because they exhibit less interaction with metal ions and high binding energy with proteins. This is because an effective inhibitor should not obstruct the binding of the other biologically relevant metals that are necessary for the activity of vital metalloenzymes53.

Molecular docking of NDM-1 (4EYL protein) with meropenem (standard compound) and 8-O-4-dehydrodiferulic acid. (A) 2D representation of the interaction between 4EYL protein and meropenem; (B) 3D representation of the interaction between 4EYL protein and meropenem; (C) 2D representation of the interaction between 4EYL protein and 8-O-4-dehydrodiferulic acid; (D) 3D representation of the interaction between 4EYL protein and 8-O-4-dehydrodiferulic acid.
Secondly, Emerixanthone B with a docking score of −6.45 kcal/mol forms hydrogen bonds with 4EYL (Figure S1A, B). In these interactions, the oxygen atom of the ligand at position 56 shows H-donor interaction with the HIS-122 residue at a 2.92 Å distance and − 0.9 kcal/mol energy. HIS-122 is a key active site residue that performs a crucial role by coordinating with zinc ions and aiding in the hydrolysis of beta-lactam antibiotics41. Another oxygen atom at position 20 forms H-acceptor interactions with the GLN-123 residue at a 3.22 Å distance with − 1.5 kcal/mol bond energy. GLN-123 plays a vital role in the stability of NDM-1 and the hydrolysis of antibiotics22. Here, also, some other interactions between zinc atoms and receptor residues were seen. The ZN-303 atom indicates metal interactions with HIS-120 and HIS-189 residues at 2.33 and 2.24 Å distances and − 3.8 and − 4.7 kcal/mol bond energies, respectively. In addition, ZN-303 also shows ionic interactions with ASP-124 and HIS-189 residues at distances of 3.31 and 2.24 Å with − 2.7 and − 12.1 kcal/mol bond energies, respectively. Furthermore, another interaction in the PyMOL software was also shown between the oxygen atom of the ligand at position 56 and the ASN-220 residue, they form hydrogen bonds at a distance of 2.4 Å (Table 2). ASN-220 is an important residue involved in the binding of substrate and integrity of intermediate, as the hydrophilic portion of the molecule forms hydrogen bonds with ASN-22046.
Similarly, Trichaspside B with a docking score of −6.20 kcal/mol shows a hydrogen bond and H-pi interactions with 4EYL (Figure S1C, D). In these interactions, the oxygen atom of the ligand at position 54 forms an H-donor interaction with the GLU-152 residue at a distance of 2.94 Å with − 1.4 kcal/mol energy. GLU-152, an essential NDM-1 residue, aids in the creation of hydrogen bonds between NDM-1 and antibiotics. In addition, the carbon atom of the ligand at position 51 indicates H-pi interactions with the HIS-122 residue at a 3.93 Å distance and − 1.0 kcal/mol bond energy (Table 2). HIS-122 is an active key site residue that collaborates with the zinc ion to facilitate the hydrolysis of beta-lactam antibiotics41.
In the NDM-1-Emerixanthone A complex, Emerixanthone A has a docking score of −6.12 kcal/mol and forms a hydrogen bond with 4EYL (Figure S2A, B). In this interaction, the oxygen atom of the ligand at position 20 forms an H-acceptor interaction with the GLN-123 residue at a distance of 3.20 Å and − 1.6 kcal/mol bond energy. GLN-123 is involved in the stability of NDM-1 and helps in the formation of hydrogen with antibiotics22. Other than ligand-receptor interactions, additional interactions between ZN-303 atoms and receptor residues were also seen. ZN-303 forms metal interactions with HIS-120 and HIS-189 residues at distances of 2.32 and 2.26 Å, with bond energies of −3.9 and − 5.0 kcal/mol, respectively. Likewise, ZN-303 forms ionic interactions with ASP-124 and HIS-189 residues at 3.31 and 2.26 Å distances with − 2.7 and − 11.9 kcal/mol bond energy, respectively. Some extra interactions in PyMOL were seen. The oxygen atom of the ligand at position 56 forms a hydrogen bond with the ASN-220 residue at a 2.4 Å distance. In addition, another oxygen atom forms a hydrogen bond with the HIS-122 residue at a 3.6 Å distance, an active key site residue (Table 2).
The next inhibitor, Tajixanthone hydrate (Figure S2C, D), has a docking score of −6.10 (kcal/mol) and forms a hydrogen bond (H-donor) with the ASP-223 residue at a distance of 3.03 Å and − 2.1 (kcal/mol) bond energy. Likewise, pi-H interactions between the 6-ring structure of the ligand and ASN-220 residue at 3.85 Å and − 0.7 kcal/mol bond energy, as we have discussed above, play an important role during substrate hydrolysis (Table 2).
Trichaspside A, with a docking score of −6.08 kcal/mol, forms a hydrogen bond (H-acceptor) with ASN-220 residue. In this interaction, the oxygen atom of the ligand at position 56 forms an H-acceptor interaction with the ASN-220 residue at a distance of 2.93 Å and − 1.3 kcal/mol of bond energy (Figure S3A, B). Other metal and ionic interactions between ZN-303 and receptor residues were seen except for ligand-receptor interaction. ZN-303 forms metal interactions with HIS-120 and HIS-189 residues at 2.31 and 2.24 Å distances with bond energies of −3.9 and − 4.8 kcal/mol, respectively. In addition, ZN-303 also forms ionic interactions with the ASP-124 and HIS-189 residues at 3.31 and 2.24 Å distances and − 2.7 and − 12.2 kcal/mol bond energies, respectively (Table 2).
Aspergixanthone-K achieved a docking score of −5.99 (kcal/mol) and displays a hydrogen bond with 4EYL (Figure S3C, D). In these interactions, the oxygen atom of the ligand at position 60 forms a hydrogen bond (H-donor) with the HIS-122 residue at a distance of 3.08 and − 0.8 kcal/mol bond energy. Likewise, the oxygen atom of the ligand at position 20 forms a hydrogen bond (H-acceptor) with the GLN-123 residue at a 3.21 Å distance and − 1.7 kcal/mol bond energy. Except for ligand-receptor interactions, some other interactions between ZN-303 and receptor residues were also seen. ZN-303 shows metal interactions with HIS-120 and HIS-189 residues at 2.32 and 2.24 Å distances with − 3.9 and − 4.7 kcal/mol bond energy, respectively. In addition, ZN-303 shows ionic interactions with ASP-124 and HIS-189 residues at 3.30 and 2.24 Å distances with − 2.8 and − 12.1 kcal/mol bond energy, respectively. Another interaction shown in PyMOL was the hydrogen bond between the oxygen atom of the ligand and the ASN-220 residue. In this interaction, the oxygen atom of the ligand at position 60 forms a hydrogen bond with the ASN-220 residue at a 2.5 Å distance (Table 2). The interactions, along with the good docking scores of the top-selected metabolites against NDM-1, indicate that they have the potential to be highly effective.
Most of the top-selected metabolites form significant interactions with metallo beta-lactamase, including the H-bonds with GLU-152, GLN-123, and ASN-220, which ultimately blocks the hydrolysis of antibiotics, while the blocking with the hydrophobic portion of ASN-220 indirectly decreases NDM-1 action. Moreover, hydrogen bonding and H-pi interactions with residues like HIS-122 block the metal coordination bond with Zn 302, also leading to the inhibition of the hydrolytic action of NDM-1. Overall, the diversity of observed interactions between our top metabolites and these aforementioned important residues is enough for the complete inhibition of NDM-1, indicating the potential of our top metabolites to interfere with bacterial resistance mechanisms and effectively stop the action of this protein. Therefore, these metabolites have a significant potential to develop into therapeutic medicines due to their broad interactions and substantial binding patterns, which present interesting paths for treating antibiotic-resistant bacterial variants.
Pharmacophore analysis
We created comprehensive pharmacophore models for the top three metabolites, namely 8-O-4-dehydrodiferulic acid, Emerixanthone B, and Trichaspside B, highlighting the key interacting functional sites for inhibiting 4EYL. In the detailed interaction analysis, although we observed metal coordinate bonds in which zinc ions interacted with the active site itself (Fig. 4), we have only shown those interactions (both 2D and 3D models) that were observed directly between ligands and the protein active site. Don&Acc (hydrogen bond donor and acceptor), Acc (hydrogen bond acceptor), and Hyd (hydrophobic) features were observed. The 8-O-4-dehydrodiferulic acid model displayed one key feature, F1, indicating Don&Acc. This feature with a radius of 0.50 describes the capability of donating and accepting the hydrogen bonds (Fig. 4A–C). This key hydroxyl group is significant for its binding with GLU-152, as can be seen from the interaction pattern (Fig. 3B and C). This interaction biologically blocks the hydrolysis of antibiotics by the metallo beta-lactamase.

Pharmacophoric features of the top 3 metabolites and their 2D and 3D interactions shown only for the key residues of metallo beta-lactamase (A–C) Emerixanthone B, (D–F) Trichaspside B, and (G–I) 8-O-4-dehydrodiferulic acid. This section indicates the illuminated active components that are important for binding with metallo beta-lactamase protein.
The Emerixanthone B model illustrated three key components presented by two features shown as F1, indicating Don&Acc with a radius of 0.50 (for both interacting oxo and hydroxyl groups with HIS-122 and GLN-123, respectively), and F2 indicating Acc with a greater radius of 0.82 (for the interacting hydroxyl group with ASN-220) (Fig. 4D–F). Finally, the Trichaspside B model reflects two features: F1 indicating Hyd and F2 indicating Don&Acc. The features F1 with a radius of 0.50 express a hydrophobic interaction with the key active site residue HIS-122, while F2 with a radius of 0.50 uses a hydroxyl group to form hydrogen bond donor and acceptor interactions with GLU-152 (Fig. 4G–I). As aforementioned, these features are significant in blocking the hydrolysis of antibiotics by metallo beta-lactamase.
Overall, our top anti-fungal-derived antibiotics can form diverse interactions with the active components of the metallo beta-lactamase enzyme. These diverse interactions boost the inhibitory activity of our top compounds. This in-depth knowledge of the binding mechanics of our top compounds encourages the creation of more powerful derivatives or comparable compounds with better pharmacological profiles from additional atomic-level research.
Spatial integrity of the ligand-protein complexes
A time-resolved and dynamic view of biomolecular systems is offered by MD simulations, which show how proteins and ligands interact in an environment that closely resembles physiological ones (Figure S4). The stability, flexibility, and interaction dynamics of the 4ELY protein in combination with three ligands were evaluated in this study using 200 ns MD simulations. Energy minimization, equilibration, and production runs were all part of the computational process, which made sure that the system reached equilibrium and produced reliable trajectory data for in-depth analysis. These analyses were done for 8-o-4-dehydrodiferulic acid (Compound 1), Emerixanthone B (Compound 2), and Trichaspside B (Compound 3). The root mean squared deviation (RMSD), radius of gyration (Rg), and solvent accessible surface area (SASA) plots were used as a tool to evaluate the spatial integrity of the ligand-protein complexes (Fig. 5).
Root mean squared deviation
The overall structural stability of the protein-ligand complexes is tracked by the RMSD plots (Fig. 5A). All three complexes exhibited stable RMSD values of approximately 2–3 Å after an initial equilibration phase (~ 20 ns), which indicates that the ligands remained effectively bound in the active site. The protein did not undergo significant conformational rearrangements. Slightly larger fluctuations were seen between 80 and 160 ns time points, showing temporary structural rearrangements in the binding pocket to improve the interactions between the protein and the ligand.
Radius of gyration (Rg)
The consistent Rg values during the simulation (~ 16.6 Å) across all three complexes highlight the protein’s retention of structural integrity during the ligand binding (Fig. 5B). Slight reductions in Rg values during simulation suggest localized conformational tightening, possibly enhancing ligand binding stability.
Solvent accessible surface area
SASA measures the protein’s solvent-exposed surface area and can reveal structural adjustments during ligand binding. A decrease in SASA values in the ligand-bound states compared to the apo state suggests that ligand binding induced a more compact protein conformation, reducing solvent exposure (Fig. 5C). This behavior is typically associated with stable protein-ligand complexes.
Dynamic behavior of ligands in protein systems
The dynamic behavior of ligands within the protein binding pocket was further investigated using root mean square fluctuation (RMSF) plots and structural visualization.
Root mean square fluctuation
The RMSF plot of the Cα carbon atoms of the protein in the apo state as well as in the bound state with the three ligands provides a residue-level analysis of flexibility across the protein (Fig. 5D). The loop regions (loop1 and loop2) close to the binding pocket showed notable oscillations. The dynamic behavior exhibited by these regions was crucial for ligand accommodation and stabilization. Lower RMSF values were shown by the core residues encircling the ligands, suggesting stability and rigidity in these crucial interaction zones (Fig. 5E).
Structural visualization
Structural visualization of the ligand binding site in the protein depicted valuable insights (Fig. 5E). Over the course of the simulation, it was discovered that the ligands were always situated between loops 1 and 2. This illustration shows how the flexibility of the loops promotes steady binding interactions, guaranteeing that the ligands stay firmly docked.

(A) Root Mean Square Deviation (RMSD) plot of the 4ELY protein bound to three compounds, indicating the structural stability of each protein-ligand complex over the simulation period. (B) Radius of Gyration (Rg) plot of 4ELY with the three compounds, representing the compactness of the protein structure throughout the simulation. (C) Solvent Accessible Surface Area (SASA) plot of 4ELY with the three compounds, depicting changes in the protein’s solvent-exposed surface area during the simulation. (D) Root Mean Square Fluctuation (RMSF) plot illustrating residue-level flexibility, with significant fluctuations observed in loop1 and loop2 regions. (E) Visualization of the ligand binding site situated between loop 1 and loop 2, highlighting the stable interactions maintained throughout the 200 ns simulation.
Correlation plots of Zn–O2 ligand distances and ligand RMSD
Figure 6 highlights the relationship between the stability of the ligand within the protein’s binding site and the coordination environment of the Zn ion, a central feature of the protein-ligand complex. The plots depict the distances between Zn and the ligand’s O2 atom (a key coordinating atom) and the RMSD of the ligand over the simulation period for each complex (4ELY-Compound 1, 2, and 3). This dual analysis provides an understanding of how well the ligand maintains its binding orientation and how it gets flipped to maintain the protein-ligand interaction. Figure 6A shows compound 1 RMSD and the distance of its O2 atoms from the Zn ion of the protein throughout the simulation. We notice that after 40 ns (the first dotted line from the left in Fig. 6A), the ligand gets flipped from its initial orientation, and because of that, its distance from the Zn atom increases. This flipping of the ligand was seen visually during the simulation as well, and it is corroborated by the corresponding increased value in the RMSD plot. Further, this flipping of the ligand is not permanent, and the ligand again gets flipped back to its original orientation around 100 ns (the second dotted line from the left in Fig. 6A), which is reflected in the distance and RMSD plots where the distance and RMSD values attain their initial values. This to-and-fro flipping of the ligand describes the protein-ligand binding in two prominent modes. Similar flipping behavior was observed for compounds 2 and 3 also, around 60 and 20 ns during the simulation, but the flipping of compounds 2 and 3 was found to be different than that of compound 1 in the sense that compound 1 regains its original conformation around 100 ns, but compounds 2 and 3 do not (Fig. 6B and C). Further, compound 3 again changes its orientation around 140 ns (shown with the second dotted line from the left in Fig. 6C), which further increases its RMSD but does not result in any distance change from the Zn ion. The correlation plots effectively demonstrate that Zn–O2 distance stability and ligand RMSD are reliable indicators of a ligand’s binding strength and stability within the protein’s active site. This analysis describes that Compound 1 is mostly present in only one conformation, whereas Compounds 2 and 3 change their orientations, resulting in more than one prominent conformation.

Plots showing correlation of Zn–O2 ligand distances and ligand RMSD for (A) 4ELY–compound 1, (B) 4ELY–compound 2, and (C) 4ELY–compound 3. All distances are reported in Å, highlighting the relationship between ligand stability and the coordination environment of the Zn ion. The dotted straight line shows the orientation changes of ligands.
Mapping ligand clusters within protein structures
Clustering analysis was carried out to categorize the conformations of the protein-ligand complexes based on structural similarity. As discussed in the last subsection, compound 1 is mostly present in only one conformation, but compounds 2 and 3 explore more conformational spaces during the simulations. We analyzed the clusters for compound 3 (which changed its orientation multiple times) in detail, which has been shown in Fig. 7A–E. Most Populated, Cluster 1 (Fig. 7A) represents the dominant conformation observed during the simulation. Cluster 2 (Fig. 7B) and Cluster 3 (Fig. 7C) depict alternative conformations that are less frequently sampled but provide the ligand’s flexibility insights within the binding site. Least Populated, cluster 4 (Fig. 7D), represents rarely occurring conformational states that reflect transient binding poses. The superimposition of all the clusters (Fig. 7E) illustrates the ligand’s ability to flip in multiple conformations within the binding pocket. This ability of the ligand is important for the ligand’s function and to act as a potential drug candidate. Consistent with the last subsection discussion, the RMSD distribution plot shown in Fig. 7F depicts that compound 1 has only one prominent peak in the distribution (with another small peak corresponding to ligand flipping for some time), while compound 2 has one prominent peak along with a small narrow and a small dispersed peak (showing the flipping of the ligand). On the other hand, the RMSD distribution plot of compound 3 has been depicted with multiple peaks, coming from its multiple flipping.

Clustering of trajectories for 4ELY–compound 3 into four distinct clusters based on structural similarity. (A) Most populated cluster (Cluster 1), representing the dominant conformation during the simulation. (B) Cluster 2, highlighting the second most populated conformation. (C) Cluster 3, showing an alternative conformational state. (D) Cluster 4, representing the least populated conformation. (E) Superimposition of all identified clusters to illustrate their structural diversity. (F) RMSD of the ligand across the simulation, indicating its conformational stability within the protein binding site.
Determining binding free energies in molecular complexes
The binding free energies for the three protein-ligand complexes were calculated using the Molecular Mechanics/Generalized Born Surface Area (MMGBSA) method, and the results are summarized in Table 3. The overall binding free energy of Complex 1, Complex 2, and Complex 3 was calculated as −16.6 ± 2.0, −16.9 ± 3.0, and − 12.2 ± 3.5 kcal/mol, respectively. Binding free energy component analysis (Table 3) showed favorable binding affinities for all three compounds, with electrostatic and van der Waals contributions predominating the binding interactions. When the complexes were compared based on overall binding free energy values, Complex 1 and 2 displayed the best binding affinities (considering the corresponding standard deviation values) towards 4ELY. This finding was also supported by the hydrogen bond analysis, which has been discussed in the later subsection. Despite being stable, Complex 3’s higher fluctuations and less compact binding poses resulted in somewhat weaker binding.
Gibbs free energy landscape
Gibbs free energy landscapes for the protein 4ELY in its apo state (Fig. 8A) and when bound to the three ligands—Compound 1 (Fig. 8B), Compound 2 (Fig. 8C), and Compound 3 (Fig. 8D —have been discussed. The free energy landscapes, generated through MD simulation trajectories, depict the stability and conformational preferences of the protein-ligand complexes. Blue regions represent low-energy, more stable conformational states, while red regions indicate high-energy, less stable states. The energy landscape of the apo state (Fig. 8A) exhibits many lower energy basins, indicating the protein’s intrinsic flexibility. The broadly spread blue regions reflect the protein’s ability to sample a wider range of conformational states without structural constraints imposed by any ligand binding. The Gibbs free energy landscape for Compound 1 (Fig. 8B) is dominated primarily by a single, deep blue basin, which indicates a stable conformational state. The narrow spread of lower energy regions suggests that the ligand restricts the protein’s flexibility significantly, stabilizing it in a specific conformation. This tight clustering correlates with the stable Zn–O2 coordination and low ligand RMSD observed in Fig. 6A, reinforcing Compound 1’s strong binding affinity. The energy landscape for Compound 2 (Fig. 8C) also shows a predominant low-energy basin, though it is slightly broader than that of Compound 1. This indicates a stable binding state, with minor flexibility in the protein-ligand complex, allowing the system to explore neighboring conformations. The energy distribution aligns with the similar stability in the Zn–O2 distance and ligand RMSD for Compound 2, highlighting its strong, flexible binding. The Gibbs free energy landscape for Compound 3 (Fig. 8D) differs, where multiple lower energy basins of almost similar depth are scattered across the plot. This sparse distribution depicts the ligand’s reduced binding ability and the protein’s tendency to sample diverse conformational states in the presence of Compound 3. The higher-energy red regions suggest that Compound 3 has less ability to fully stabilize the protein, consistent with its weaker coordination and higher RMSD. A concentrated blue region, as seen in Compounds 1 and 2, reflects a ligand’s ability to stabilize the protein in a specific functional conformation. In contrast, the sparse lower energy regions corresponding to Compound 3 indicate its lower binding ability towards the protein 4ELY, reducing its potential as a strong binder.

Gibbs free energy 2D plots for the three ligand-bound complexes of 4ELY. (A) Alone 4EYL protein (B) Complex 1, (C) Complex 2, and (D) Complex 3. The plots depict the conformational free energy landscapes, with blue regions indicating more stable conformations with highly negative binding energies and red regions representing less stable, higher-energy states.
Principal component analysis
Principal Component Analysis (PCA) is a dimensionality reduction technique used to study the dominant motions in MD simulations. 2D projections of the simulation trajectories of the 4ELY protein in the apo state (Fig. 9A) and when bound to three ligands (Compounds 1, 2, and 3) (Figs. 9B-D) onto the first two principal components (PC1 and PC2) are observed. These principal components represent the largest variations in the complex’s conformational space, which provides insights into the structural flexibility. The broader PCA of the apo state of the protein 4ELY (Fig. 9A) depicts the protein’s intrinsic flexibility in the absence of any ligand. The PCA of compound 1 (Fig. 9B) bound complex shows that the corresponding trajectory is tightly clustered, forming a single prominent region of the conformational space. This reflects the higher stability of the protein-ligand-bound complex with small structural variations. The limited exploration of conformational space suggests that Compound 1 significantly constrains the protein, locking it into a particular low-energy conformation. The PCA plot corresponding to the Compound 2 (Fig. 9C) bound complex also shows a similar narrow distribution as Compound 1, with a primary cluster. The PCA plot for the Compound 3 (Fig. 9D) bound complex exhibits a more dispersed pattern, with multiple clusters scattered across the conformational space. This distribution suggests significant structural variability, indicating weaker ligand binding and less stability in the protein-ligand complex. The scattered clustering aligns with the higher RMSD and fragmented Gibbs free energy landscape observed for Compound 3. These findings align with other analyses, reinforcing the interpretation of binding stability and ligand efficacy.
Ligand-protein interaction analysis: H bonding dynamics
Hydrogen bonding is essential to stabilize the protein-ligand interactions. The hydrogen bond (H-bond) analysis was performed, showing the number of hydrogen bonds formed between the protein 4ELY and the three ligands (compounds 1, 2, and 3) throughout the simulation (Fig. 9E). The plot shows that compounds 2 and 3 formed four to six H-bonds with the protein in the complexes, while compound 1 formed no H-bond. Despite no H-bond, higher binding affinity of compound 1 results from its strong VDW and electrostatic interactions with the protein.

Principal Component Analysis. The 2D projections of trajectories on eigenvectors showed different projections of apo 4EYP (A) and 4EYP-complexes with compounds 1–3 (C-D). The number of hydrogen bonds between protein systems and ligands (E).
Pharmacokinetic analysis
The results of pharmacokinetic analysis exhibited that the top 7 selected metabolites possessed high GI absorption, which implies that these metabolites possess desirable characteristics for oral medications, the most preferred method of drug administration. Additionally, the top metabolites cannot pass through the BBB, which indicates that they perform their desired action without causing any side effects to the central nervous system (CNS)38. Except for 8-O-4-dehydrodiferulic acid and Emerixanthone B, all other metabolites showed substrate prediction for pg-p (Table S2), which implies that these metabolites have the probability to be easily effluxed from the brain, which further indicates fewer chances for brain toxicity by our metabolites54.
Our top metabolites are intended for use in combination with β-lactam antibiotics. To minimize the risk of metabolic interactions or interference, we evaluated their effects on cytochrome P450 (CYP) enzymes and compared the results with those of five commonly used β-lactam antibiotics (Fig. 10). We used ADMET LAB 3.0 and SwissADME to check both substrate/non-substrate and inhibitor/non-inhibitor characteristics. During this comparison, it is essential that when β-lactam antibiotics act as substrates for CYP enzymes, the top-selected metabolites do not function as CYP inhibitors. This implies that the beta-lactamase inhibitor will effectively block the beta-lactamase enzyme without interacting with the metabolism or functionality of beta-lactam antibiotics. In contrast, if the beta-lactam antibiotics operate as CYP inhibitors, our metabolites must not be CYP substrates. This prevents any metabolic interactions that can inhibit antibiotic efficacy. We took five beta-lactam antibiotics from the penicillin group: benzathine, benzylpenicillin, phenoxymethylpenicillin, procaine benzylpenicillin, and phenethicillin55. Among the top-selected metabolites—8-O-4-dehydrodiferulic acid, Emerixanthone B, and Trichaspside B—none serve as a substrate or inhibitor of CYP. Consequently, all beta-lactam antibiotics can be used in combination with these metabolites. Emerixanthone A, Tajixanthone hydrate, and Trichaspside A are CYP2B6 inhibitors, so any beta-lactam antibiotic that is being used in their combination must not be a CYP2B6 substrate. All the antibiotics discussed in our study are non-substrates for CYP2B6. Aspergixanthone–K is a substrate for CYP3A4; therefore, the antibiotics being administered in combination with this metabolite must not be CYP3A4 inhibitors. Fortunately, all the antibiotics included in our study were predicted to be non-inhibitors of CYP3A4, indicating that they are suitable for combination use. Consequently, our selected metabolites can be co-administered with the aforementioned β-lactam antibiotics without risk of CYP3A4-mediated interactions.

(A) Graphical representation of CYPs substrate, inhibitor profile of some beta-lactam antibiotics of the penicillin group. (B) Graphical representation of CYPs substrate, inhibitor profile of top selected metabolites.
Toxicity analysis
A ProTox II server estimated the toxicity of the top seven hits39. According to the prediction, all of the hits are non-cytotoxic. Analysis of hepatotoxicity showed that 8-O-4-dehydrodiferulic acid has a 53% probability of not being a hepatotoxic agent, while all other metabolites show more than 64% probability. LD50 values showed that all the metabolites lie in Class IV and the non-toxic range (Table 4). To summarize, our top hits are non-toxic, which guarantees patient safety. Additionally, our hits have a higher chance of moving through preclinical research and clinical trials successfully, which lowers development costs and failure rates.
Medicinal chemistry
In the context of medicinal chemistry, the hits indicated no PAINS alerts, which implies the absence of potentially problematic fragments and accurate experimental results. Moreover, our hits show 1 or 2 Brenk alterations (except Emerixanthone A, which shows 3 Brenk alerts), which can be problematic; therefore, there is a need for structural optimization to eliminate this issue while preserving their therapeutic properties. The overall synthetic accessibility of top-selected metabolites was in the range of 3.17 to 5.89. In the final assessment of lead likeness, the top selected hits exhibited 2 violations comprising MW and lipophilicity (Table 5). To summarize, although our hits are non-toxic, they may need structural optimization to ensure safety.
Target fishing analysis
Off-target drug effects can trigger adverse reactions; therefore, it is essential to understand the complex network of interaction between bioactive compounds and various biological targets56. Target fishing analysis is a powerful tool to identify possible off-target drug effects. The top leads were analyzed by SWISS target prediction to determine if they might inhibit human proteins other than our target protein. Fortunately, there was no target predicted for Trichaspside B, which shows its high degree of selectivity towards NDM-1. Moreover, except for Emerixanthone A and B, all identified hits exhibited a very low probability of interacting with human targets. This reduces the likelihood of drug–drug interactions when used in combination therapies and significantly lowers the risk of adverse effects on human cells. Whereas, Emerixanthone A and B show a high probability (52% and 63% respectively) of interacting with human targets, including Calmodulin which is involved in regulating processes related to skeleton muscle movement and energy production57. It implies that there might be interference by Emerixanthone A and B in such processes. This is important in terms of the safety and specificity of compounds.
8-O-4-dehydrodiferulic acid (Figure S5), Tajixanthone hydrate, Trichaspside A, and Aspergixanthone–K anticipated extremely low probabilities (10-11%) to interact with the receptors and enzymes (Table S3). The majority of these enzymes and receptors regulate a variety of cellular functions, including cell division and proliferation, while some other notable targets include the proteasome macropain subunit and 26 S proteasome, which are involved in protein degradation. Sodium/glucose co-transporters 1 and 2 regulate blood glucose levels. Interleukin-1 receptor-associated kinase 4 and MAP kinase p38 beta have been related to immunological responses. Angiotensin-converting enzyme and Neprilysin regulate blood pressure, fluid balance, and cardiovascular function. The potential for a positive safety profile resulting from such specificity renders Trichaspside B a viable option for additional development. These findings indicated that our hits are adaptable and useful in the treatment of infectious disorders since they may be used for a wider range of patients.
Prediction of activity spectra
We applied the PASS prediction analysis with strict selection criteria to ensure that only characteristics with a probability greater than 0.7% were evaluated. Alongside their main antibacterial action, we concentrated our efforts on assessing a range of other biological activities. Predicted biological activities for 8-O-4-dehydrodiferulic acid include membrane integrity agonist, antimutagenic, TNF expression inhibitor, membrane permeability inhibitor, and free radical scavenger, which show its activity as promoting the stability and health of the cell membrane, preventing or reducing the occurrence of mutations in genetic material, reducing the production of tumor necrosis factor, and helping to neutralize and remove harmful free radicals. The ability of Emerixanthone B and Aspergixanthone K to inhibit or destroy the growth of cancer cells, as well as to prevent substances from crossing the cell membrane, is demonstrated by their antineoplastic and membrane permeability inhibitory qualities. Similarly, Trichaspside B, functions as a substrate for the CYP2H enzyme in addition to exhibiting immunostimulant and immunosuppressant characteristics. Emerixanthone A is another top metabolite that has the power to stop or eradicate the growth of cancer cells. For UDP-glucuronosyltransferase, an enzyme involved in several metabolic processes, Tajixanthone hydrate serves as a substrate. Additionally, it may also prevent membrane permeability. Trichaspside A is a unique metabolite that exhibits a variety of effects. It shows properties as an immunostimulant, as it can strengthen your immune system; antifungal and antibacterial; also acts as a respiratory analeptic, as it helps with respiratory issues; an immunosuppressant; and analeptic, which refers to its activity as stimulating or restoring normal body functions (Table S4). In addition, previous studies have reported more evidence and findings regarding their inhibitory effects, like the metabolite 8-O-4-dehydrodiferulic acid, which was derived from the fungus Aspergillus sp., which was isolated from the sponge Tethyaaurantium in the Adriatic Sea. It exhibited significant activity against the marine fouling bacterium Vibrio proteolyticus, with a minimum inhibitory concentration (MIC) value of 0.100 µg/mL58. Trichaspside A and B were obtained from the marine-alga endophytic fungus Trichoderma asperellum cf44-2. They exhibited promising activities against various Vibrio species, including V. anguillarum, V. harveyi, V. parahaemolyticus, and V. splendidus, with inhibition zones ranging from 6.10 to 6.40 mm. This activity is attributed to the presence of 2-acetamido-2-deoxy-α-D-glucopyranosyl59. Emerixanthone A and B, which were obtained from the deep-sea fungus Emericella sp. SCSIO 05240 in the South China Sea (at a depth of 3258 m), have been identified. These molecules demonstrate inhibitory effects against Escherichia coli (ATCC 29922), Klebsiella pneumoniae (ATCC 13883), S. aureus (ATCC 29213), E. faecalis (ATCC 29212), Acinetobacter baumannii (ATCC 19606), and Aeromonas hydrophila (ATCC 7966)60. Based on the study, it is suggested that these can strongly bind to the active site of NDM-1. This indicates that they could serve as potential inhibitors. With MIC values ranging from 1.56 to 25.0 µM, Aspergixanthone-K and Tajixanthone hydrate demonstrated antagonistic activity against various Vibrio species, such as V. alginolyticus, V. anguillarum, and V. parahaemolyticus. Aspergixanthone–K and Tajixanthone hydrate were obtained from marine sediment fungus Aspergillus sp. ZA-0161.
Conclusion
The emergence of drug resistance in bacteria is primarily driven by the activity of the metallo-beta-lactamase enzyme (NDM-1), and our research highlights the potential of fungal-derived secondary metabolites as inhibitors to effectively target and block this enzyme. Using computational techniques, seven therapeutic metabolites, namely, 8-O-4-dehydrodiferulic acid, Emerixanthone B, Trichaspside B, Emerixanthone A, Tajixanthone hydrate, Trichaspside A, and Aspergixanthone–K, were identified from marine-derived fungi that can combat the antibiotic resistance by NDM-1. These secondary metabolites may effectively inhibit NDM-1 due to their extensive interactions and strong binding patterns with key amino acid residues of the 4EYL protein, including hydrogen bonds with GLU-152, GLN-123, and ASN-220, as well as disruption of the metal coordination bond with Zn302. The pharmacophoric features highlighted the important interacting parts of the top metabolites. The comprehensive MD simulation analyses, including RMSD, RMSF, Rg, SASA, hydrogen bond analysis, binding free energy by MMGBSA, PCA, and FEL, revealed that all top three metabolites displayed stable behavior, with one metabolite as the most favorable candidate. Drug-likeness, medicinal, pharmacokinetic, and toxicity analyses guaranteed a non-toxic and safe profile of the metabolites. Almost all of the metabolites demonstrated a very low probability of interacting with human targets, reducing the risk of drug–drug interactions when used in combination therapies and significantly minimizing the potential for off-target effects on human cells. These findings support the need for experimental validation to assess the efficacy and suitability of these metabolites in the development of effective antibiotic agents.
Data availability
All data generated or analysed during this study are included in this article and its supplementary information files.
References
Jagyasi, A. Molecular modeling and Docking analysis of novel drug like compounds for NDM-1. National Seminar on Application of Artificial Intelligence in Life Sciences 47–54 (2013).
Rolain, J. M., Parola, P. & Cornaglia, G. New Delhi metallo-beta-lactamase (NDM-1): towards a new pandemia ? Clin. Microbiol. Infect. 16, 1699–1701 (2010).
Nordmann, P., Poirel, L., Mark, A., Timothy, R. & T. & Does broad-spectrum β-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by Gram-negative bacteria? J. Antimicrob. Chemother. 66, 689–692 (2011).
Mag, P. et al. and B are Potential Inhibitors of New Delhi Metallo β Lactamase 1 (NDM 1): A Computational Approach. 1, 1–10 (2022).
de Viana Marques, D. Production of β-lactamase inhibitors by Streptomyces species. Antibiotics 7, 1–26 (2018).
Feng, H. et al. Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J. Am. Chem. Soc. 136, 14694–14697 (2014).
Tn, C., Pereira, M. D. O., Silva, M., Magagnin, M. & Leiroz K India 59, 7387–7395 (2015).
An, J. et al. NDM-producing Enterobacteriaceae in a Chinese hospital, 2014–2015: identification of NDM-producing Citrobacter werkmanii and acquisition of blaNDM-1-carrying plasmid in vivo in a clinical Escherichia coli isolate. J. Med. Microbiol. 65, 1253–1259 (2016).
King, D. T., Worrall, L. J., Gruninger, R. & Strynadka, N. C. J. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and Inhibition. J. Am. Chem. Soc. 134, 11362–11365 (2012).
Klingler, F. M. et al. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: strategy to combat multidrug-resistant bacteria. J. Med. Chem. 58, 3626–3630 (2015).
King, A. M. et al. Aspergillomarasmine A overcomes metallo-Î 2-lactamase antibiotic resistance. Nature 510, 503–506 (2014).
Docquier, J. D. et al. High-resolution crystal structure of the subclass B3 metallo-β- lactamase BJP-1: rational basis for substrate specificity and interaction with sulfonamides. Antimicrob. Agents Chemother. 54, 4343–4351 (2010).
Whinfield, B. Production of penicillin by germinating conidia of penicillium notatum [23]. Nature 157, 773 (1946).
Sychantha, D., Rotondo, C. M., Tehrani, K. H. M. E., Martin, N. I. & Wright, G. D. Aspergillomarasmine A inhibits metallo-β-lactamases by selectively sequestering Zn2+. J. Biol. Chem. 297, 100918 (2021).
Shaker, B., Ahmad, S., Lee, J., Jung, C. & Na, D. In Silico methods and tools for drug discovery. Comput. Biol. Med. 137, 104851 (2021).
Devi, R. et al. Fungal Secondary Metabolites and their Biotechnological Applications for Human Health. New and Future Developments in Microbial Biotechnology and Bioengineering: Trends of Microbial Biotechnology for Sustainable Agriculture and Biomedicine Systems: Perspectives for Human Health (Elsevier Inc., 2020). https://doi.org/10.1016/B978-0-12-820528-0.00010-7
Kar, B., Kundu, C. N. & Pati, S. Discovery of phyto-compounds as novel inhibitors against NDM-1 and VIM-1 protein through virtual screening and molecular modelling. J. Biomol. Struct. Dyn. 0, 1–14 (2023).
Shah, M. et al. Computational analysis of Plant-Derived terpenes as α -glucosidase inhibitors for the discovery of therapeutic agents against type 2 diabetes mellitus. South. Afr. J. Bot. 143, 462–473 (2021).
Shah, M. et al. Computer-aided identi Fi cation of Mycobacterium tuberculosis resuscitation-promoting factor B (RpfB) inhibitors from Gymnema sylvestre natural products. 1–18 (2023). https://doi.org/10.3389/fphar.2023.1325227
King, D. & Strynadka, N. Crystal structure of new Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 20, 1484–1491 (2011).
Khan, A. U. & Rehman, M. T. Role of Non-Active-Site residue Trp-93 in the function and stability of new Delhi Metallo-β-Lactamase 1. Antimicrob Agents Chemother 60(1), 356–360 (2016).
Ali, A., Gupta, D. & Khan, A. U. Role of non-active site residues in maintaining new Delhi metallo-β-lactamase-1(NDM-1) function: an approach of site-directed mutagenesis and Docking. FEMS Microbiol. Lett. 368, 1–7 (2021).
Shah, M. et al. In-silico evaluation of natural alkaloids against the main protease and Spike glycoprotein as potential therapeutic agents for SARS-CoV-2. PLoS One. 19, e0294769 (2024).
Khalid, H. et al. Phytobioinformatics screening of ayurvedic plants for potential α-Glucosidase inhibitors in diabetes management. Curr. Plant. Biol. 40, 100404 (2024).
Ahmad, I. et al. Identification of novel quinolone and Quinazoline alkaloids as phosphodiesterase 10A inhibitors for parkinson’s disease through a computational approach. ACS Omega. 9, 16262–16278 (2024).
Yamin, R. et al. Identifying plant-derived antiviral alkaloids as dual inhibitors of SARS-CoV-2 main protease and Spike glycoprotein through computational screening. Front Pharmacol 15, 1369659 (2024).
Khan, F. et al. Exploring the genomic potential of Kytococcus schroeteri for antibacterial metabolites against multi-drug resistant Mycobacterium tuberculosis. J. Infect. Public. Health. 18, 102598 (2025).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Singh, R., Mishra, V. K. & Das, A. K. Crystal structure of a thiolase from archaeal Pyrococcus furiosus and its in Silico functional annotation. Biochem. Biophys. Res. Commun. 693, 149377 (2024).
Mishra, V. K. & Mishra, S. Flipped regiospecificity in L434F mutant of 8-lipoxygenase. Phys. Chem. Chem. Phys. 22, 16013–16022 (2020).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Roe, D. R. & Cheatham, T. E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Turner, P., XMGRACE & Version 5.1. 19. Center for coastal and land-margin research, Oregon Graduate Institute of Science and Technology, Beaverton, OR 2, 19 (2005).
Massova, I. & Kollman, P. A. Combined molecular mechanical and continuum solvent approach (MM- PBSA/GBSA) to predict ligand binding. Perspect. Drug Discovery Des. 18, 113–135 (2000).
Ja’afaru, S. C. et al. Computational modeling and molecular dynamics studies of Methyl sulfonyl acetate derivatives as potent SmTGR inhibitors: insights into binding interactions. Mol. Simul. 50, 1274–1291 (2024).
Ja’afaru, S. C. et al. Virtual screening and molecular dynamics studies of novel small molecules targeting Schistosoma mansoni DHODH: identification of potential inhibitors. Silico Pharmacol. 12, 113 (2024).
Daina, A., Michielin, O., Zoete, V. & SwissADME A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 1–13 (2017).
Honorio, M., Moda, K. L., Andricopulo, D. & T. & Pharmacokinetic properties and in Silico ADME modeling in drug discovery. Med. Chem. (Los Angeles). 9, 163–176 (2013).
Banerjee, P., Eckert, A. O., Schrey, A. K. & Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263 (2018).
Sanders, M. P. A. et al. Comparative analysis of pharmacophore screening tools. J. Chem. Inf. Model. 52, 1607–1620 (2012).
Zhang, H. M. & Hao, Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 25, 2574–2582 (2011).
Krajnc, A. et al. Bicyclic boronate VNRX-5133 inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 62, 8544–8556 (2019).
Skagseth, S. et al. Metallo-β-lactamase inhibitors by bioisosteric replacement: preparation, activity and binding. Eur. J. Med. Chem. 135, 159–173 (2017).
Azumah, R. et al. In vitro evaluation of metal chelators as potential metallo- β -lactamase inhibitors. J. Appl. Microbiol. 120, 860–867 (2016).
Ma, J. et al. Real-time monitoring of new Delhi metallo-β-lactamase activity in living bacterial cells by 1HNMR spectroscopy. Angewandte Chemie - Int. Ed. 53, 2130–2133 (2014).
Chen, A. Y. et al. Dipicolinic acid derivatives as inhibitors of new Delhi Metallo-β-lactamase-1. J. Med. Chem. 60, 7267–7283 (2017).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv Rev. 64, 4–17 (2012).
Ghose, A., … V. V.-J. of combinatorial & 1999, undefined. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1.A qualitative and quantitative characterization of. ACS Publications 1, 55–68 (1999).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 (2002).
Egan, W. J., Merz, K. M. & Baldwin, J. J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 43, 3867–3877 (2000).
Muegge, I., Heald, S. L. & Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 44, 1841–1846 (2001).
Barber, R. D. Software to Visualize Proteins and Perform Structural Alignments. Current Protocols vol. 1 Preprint at (2021). https://doi.org/10.1002/cpz1.292
Wade, N. et al. Mechanistic investigations of Metallo-β-lactamase inhibitors: strong zinc binding is not required for potent enzyme Inhibition**. ChemMedChem 16, 1651–1659 (2021).
Raub, T. J. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharm. 3, 3–25 (2006).
Antibiotic and Chemotheraphy. Postgraduate Medical Journal vol. 40 (1964).
Scheiber, J. et al. Gaining insight into off-target mediated effects of drug candidates with a comprehensive systems chemical biology analysis. J. Chem. Inf. Model. 49, 308–317 (2009).
Walsh, M. P. Review Article calmodulin and its roles in skeletal muscle function. Can. Anaesth. Soc. J. 30, 390–398 (1983).
Zhou, Y. et al. Marine bacterial inhibitors from the sponge-derived fungus. Tetrahedron Lett. 55, 2789–2792 (2014).
Song, Y. et al. Phytochemistry Bisabolane, cyclonerane, and harziane derivatives from the marine- alga-endophytic fungus Trichoderma asperellum cf44-2. Phytochemistry 152, 45–52 (2018).
Fredimoses, M. et al. New prenylxanthones from the Deep-Sea derived fungus emericella Sp. SCSIO 05240, 3190–3202. https://doi.org/10.3390/md12063190 (2014).
Zhu, A. et al. Aspergixanthones I–K, new anti-vibrio prenylxanthones from the marine-derived fungus Aspergillus sp. ZA-01. Mar. Drugs. 16, 1–8 (2018).
Acknowledgements
This study was funded by the Doctoral Research Fund awarded to Ke Chen. The authors also express their appreciation to the Deanship of Research and Graduate Studies at King Khalid University, Saudi Arabia, for supporting this study through large research project under grant number 675/46. The authors are also thankful to the Bahauddin Zakariya University, Multan, Pakistan, for providing the necessary infrastructure to perform this research.
Author information
Authors and Affiliations
Contributions
Farhat Shabbir: Formal analysis, Investigation, Writing – original draft. Vipin Kumar Mishra: Formal analysis, Investigation, Software, Writing – original draft. Sana Noor: Formal analysis, Investigation. Subrata Nath: Formal analysis, Investigation. Muhammad Umer Khan: Investigation, Software, Writing – original draft. Umar Nishan: Visualization, Validation, Writing–review & editing. Hanna Dib: Software, validation, Writing – original draft. Khaled Fahmi Fawy: Funding acquisition, validation, Writing – original draft. Mohibullah Shah: Conceptualization, Supervision, Resources, Writing–review & editing. Ke Chen: Conceptualization, validation, Funding acquisition, Writing – original draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shabbir, F., Mishra, V.K., Noor, S. et al. Computer-aided screening of marine fungal metabolites as potential inhibitors of new Delhi metallo-beta-lactamase-1. Sci Rep 15, 33997 (2025). https://doi.org/10.1038/s41598-025-12523-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-12523-4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}