
Abstract
The potential of octreotide (OCT), an octapeptide synthetic somatostatin agonist, to mitigate addiction, specifically morphine dependence, was evaluated in a comprehensive approach involving behavioral, molecular, histological, and docking studies. OCT decreased addiction-related behaviors in Wistar rats previously exposed to morphine, normalizing their exploratory activities and reducing drug-seeking behaviors. Histological assessments revealed octreotide-induced reductions in opioid-induced neuronal damage, suggesting a neuroprotective function. Octreotide’s behavioral and histopathological effects were associated with regulation of molecular pathways known to be critical in morphine dependence processes, including TLR4, BDNF, SIRT1, and mTOR. Supplementary docking studies elucidated octreotide’s high affinity for addiction-related targets, suggesting that a biological interaction between OCT and players in pathways known to play a role in addiction could be involved in behavioral and cellular effects noted. When our results are taken together, we conclude that OCT could be a promising candidate for opioid dependence treatment and underscore the necessity for further preclinical and clinical investigations.
Similar content being viewed by others


Introduction
Addiction is a chronic brain disorder marked by compulsive drug use, relapse, and changes in brain circuits tied to reward, stress, and self-control1,2. Prolonged opioid exposure induces physical dependence, tolerance, and withdrawal, fueling addiction through neuroadaptations in the mesolimbic dopamine system, prefrontal cortex (PFC), and hippocampus3. The PFC regulates executive function, while the hippocampus supports memory and drug-cue associations, making them critical targets for studying dependence 4,5. Moreover, neuroinflammation is shown to play a role in addiction6, and the modulation of inflammatory and immune pathways has gained considerable attention in the pathophysiology and treatment of drug abuse7,8. Neuroinflammation, driven by toll-like receptor 4 (TLR4) activation, exacerbates neuronal damage9. Synaptic plasticity deficits, mediated by reduced brain-derived neurotrophic factor (BDNF) and mammalian target of rapamycin (mTOR) signaling, alongside oxidative stress countered by sirtuin 1 (SIRT1), further underpin dependence8,9,10,11. Therefore, while behavioral strategies certainly play a crucial role in abstinence from drug use, the complex alterations in the brain underscore the likely necessity of use of pharmacological interventions to target functional changes in neural circuits and to reduce neuroinflammation for effective treatment of drug abuse. While pharmacological treatments for drug addiction are available, their success is limited and therefore there remains a high need to explore novel therapeutic approaches to management of drug addiction.
Octreotide (OCT), which is a synthetic analog of somatostatin, an inhibitory hormone with diverse biological effects, is characterized by an extended duration of action and has exhibited a wide range of therapeutic actions12,13. OCT is used clinically to manage acromegaly14, gastrointestinal disorders15,16, and neuroendocrine tumors17. Further, the use of OCT in targeted drug delivery strategies for diagnosis or treatment of tumors has been suggested18. While the mechanisms of action of OCT are incompletely understood, OCT targets several molecular pathways involved in neural plasticity and inflammation including BDNF19, SIRT120, TLR421, and mTOR22. OCT acts via somatostatin receptors (SSTR2, SSTR3, and SSTR5), inhibiting adenylate cyclase and modulating ion channels23. The molecular pathways known to be regulated by OCT have been implicated in the pathophysiology of addiction23,24,25,26,27,28,29, which raises the possibility that OCT could exhibit therapeutic effects in addiction. However, to the best of our knowledge, while a role for somatostatin in drug addiction has been proposed30, there has been no study investigating the therapeutic potential of OCT in the context of addiction to date. In this study, we explored the potential of OCT to manage drug addiction by evaluating effects of OCT on opioid -induced animal behaviors, changes in histological features seen following opioid exposure, and alterations in molecular players associated with opioid- addiction. Further, we utilized in silico docking studies to analyze the interaction and affinity of OCT with potential protein targets involved in the effects of OCT.
Results
Open field test (OFT)
In the OFT, the morphine group exhibited a significantly lower number of rearing events compared to the saline and OCT-treated groups (F (3, 28) = 6.24, p = 0.01, Fig. 1A). Moreover, the morphine group exhibited significantly higher grooming events compared to other groups (F (3, 28) = 2.85, p = 0.048, Fig. 1B). These findings suggest that OCT administration in morphine-treated rats resulted in normalizing the number of rearing and grooming events to a level comparable to control groups. Evaluation of the total distance moved within the open field apparatus demonstrated that the morphine group traveled a significantly lower distance when compared to that seen in the other groups. However, the morphine + OCT group moved a distance that did not differ from that exhibited by both saline and OCT groups (F (3, 28) = 3.14, p = 0.05, Fig. 1C), suggesting that OCT administration in morphine-treated animals enhanced exploratory activity. Furthermore, our findings showed a significantly lower time spent in the inner zone in the morphine group, while the morphine + OCT group did not show any difference with the control groups (F (3, 28) = 3.26, p = 0.041, Fig. 1E). There were no significant differences in immobility duration (F (3, 28) = 0.46, p = 0.7, Fig. 1D) and time spent in the outer zone (F (3, 28) = 1.08, p = 0.37, Fig. 1F) between the groups.

Characterization of exploratory activity in the open-field test. (A) rearing number, (B) number of grooming events, (C) total distance moved, (D) duration of immobility, and (E) the time spent in inner and (F) time spent in outer zones of the test arena. Data are expressed as the mean ± SEM. (* in comparison to the Saline group, # in comparison to the OCT group, × in comparison to the Morphine group.
Elevated plus maze
Findings of the elevated plus maze test did not show any significant differences between the groups in the time spent in the open arm (F (3, 28) = 1.06, p = 0.38, Fig. 2A) and closed arm (F (3, 28) = 0.025, p = 0.99, Fig. 2B).

The plus-maze test included the duration of time that was spent in the closed and open arms for the four groups measured in seconds.
Passive avoidance learning and memory
Findings of the passive avoidance test (PAT) demonstrated that the shock number did not differ significantly between the study groups (F (3, 28) = 0.35, p = 0.78, Fig. 3A). However, the morphine group demonstrated a significantly lower STL compared to other groups, suggesting an impaired passive avoidance memory. Notably, the step through latency (STL) was normalized in the morphine + OCT group, highlighting the beneficial effects of OCT in ameliorating the morphine-induced deficits in passive avoidance memory (F (3, 28) = 1.56, p = 0.22, Fig. 3B).

The passive avoidance latency test (PAT) included the shock number and step-through latency (s) of the four experimental groups. Data are expressed as the mean ± SEM (*P < 0.05) compared to the control group, and (#P < 0.05) in comparison to the OCT group.
OCT reverses morphine preference in rats during the conditioned place preference test
On the first day of the test, all groups spent nearly the same amount of time in the non-preferred chamber. While the duration of time for both the saline and OCT groups remained consistent, the time the morphine group spent in this chamber increased significantly over the course of two weeks (Fig. 4A). However, the morphine + OCT group exhibited a preference for morphine in day 13, which was comparable to saline and OCT groups. Pretest data indicates that neither group exhibited a strong preference for either compartment prior to conditioning. Post-saline and OCT groups, while thetest results demonstrate that the morphine-exposed group developed a significantly higher CPP score compared to the saline and OCT groups, while the conditioned place preference (CPP) of the morphine + OCT group was notably reduced. A two-way repeated-measures ANOVA revealed significant main effects of group (F (3, 56) = 9.9, P = 0.04, Fig. 4B) and conditioning (F (3, 56) = 41.1, P < 0.001), along with a significant interaction effect (F (1, 56) = 5.1, P = 0.02, Fig. 4B). These findings suggest that OCT administration can reverse the morphine preference behavior in morphine-exposed rats.

Impact of OCT administration on morphine preference behavior in rats exposed to morphine. (A) Duration of time that each group spent in the non-preferred chamber (s) on the 7th, 13th, and 20th days. (B) A two-way repeated-measures ANOVA of the Posttest results reveals that the morphine-exposed group exhibited a significantly higher CPP score compared to the saline and OCT groups, whereas the CPP score in the morphine + OCT group was significantly reduced. Data are expressed as the mean ± SEM (*P < 0.05) compared to control group and (#P < 0.05) other groups.
PCR
The qRT-PCR analysis of TLR4, BDNF, SIRT1, and mTOR gene expression, as illustrated in Figs. 5 and 6, revealed significant differences in gene expression patterns across different brain regions and experimental groups.

The expression of TLR4 and BDNF genes in frontal and hippocampus regions in four experimental groups. Data are expressed as the mean ± SEM (*P < 0.05). *The significance compared to the saline group, #The significance compared to the OCT group, XThe significance compared to the morphine group. Expression of mTOR was significantly higher in the morphine + OCT group than that in the morphine group.

The expression of SIRT1 and MTOR genes in PFC and hippocampus regions in the four experimental groups. Data are expressed as the mean ± SEM (*P < 0.05). *The significance compared to the saline group, #The significance compared to the OCT group, XThe significance compared to the morphine group.
TLR4 gene expression
In the PFC, TLR4 was expressed differentially among the groups [H (3) = 10.5, P < 0.001, Fig. 5A]. The OCT and morphine groups exhibited significantly, higher and lower TLR4 expression, respectively, compared to the saline group (Fig. 5A, p < 0.05). Additionally, expression in the morphine and morphine + OCT groups was significantly lower in the PFC compared to the OCT group (p < 0.05). Moreover, TLR4 was expressed significantly higher in the morphine + OCT group compared to the morphine group in the PFC (p < 0.05). In the hippocampal area, TLR4 was also expressed differentially among the groups [H (3) = 8.05, P < 0.05, Fig. 5B]. In the OCT group, TLR4 was expressed significantly higher when compared to expression in the saline group (Fig. 5B, p < 0.05). Additionally, TLR4 in the morphine and morphine + OCT groups were expressed significantly lower in the hippocampus compared to that seen in the OCT group (p < 0.05).
BDNF gene expression
In the PFC, BDNF was expressed differentially among the groups [H (3) = 9.5, P < 0.01, Fig. 5C]. In the morphine and morphine + OCT groups, BDNF were expressed significantly lower in the PFC when compared to that seen in the saline group (Fig. 5C, p < 0.05). However, in the hippocampus, there was no significant difference among groups [H (3) = 8.6, P < 0.01, Fig. 5D].
SIRT1 gene expression
In the PFC, SIRT1 expression varied significantly among the groups [H (3) = 9.4, P < 0.01, Fig. 6A]. SIRT1 levels were significantly higher in the morphine group compared to the saline group (Fig. 6A, p < 0.05). Conversely, SIRT1 was expressed significantly lower in the morphine + OCT group compared to the morphine group (p < 0.05). In the hippocampus, SIRT1 was also expressed differentially among various groups [H (3) = 9.6, P < 0.01, Fig. 6B]. SIRT1 expressed significantly higher in the morphine group compared to expression in the saline group (Fig. 6B, p < 0.05). However, SIRT1 was expressed significantly lower in the morphine + OCT group when compared to expression in the morphine group (p < 0.05).
mTOR gene expression
In the PFC, expression of mTOR varied significantly among the groups [H (3) = 10.53, P < 0.001, Fig. 6C]. In the OCT group, a significantly higher mTOR expression was seen compared to that in the saline group Fig. 6C). In contrast, both the morphine and morphine + OCT groups showed considerably lower mTOR expression than that in the OCT group. In the hippocampus, mTOR expression was significantly higher in the OCT and morphine groups compared to the saline group [H (3) = 9.5, P < 0.01, Fig. 6D].
Histological examination
Histological analysis of H&E stained PFC and hippocampal tissue slices revealed differences in structural integrity between the experimental groups. In the PFC (Fig. 7, Top panel), (prefrontal saline (PS)) displayed healthy, definitions between groups (F (3, 8) = 11.2, p < 0.001). Quantitative analysis revealed significantly more intact neurons in the saline group when compared to the other groups (one-way ANOVA, p < 0.001). Indicative of damage by morphine, the most prominent neurodegeneration was seen in the morphine group (prefrontal morphine (PM); n = 50), with darkly stained, fragmented cells occupying over 60% of the observed field. Neurons in the OCT treatment group (n = 30) mainly resembled controls, though some mild phenotypic changes were noted in approximately 25% of cells. Notably, co-administration of OCT with morphine (prefrontal OCT + morphine (POM); n = 40) was associated with predominantly normal neuronal morphology, with crisp borders and nuclei. Quantification revealed the combination of OCT and morphine was associated with significantly more healthy neurons in the PFC than that seen in the group treated with morphine (Post-hoc Tukey, p < 0.001), and the numbers of healthy neurons was not statistically different than the numbers seen in the groups treated with saline or OCT, supporting the conclusion that OCT exhibits neuroprotection against morphine-induced neuronal damage in the PFC.

Micrographs showing histopathological results in the PFC (UP panel). (A) Saline group, (B) OCT group, (C) morphine group, (D) morphine + OCT group. Histopathological micrographs of hippocampal tissue slices (Down panel). Quantitative measures of PFC and hippocampal tissues among different histopathological study groups. Data are expressed as the mean ± SEM (#P < 0.05, ### P < 0.001). #: The significance compared to the saline group, *: The significance compared to the morphine + OCT group. HS, hippocampal saline group; HO, hippocampal OCT group; HOM, hippocampal morphine + OCT group; HM, hippocampal morphine group; PS, prefrontal saline group; PO, prefrontal OCT group; POM, prefrontal morphine + OCT group; PM, prefrontal morphine group.
A similar pattern was observed in the hippocampus (Figs. 7A–D, lower panel). As expected, neuronal integrity was high in the saline group (n = 18), whereas indicators of compromised cell health were seen in over 50% of cells in the morphine group (n = 50). Neurons in the group treated with OCT (n = 23) exhibited profiles similar to those seen in the saline group however, mild changes were noted in approximately 30% of neurons. Importantly, the combination of OCT and morphine (n = 21) was associated with neuronal profiles predominantly indicating intact neuronal morphology which resembled that seen in the saline group. Quantification of neuronal degeneration showed that the treatment with OCT + morphine resulted in significantly more healthy neurons than that seen in the morphine treated group (one-way ANOVA with Tukey, p < 0.01). This provides further indication of the potential for OCT to blunt morphine-mediated neurodegeneration. Taken together, these histological findings provide evidence of OCT’s neuroprotective capabilities in two neuronal regions important in addiction and shown to be affected by morphine exposure.
Docking
In the TLR4 docking study, OCT demonstrated a docking score of − 10.2, indicating strong binding affinity to the TLR4 receptor. The analysis revealed specific interactions between OCT and key amino acid residues in TLR4. The amine group of D-Phe in OCT formed an ionic bond with Asp208, demonstrating a robust electrostatic interaction. Additionally, this amine group engaged in both ionic and hydrogen bonding interactions with Asp180, contributing to the stabilization of the ligand within the TLR4 binding pocket. The amide nitrogen of Cys2 in OCT formed a hydrogen bond with Asp180, further strengthening the ligand-receptor interaction. Furthermore, the carbonyl group of Cys2 formed a hydrogen bond with Asn155, enhancing the overall binding affinity. The D-Trp residue in OCT formed a hydrogen bond with Ser85 via the nitrogen atom within the heterocyclic ring, adding to the stability of the ligand-receptor complex. Moreover, the amine group of the lysine side chain in OCT played a crucial role in the binding interaction as it formed hydrogen bonds and ionic interactions with Asp59 and Asp83, contributing to the overall stability of the ligand-receptor complex. Finally, one of the hydroxyl groups within the Thr residue of OCT formed a hydrogen bond with Lys153, further strengthening the ligand-receptor complex through a polar interaction (Fig. 8A).

OCT in the binding site of TLR4 (A) and TRKb (B).
In the TRKb, which is BDNF’s receptor31 in, our docking study, OCT displayed a docking score of − 9.96, indicating a strong binding affinity with the TRKb receptor. The analysis revealed specific and meaningful interactions between OCT and key amino acid residues in TRKb. The amine group of D-Phe in OCT formed both a hydrogen bond and an ionic bond with Glu604, establishing a stable and favorable interaction at the binding site. Additionally, the side chain amine of Lys interacted with Asp600, contributing to the overall stability of the ligand-receptor complex. Notably, Asp600 also engaged in a hydrogen bond with the hydroxyl group of the side chain of Thr, further reinforcing the binding interaction. Furthermore, the carbonyl part of Cys7 in OCT formed hydrogen bonds with Lys588 and Phe565, likely facilitating additional favorable interactions and enhancing the overall stability of the ligand-receptor complex. Moreover, the nitrogen atom of threoninol in OCT participated in a hydrogen bond formation with Glu604, further strengthening the binding between the ligand and TRKb (Fig. 8B).
In the mTOR docking study, OCT exhibited a docking score of − 7.37, indicating a favorable binding affinity with the mTOR protein. The analysis revealed several key interactions between OCT and specific amino acid residues in mTOR. The amine group of D-Phe in OCT formed a hydrogen bond with Glu315, contributing to a stable interaction at the binding site. Additionally, the aromatic ring of Phe formed a hydrogen bond with Glu315 through the nitrogen atom in its amide group. The D-Trp residue in OCT engaged in pi–pi stacking with Tyr327 and formed pi–cation interactions with Arg274, further stabilizing the ligand-receptor complex. Lysine interacted with Tyr273 through pi–cation interactions and also formed ionic interactions with Asp275. Moreover, Lysine formed a hydrogen bond with Asp293. Thr313 in OCT formed a hydrogen bond with Asp275 through its side chain hydroxyl group. Additionally, the carbonyl group of Thr formed a hydrogen bond with Lys277. Furthermore, one of the hydroxyl groups of threoninol in OCT formed a hydrogen bond with Thr313, while the other hydroxyl group formed a hydrogen bond with Leu296 (Fig. 9A).

OCT in the binding site of mTOR (A) and TAF15 (B).
In the TAF15, which is SIRT1’s receptor20, in our docking study, OCT achieved a docking score of − 5.52, indicating a moderate binding affinity with the TAF15 protein. The analysis revealed several interactions between OCT and specific amino acid residues in TAF15. One notable interaction involved the hydrogen bonding between the amine group of D-Phe in OCT and Thr275 in TAF15, contributing to the stabilization of the ligand-receptor complex. Additionally, the side chain amine of Lys interacted with Glu323 in TAF15 through both an ionic interaction and a hydrogen bond formed by the amide nitrogen, further contributing to the binding of OCT to TAF15. Furthermore, Lys268 formed a hydrogen bond with one of the hydroxyl groups of OCT’s threoninol, adding to the overall binding interaction. Another interaction involved threoninol in OCT forming a hydrogen bond with Asn272 in TAF15 through its amide nitrogen, contributing to the stability of the ligand-receptor complex (Fig. 9B).
Discussion
This study explores the potential of OCT in treating addiction, emphasizing its effects on morphine-dependent behaviors while analyzing morphine-induced degeneration and molecular changes in the prefrontal cortex and hippocampus. We also complemented our findings with in silico docking studies. OCT decreased morphine dependence behaviours, as well as exerted neuroprotective effects, and regulated addiction-related gene expression in the PFC and hippocampus. Docking studies revealed a high binding affinity of OCT to proteins implicated in drug addiction. OCT administration markedly alleviated opioid-induced behavioral abnormalities in animal models, repaired structural damage caused by prolonged opioid use, and regulated key addiction-related pathways and biomarkers. In silico docking studies demonstrated strong binding affinities to crucial protein targets, underscoring its molecular interactions and pharmacological potential. These results position OCT as a promising therapeutic candidate for opioid dependency, meriting further exploration.
Behavioral and histopathological evaluations
As expected, we noted degenerated histopathology in the PFC and hippocampus of the morphine treated groups. Treatment with OCT + morphine resulted in degeneration profiles similar to those seen in groups exposed to saline and OCT alone, suggesting a neuroprotective effect. Rescue of histopathology only in the hippocampal OCT + morphine groups in the quantified histopathological examinations. OCT reduced drug reward (delta time in CPP), anxiety-like behavior, memory deficits, and neuronal damage in the PFC and hippocampus, while modulating TLR4, BDNF, SIRT1, and mTOR pathways, supported by molecular docking. These findings suggest OCT’s potential anti-inflammatory and neuroprotective mechanisms32,33. OCT is thought to influence neurotransmitters such as dopamine33, which helps regulate mood and alleviate withdrawal symptoms. It also promotes neurogenesis, reduces neuroinflammation, and stimulates neurotrophic factors 19,21,22 like BDNF, aiding in the repair of damage caused by prolonged opioid use.
Behavioral tests demonstrated restoration of exploratory and anxiety-like behaviors in the OCT + morphine group from behavior seen in the morphine group in the OFT. The reduced rearing behavior, shorter total traveling distance, and decreased time spent in the central zone observed in the MOR group suggest morphine-induced anxiety, which appears to be alleviated by OCT. The enhanced rearing behavior, increased traveling distance, and greater time spent in the central zone in the MOR + OCT group further confirm the anxiolytic-like effects of OCT in counteracting morphine-induced anxiety. These findings underscore OCT’s potential therapeutic value in addressing opioid-related behavioral changes. A greater passive avoidance memory was seen in the OCT + morphine group compared to that seen in the morphine group, and reduced morphine preference behavior was noted in the morphine group treated with OCT compared to that seen in the morphine group when evaluated using the CPP test. Our findings align with previous cognitive-based behavioral and histopathological studies involving OCT. An anxiolytic effect of OCT associated with reduced neuronal death was previously reported in a rodent model of epilepsy34 and improvements in histopathological features including anti-apoptotic effects have been seen in several studies following OCT administration34,35. Interestingly, an enhancement of memory has been documented in older adults taking OCT36. While the underlying mechanisms on improvements in memory were not determined, somatostatin is known to enhance synaptic plasticity by facilitating long-term potentiation in hippocampus37, and thus OCT actions on cognitive functions controlled by hippocampus and PFC could involve changes in synaptic strength in these memory-related neural regions. These results indicate that OCT may play a protective role in reducing the cognitive and behavioral impairments commonly linked to morphine use. Additionally, the observed decrease in morphine preference behavior underscores the potential of OCT as a therapeutic option for addressing substance dependence. Future studies should aim to clarify the specific mechanisms driving these effects and investigate the broader potential of OCT in treating addiction and cognitive dysfunction.
Docking
TLR4
A strong binding affinity between the ligand and the TLR4 receptor was noted and specific interactions with key amino acid residues in TLR4 were detected, which contribute to the stability of the ligand-receptor complex. In the binding site of TLR4 with MD-2, there are two distinct patches: patch A and patch B. Patch A contains important amino acid residues such as Asp41, Asp83, Glu134, and His158, which are involved in ligand recognition and binding38. Docking results revealed that OCT binding to TLR4 resulted in the blocking of patch A, indicating interference with ligand or co-receptor interactions facilitated by this region. On the other hand, patch B, comprising Arg233 and Arg28838, did not show significant alterations upon OCT binding, suggesting that it remains accessible for interactions with other molecules. The blocking effect of OCT on patch A provides valuable insights into the functional consequences of its binding to TLR4. By interfering with the interactions facilitated by patch A, OCT may disrupt downstream signaling pathways triggered by TLR4 activation. This interference could potentially modulate inflammatory responses and immune signaling processes associated with TLR4-mediated pathways, and thus, our data suggest that OCT may have promising applications in the treatment of inflammatory disorders. As we saw a reduction in morphine-associated neurodegeneration with OCT and inflammation plays a role in addiction, OCT’s actions in normalizing neuronal death in morphine treated animals could involve an effect mediated through TLR4. However, further studies are needed to fully elucidate the precise mechanisms and downstream effects of OCT binding to TLR4 and its impact on inflammation-associated disorders.
TRKb
The TRKb docking study revealed a highly favorable binding affinity between OCT and the TRKb receptor, and identification of specific and meaningful interactions between OCT and key amino acid residues in TRKb were noted. When comparing our docking results to the reported binding site in the crystallographic structure of TRKb, we found that the ligand docked in almost the same position as some selective inhibitors. The reported binding site consists of amino acid residues Ala564, Phe565, Gly566, Phe633, Asp710, Phe711, Gly712, and Val61739. The alignment of OCT with these critical residues suggests that OCT may target the same binding pocket and interact with key amino acids involved in ligand recognition and signaling. The strong binding affinity and specific interactions observed in our TRKb docking study, along with the similarity to the reported binding site of selective inhibitors, provide valuable insights into the potential biological effects of OCT binding to TRKb. By occupying the binding site and interacting with specific residues, OCT has the potential to modulate TRKb-mediated signaling pathways, which play crucial roles in cellular processes such as cell survival, proliferation, and differentiation. Targeting TRKb signaling has been suggested in management of opioid addiction, and suggestion by which TRKb signaling could play a role in addiction is in neural protection40. Thus, we speculate that the binding interaction between TRKb and OCT could play a role in protective effects noted against morphine-induced neural damage. However, more studies are necessary to validate the biological impact of OCT binding to TRKb and to explore the downstream effects on cellular responses, if we wish to be harness the therapeutic potential of OCT in conditions associated with dysregulated TRKb signaling.
mTOR
The docking study of OCT with the mTOR protein revealed a strong binding affinity, and interactions noted likely influence the conformation and activity of mTOR. OCT was found to interact with specific amino acid residues within the binding pocket of mTOR, which aligns with the reported peptide binding site consisting of Glu200, Arg274, Asp293, Asp275, Phe294, Lys298, Thr309, and Glu31541. OCT’s interaction with Glu315, a key residue within the binding pocket, suggests that it may interfere with the binding of endogenous ligands or substrates to mTOR. The interactions between OCT and mTOR indicate its ability to modulate mTOR signaling, thus, it is possible that OCT could play a role in altering processes in which mTOR has been shown to play a role including cell growth, metabolism, and protein synthesis. Since dysregulation of mTOR signaling is associated with various diseases, including cancer, metabolic disorders, and neurodegenerative conditions, OCT could have implications in various disease contexts associated with dysregulated mTOR signaling. Further, alterations in mTOR signaling have been implicated in drug addiction29and thus actions seen in our study could involve changes in signaling within this pathway. Further experimental studies are necessary to validate the specific biological effects of OCT binding to mTOR and to elucidate the downstream consequences on cellular responses.
TAF15
Docking studies of OCT with TAF15 showed a moderate binding affinity, and specific interactions between OCT and key amino acid residues in TAF15 were noted. Amino acid interactions suggested enhancement of the binding affinity, an influence on conformational changes, and stabilization of the ligand-receptor complex, which indicates a potential role for OCT in modulating the functionality of TAF15. Furthermore, the docking study revealed that OCT binds in the same pocket as the SON stem-loop RNA, as observed in the structure with PDB code 2MMY. This binding pocket involves specific residues, including Lys264, Lys265, and Lys268 in the L2 loop, as well as Lys277, Lys281, and Lys283 in the L3 loop, and Arg320 and Arg321 in the C-terminal region. This finding suggests a possible competition or interaction between OCT and SON stem-loop RNA, which may have implications for the regulation of RNA function or stability42. In the context of addiction, as opioids are known to increase DNA damage in cortical regions43, actions of OCT binding to TAF15 could include amelioration of this damage ending in reduced apoptosis 20,44 and potentially reduce negative cognitively based behavioral consequence of opioid addiction. However, the biological effects of OCT binding to TAF15 and its influence on cellular processes involving TAF15 and SON stem-loop RNA remain to be further elucidated, and the role OCT binding to TAF15 could play in addiction remains highly speculative.
PCR
OCT interacts with several key pathways related to addiction, including BDNF, SIRT1, TLR4, and mTOR19,20,21,22. While effects of OCT on these pathways have been studied, actions have not been evaluated in the context of morphine dependence. In our qRT-PCR studies, frontal TLR4 was significantly upregulated in the OCT group compared to the saline group, significantly downregulated in the groups receiving morphine and OCT plus morphine compared to the OCT group, and significantly upregulated in the OCT and morphine group compared to the group receiving morphine alone. Additionally, hippocampal TLR4 was significantly upregulated in the OCT group compared to the saline group, and significantly downregulated in the morphine and OCT and morphine groups compared to the OCT group. Furthermore, mTOR in the PFC was significantly upregulated in the OCT group compared to the saline group, significantly downregulated in the morphine and OCT and morphine groups compared to the OCT group, and significantly upregulated in the OCT and morphine compared to the morphine group. Hippocampal mTOR was significantly upregulated in the OCT and morphine groups compared to the saline group. An antagonistic effect of OCT on TLR421 and mTOR22,45 pathways have been demonstrated elsewhere. Increased exposure to agonist can result in receptor downregulation and decreased exposure to agonists (increased exposure to antagonists) can result in receptor upregulation46. If OCT acts as a receptor antagonist, this could result in upregulation of TLR4 and mTOR pathways when the peptide is present, and downregulation of these pathways when morphine acts as an agonist47,48. The presence of OCT during morphine exposure appears to counteract morphine’s impact on the downregulation of TLR4 and mTOR. Additionally, BDNF levels in the PFC were markedly reduced in both the morphine group and the OCT + morphine group compared to the saline group. Our data are in line with findings that morphine reduces BDNF protein in the PFC49, however, our expression results suggest that OCT does not block this action. SIRT1 in the PFC and hippocampus were significantly upregulated in the morphine groups compared to saline groups, and significantly downregulated in the OCT and morphine groups compared to levels seen in the group treated with morphine. Upregulation of SIRT1 in the morphine group may occur as a protective mechanism against morphine-induced oxidative stress50. This could be expected if OCT functions as an antioxidant51, which might partially normalize SIRT1 upregulation. While speculative, this interpretation is in agreement with a previous study that demonstrated OCT’s stabilizing effect on SIRT1 mRNA20.
However, this approach has certain limitations, especially when paired with histological and molecular analyses, as the aversive stimuli used in behavioral testing might alter gene expression patterns. Future research could benefit from employing non-aversive cognitive tests, such as self-administration and novel object recognition studies, particularly when focusing on molecular endpoints.
Conclusion
Preclinical data suggest that OCT has promising potential in reducing morphine dependence by reversing the behavioral, molecular, and structural changes caused by morphine. Additionally, findings indicate that OCT interacts with key molecular pathways involved in the development of morphine dependence. To thoroughly assess its therapeutic effectiveness, translational applicability, and mechanisms of action, further preclinical research on OCT is essential.
Materials and methods
Animals
Wistar rats, weighing 230–250 g, were provided by Kerman University of Medical Sciences. Rats were housed under conditions of controlled humidity (55–65%), temperature (23 ± 1 °C), and lighting (lights on 08:00–20:00 h) and were given free access to food and water. All animal experiments were conducted between 9:30 a.m. and 12:30 p.m., except for the CPP experiment, which extended until 6 p.m. due to the test’s requirements and the four-hour interval between administering morphine and saline injections. The Ethics Committee of Kerman University of Medical Sciences approved this study (Ethics code: IR.KMU.REC.1400.242). All experiments were performed according to relevant guidelines.
Experimental groups
The animals were randomly assigned to two main groups, each further divided into four subgroups (8 animals per subgroup). All experimental groups received intracerebroventricular (i.c.v.) injections of either control or OCT solutions into the third ventricle of the brain. The saline group received control injections of saline. The OCT group received OCT plus saline injections. The morphine group was administered morphine injections and the 4th group received morphine and OCT injections (Morphine + OCT). The study’s design ensured clear separation between the groups assigned to the PAT and those used for the conditioned place preference (CPP) tests. For one group of animals, the CPP tests were conducted over four days, starting on day 20, immediately after the elevated plus maze (EPM) test. In separate groups, the OFT was performed first on day 20, followed by the PAT, which was divided over 2 days—day one focused on learning, while day two assessed memory. Only the animals involved in the CPP test were included in the subsequent histological and molecular analyses. The integration of the OFT, EPM, PAT, and CPP in this study design allows for a comprehensive assessment of behaviors related to anxiety, locomotion, learning, and reward. Each test focuses on specific neurobiological mechanisms associated with dysfunctions in the PFC and hippocampus. This robust will help us better understand the complexities of behavior related to substance dependence and its impact on brain function. Ultimately, this approach may inform the development of future therapeutic strategies.
Intracerebroventricular cannula insertion
As a somatostatin analog, OCT has shown limited blood-brain permeability due to its large molecular size necessitating i.c.v. administration. Accordingly, a 22-guage with an attached dummy cannula was stereotaxically implanted into the third cerebral ventricle under pentobarbital sodium (45 mg/ kg, i.p.) anesthesia as described previously52. The guide cannula was secured to the skull with bone screw anchors and dental cement. The correct positioning was confirmed through the detection of cerebrospinal fluid in the cannula. Rats were allowed 7 days recovery period. After the removal of the dummy cannula, an inner injection cannula which was connected to the micro syringe by polyethylene tubing was gently inserted. Microinjection in a volume of 5 µl was made over 15 s, and a further 15 s elapsed before withdrawal of the inner cannula. In the morphine injection period, 0.25 µg/ 5 µl OCT was injected i.c.v. once per day and for 7 consecutive days after the last morphine injection 53,54.
Induction of morphine dependence
The morphine-dependent rat model was established by subcutaneous administration of ascending doses (5, 10, 20, 40, 50, and 60 mg kg −1) of morphine i.p. 3 times per day for 6 consecutive days in all animals except those in the saline control group and the group receiving OCT only.
Behavioral tests
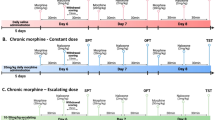
A series of behavioral tests (Fig. 10) were performed as follows:

Timeline of the study.
Open field test (OFT)
The OFT was utilized to assess the locomotor activity and anxiety-like behavior of animals. The open field consisted of an arena (90 × 90 × 30 cm) made of Plexiglas. Animals were placed at the center of the arena and their locomotor activity were recorded and analyzed within a 5 min period using offline analysis (Ethovision7.1, Noldus Information Technology, Netherlands). The following parameters were recorded for each animal: rearing and grooming events, total distance moved, immobility duration, time spent in the center and periphery of the arena55.
Elevated plus maze (EPM)
The elevated plus maze setup comprised two open and two enclosed arms with an intervening central square, elevated 50 cm above the ground. Each trial occurred in an environment illuminated by a 60 W light bulb situated centrally above the maze. Prior to testing, animals underwent a one-hour acclimation period in the experimental space without exposure to the elevated plus maze apparatus. Subsequently, the rat was situated in the central square and was allowed five minutes of unrestricted exploration of the maze. The entire session was captured by a video camera positioned overhead. Anxiety-like behavior was evaluated by measuring the percentage of time spent in the open and closed arms56.
Passive avoidance test (PAT)
The PAT was carried out to assess passive avoidance learning and memory. The experiment utilized a two-way shuttle box measuring 60 × 20 × 20 cm, featuring Plexiglas walls and a floor constructed with stainless-steel rods spaced at 1.0 cm intervals. The floor was partitioned into two equal-sized chambers by means of a wooden divider elevated 1.5 cm above the grid floor. Animals were initially placed in the box to habituate for 3 min. During the learning session, upon the mouse entering the dark compartment, a solitary electric shock of 0.5 mA intensity, 50 Hz frequency, and 2 s duration was administered57. The recorded data included the count of electric shocks administered to each animal as a deterrent from entering the dark compartment. To evaluate memory, the mouse was positioned in the light compartment 24 h post the learning session, and the door separating the compartments was opened. The duration taken to enter the dark chamber, referred to as step-through latency (STL), was subsequently measured58,59.
Conditioned place preference test
The unbiased conditioned place preference (CPP) animal model of morphine preference was utilized to evaluate the preference of animals for morphine exposure. The CPP apparatus consisted of three wooden compartments. Two of the compartments, A and B, were equal in size (30 × 30 × 40 cm) but presented distinctly different visual and tactile cues. Compartment A was painted in black with vertical white stripes with a textured floor, and compartment B was painted in black with horizontal white stripes and a smooth floor. The third compartment (C) was neutral with white walls and a smooth floor between the other two compartments. The final phase of the CPP experiments began on day 20, following the elevated plus-maze trials. This phase consisted of the final behavioral test, divided into three key stages: pre-conditioning, conditioning, and post-conditioning, all of which were recorded on video. On the first day, the animals were individually placed into compartment C, and the door was removed, allowing free access to the other compartments for 10 min. Evaluation of the time spent in each compartment was conducted to determine any individual inherent preference for any compartment. For the other four days, subcutaneous injection of morphine (10 mg/kg, i.p.) and saline was given for each animal so that morphine injection would be associated with the placement of the animals in compartment A. After morphine administration, the animal was immediately conditioned for 30 min in the morphine-paired compartment. After 4 h, the animal received saline and was placed for 30 min in the other compartment (B). The animals were confined to one compartment by closing the guillotine doors during these sessions. On the second day of conditioning, the animals received saline in the morning session and morphine in the evening session. The third day of conditioning followed the same schedule as the first day. This schedule was chosen to avoid circadian variability (morning/evening). On the seventh and thirteenth day, animals were put in the neutral compartment and the doors were removed to let the animal freely move through the compartments. To determine the CPP score, the time spent in both the saline-paired and morphine-paired compartments were recorded in both pretest and posttest. The CPP score was calculated by subtracting the time spent in the saline-paired compartment from the time spent in the morphine-paired compartment60,61.
RNA isolation and quantitative real-time-polymerase chain reaction
Total RNA was extracted from prefrontal cortex (PFC) and hippocampal frozen tissues (RNeasy mini kit, Qiagen, UK), and then, based on primary RNA integrity, the cDNA was synthesized using Script RT Kit (Qiagen, UK). qRT-PCR for TLR4, BDNF, mTOR, and SIRT1 was performed in triplicate on each sample of cDNA. The setup of the PCR system was 35 cycles of 95 °C for 40 s followed by 65 °C for 1 min. The primers used in this study are presented in Table 1. Based on the 2−∆∆CT method the relative gene expression was calculated and GAPDH was used as the housekeeping gene.
Morphological analysis
H&E staining
To evaluate the impact on neuron survival, we conducted histological examinations of neurons in hippocampus and PFC, two neuronal regions known to be altered by exposure to morphine62,63. For indicators of necrosis, which can be induced by inflammatory markers, we compromised structural integrity, swollen organelles, ruptured plasma membranes, cell lysis, darkened cytoplasm, condensed nuclei, autolytic necrosis with cytoplasmic vacuoles, and reduced, and more spherical somata. Brains (n = 3 per group) were fixed in 10% formalin post-euthanasia (Isoflurane inhalation), dissected, embedded in paraffin, and serially sectioned (4 μm). Slices were then dewaxed and heated at 120 °C for 10 min using a 600 W microwave oven. Following staining of the slices with hematoxylin–eosin (H&E) a bright-field microscope (Olympus CX31) was used to identify and evaluate neurons exhibiting clearly defined nuclei and cell bodies64. Two blinded researchers analyzed 15 fields/group/region.
Statistics
To analyse data, GraphPad Prism 8 (GraphPad Software, USA) was utilized. The Kolmogorov-Smirnov test (p > 0.05) and Levene’s test were used to evaluate data normality and homogeneity of variance. A one-way ANOVA was performed for OFT, PAT, EPM, molecular, and histological datasets, while a two-way ANOVA (group × time) with Greenhouse-Geisser corrections for sphericity violations was applied to CPP data. Results are reported with F-statistics, degrees of freedom, and exact p-values. Tukey post hoc tests followed significant ANOVA findings. If assumptions were not met, a non-parametric Kruskal–Wallis test was conducted as an alternative. Data are presented as mean ± SEM, with statistical significance set at p < 0.05.
Docking
The ligand was prepared using Schrodinger LigPrep65, which allows for the generation of different ionization states within a pH range of 5 to 9 by employing Epik66. Subsequently, energy minimization was performed using the OPLS4 force field67. The three-dimensional structures of the proteins (TLR4:2Z64 38, TRKb:4ASZ39, TAF15:2MMY42, mTOR:1GZO41) were obtained from the Protein Data Bank and chosen as they could play a role in the mechanism of action involved in the mechanism of action of the ligand. The Protein Preparation Wizard68 was used to optimize protein structures, which included adding hydrogen atoms, creating disulfide bonds, and removing water molecules located beyond a distance of 5.00 Å from HET groups. Additionally, any missing loops and side chains were filled in using the Prime tool69,70. Epik was employed to generate different protonation states for the ligand, while PROPKA was used to determine the protonated forms at a pH of 7.00. The protein structures were ultimately optimized and minimized using the OPLS4 force field. For molecular docking, Glide71,72 was utilized. A grid covering the entire protein was generated using the Glide Grid Generator, employing a blind docking approach. Glide’s Extra Precision (XP)73 docking method was then employed to account for ligand flexibility, considering various routes that accounted for the inversion of nitrogen atoms and Ring Shapers generated by Ring conf. The nonpolar regions of the ligand were adjusted by applying the van der Waals radius ratio, and a partial charge cutoff of 0.15 was set.
Data availability
The datasets used or analyzed during the current study are available from the corresponding author upon request.
Abbreviations
- OCT:
-
Octreotide
- PFC:
-
Prefrontal cortex
- TLR4:
-
Toll-like receptor 4
- BDNF:
-
Brain-derived neurotrophic factor
- SIRT1:
-
Sirtuin 1
- mTOR:
-
Mammalian target of rapamycin
- CPP:
-
Conditioned place preference
- OFT:
-
Open field test
- EPM:
-
Elevated plus maze
- PAT:
-
Passive avoidance test
- H&E:
-
Hematoxylin and eosin
- qRT-PCR:
-
Quantitative real-time PCR
References
Cheron, J. & Kerchove d’Exaerde, A. Drug addiction: from bench to bedside.Transl. Psychiatry. 11, 424 https://doi.org/10.1038/s41398-021-01542-0 (2021).
Goldstein, R. Z. & Volkow, N. D. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 12, 652–669. https://doi.org/10.1038/nrn3119 (2011).
Everitt, B. J. & Robbins, T. W. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 8, 1481–1489. https://doi.org/10.1038/nn1579 (2005).
Volkow, N. D., Wang, G. J., Fowler, J. S. & Tomasi, D. Addiction circuitry in the human brain. Annu. Rev. Pharmacol. Toxicol. 52, 321–336. https://doi.org/10.1146/annurev-pharmtox-010611-134625 (2012).
Koob, G. F. & Le Moal, M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129. https://doi.org/10.1016/s0893-133x(00)00195-0 (2001).
Kohno, M. et al. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol. Biochem. Behav. 179, 34–42 (2019).
Xiaoshan, T. et al. Immunotherapy for treating methamphetamine, heroin and cocaine use disorders. Drug Discov. Today. 25, 610–619 (2020).
Berríos-Cárcamo, P. et al. Oxidative stress and neuroinflammation as a Pivot in drug abuse. A focus on the therapeutic potential of antioxidant and anti-inflammatory agents and biomolecules. Antioxidants. 9, 830 (2020).
Wang, X. et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. U. S. A. 109, 6325–6330. https://doi.org/10.1073/pnas.1200130109 (2012).
Russo, S. J. et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 33, 267–276. https://doi.org/10.1016/j.tins.2010.02.002 (2010).
Hutchinson, M. R. et al. Non-stereoselective reversal of neuropathic pain by Naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29. https://doi.org/10.1111/j.1460-9568.2008.06321.x (2008).
Kumar, U. & Grant, M. Somatostatin and somatostatin receptors. Cell. Pept. Hormone Synth. Secret. Pathways. 97–120 (2010).
Murphy, E., Prommer, E. E., Mihalyo, M. & Wilcock, A. Octreotide. J. Pain Sympt. Manag. 40, 142–148 (2010).
Cozzi, R. & Attanasio, R. Octreotide for acromegaly. Expert Rev. Endocrinol. Metab. 2, 129–145 (2007).
Khedr, A. et al. Role of octreotide in small bowel bleeding. World J. Clin. Cases. 10, 9192 (2022).
Mas, E. et al. Drugs in focus: octreotide use in children with gastrointestinal disorders. J. Pediatr. Gastroenterol. Nutr. 74, 1–6 (2022).
La Salvia, A. et al. Targeting neuroendocrine tumors with octreotide and lanreotide: key points for clinical practice from NET specialists. Cancer Treat. Rev. 102560 (2023).
Fan, M., Huang, Y., Zhu, X., Zheng, J. & Du, M. Octreotide and Octreotide-derived delivery systems. J. Drug Target. 1–28 (2023).
Jiang, R. & Wei, H. Beneficial effects of octreotide in alcohol-induced neuropathic pain. Role of H 2S, BDNF, TNF-α and Nrf2. Acta Cir. Bras. 36, e360408. https://doi.org/10.1590/acb360408 (2021).
Zou, S. F. et al. Octreotide ameliorates hepatic ischemia-reperfusion injury through SNHG12/TAF15-mediated Sirt1 stabilization and YAP1 transcription. Toxicol. Appl. Pharmacol. 442, 115975. https://doi.org/10.1016/j.taap.2022.115975 (2022).
El-Sisi, A. E. E., Sokar, S. S., Shebl, A. M., Mohamed, D. Z. & Abu-Risha, S. E. Octreotide and melatonin alleviate inflammasome-induced pyroptosis through Inhibition of TLR4-NF-κB-NLRP3 pathway in hepatic ischemia/reperfusion injury. Toxicol. Appl. Pharmacol. 410, 115340. https://doi.org/10.1016/j.taap.2020.115340 (2021).
Zhang, S., Tang, C. & Wang, X. Octreotide activates autophagy to alleviate lipopolysaccharide-induced human pulmonary epithelial cell injury by inhibiting the protein kinase B (AKT)/mammalian target of Rapamycin (mTOR) signaling pathway. Bioengineered. 13, 217–226. https://doi.org/10.1080/21655979.2021.2012908 (2022).
Theodoropoulou, M. & Stalla, G. K. Somatostatin receptors: from signaling to clinical practice. Front. Neuroendocrinol. 34, 228–252. https://doi.org/10.1016/j.yfrne.2013.07.005 (2013).
Ferguson, D. et al. Essential role of SIRT1 signaling in the nucleus accumbens in cocaine and morphine action. J. Neurosci. 33, 16088–16098. https://doi.org/10.1523/jneurosci.1284-13.2013 (2013).
Wei, L. et al. Sirtuin 1 inhibitor EX527 suppresses morphine-induced behavioral sensitization. Neurosci. Lett. 744, 135599. https://doi.org/10.1016/j.neulet.2020.135599 (2021).
Xia, B. et al. Effect of Sirtuin-1 on synaptic plasticity in nucleus accumbens in a rat model of heroin addiction. Med. Sci. Monit. 24, 3789–3803. https://doi.org/10.12659/msm.910550 (2018).
Barker, J. M., Taylor, J. R., De Vries, T. J. & Peters, J. Brain-derived neurotrophic factor and addiction: pathological versus therapeutic effects on drug seeking. Brain Res. 1628, 68–81. https://doi.org/10.1016/j.brainres.2014.10.058 (2015).
Liu, J., Li, J. X. & Wu, R. Toll-like receptor 4: A novel target to tackle drug addiction?? Handb. Exp. Pharmacol. 276, 275–290. https://doi.org/10.1007/164_2022_586 (2022).
Ucha, M., Roura-Martínez, D., Ambrosio, E. & Higuera-Matas, A. The role of the mTOR pathway in models of drug-induced reward and the behavioural constituents of addiction. J. Psychopharmacol. 34, 1176–1199. https://doi.org/10.1177/0269881120944159 (2020).
Robinson, S. L. & Thiele, T. E. A role for the neuropeptide somatostatin in the neurobiology of behaviors associated with substances abuse and affective disorders. Neuropharmacology. 167, 107983. https://doi.org/10.1016/j.neuropharm.2020.107983 (2020).
Lu, B. Pro-region of neurotrophins: role in synaptic modulation. Neuron. 39, 735–738. https://doi.org/10.1016/s0896-6273(03)00538-5 (2003).
Chen, L. et al. The protection by octreotide against experimental ischemic stroke: up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-κB expression. Brain Res. 1475, 80–87. https://doi.org/10.1016/j.brainres.2012.07.052 (2012).
Al-Shorbagy, M. Y. & Nassar, N. N. Octreotide ameliorates inflammation and apoptosis in acute and kindled murine PTZ paradigms. Naunyn Schmiedebergs Arch. Pharmacol. 390, 61–68. https://doi.org/10.1007/s00210-016-1303-x (2017).
Shayan, T. K., Abdolmaleki, A., Asadi, A. & Hassanpour, H. Neuroprotective anticonvulsant and anxiolytic effects of octreotide in Wistar rats. J. Chem. Neuroanat. 132, 102320. https://doi.org/10.1016/j.jchemneu.2023.102320 (2023).
Dundar, S. et al. The effects of octreotide acetate on spinal cord ischemia/reperfusion injury. Neurosurg. Q. 25 (2015).
Watson, G. S. et al. Effects of insulin and octreotide on memory and growth hormone in alzheimer’s disease. J. Alzheimers Dis. 18, 595–602. https://doi.org/10.3233/jad-2009-1165 (2009).
Matsuoka, N., Kaneko, S. & Satoh, M. Somatostatin augments long-term potentiation of the mossy fiber-CA3 system in guinea-pig hippocampal slices. Brain Res. 553, 188–194. https://doi.org/10.1016/0006-8993(91)90823-e (1991).
Kim, H. M. et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 130, 906–917 (2007).
Bertrand, T. et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J. Mol. Biol. 423, 439–453 (2012).
Lim, S. Y. (ed Cengiz, P.) Opioid tolerance and opioid-induced hyperalgesia: is TrkB modulation a potential Pharmacological solution? Neuropharmacology. 220 109260 https://doi.org/10.1016/j.neuropharm.2022.109260 (2022).
Yang, J. et al. Molecular mechanism for the regulation of protein kinase b/akt by hydrophobic motif phosphorylation. Mol. Cell. 9, 1227–1240 (2002).
Kashyap, M., Ganguly, A. K. & Bhavesh, N. S. Structural delineation of stem-loop RNA binding by human TAF15 protein. Sci. Rep. 5, 1–14 (2015).
Wang, Y. et al. Opioid induces increased DNA damage in prefrontal cortex and nucleus accumbens. Pharmacol. Biochem. Behav. 224, 173535. https://doi.org/10.1016/j.pbb.2023.173535 (2023).
Singh, A. K., Kapoor, V., Thotala, D. & Hallahan, D. E. TAF15 contributes to the radiation-inducible stress response in cancer. Oncotarget. 11, 2647–2659. https://doi.org/10.18632/oncotarget.27663 (2020).
Grozinsky-Glasberg, S. et al. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell line. Neuroendocrinology. 87, 168–181. https://doi.org/10.1159/000111501 (2008).
Bittner, E. A. & Martyn, J. A. J. In Pharmacology and Physiology for Anesthesia, 2nd edn (eds Hugh C. Hemmings & Talmage D. Egan) 412–427 (Elsevier, 2019).
Liu, X. et al. Morphine promotes the malignant biological behavior of non-small cell lung cancer cells through the mor/src/mtor pathway. Cancer Cell Int. 21, 622. https://doi.org/10.1186/s12935-021-02334-8 (2021).
Gabr, M. M. et al. Interaction of opioids with TLR4-Mechanisms and ramifications. Cancers (Basel). https://doi.org/10.3390/cancers13215274 (2021).
Chen, S. L. et al. Low-dose memantine attenuated morphine addictive behavior through its anti-inflammation and neurotrophic effects in rats. J. Neuroimmune Pharmacol. 7, 444–453. https://doi.org/10.1007/s11481-011-9337-9 (2012).
Skrabalova, J., Drastichova, Z. & Novotny, J. Morphine as a potential oxidative stress-causing agent. Mini Rev. Org. Chem. 10, 367–372. https://doi.org/10.2174/1570193x113106660031 (2013).
Cavdar, S. et al. Effect of octreotide on oxidative stress in the erythrocyte and kidney tissue in adriamycin-induced experimental nephrotic syndrome model. J. Bras. Nefrol. 46, 18–28. https://doi.org/10.1590/2175-8239-JBN-2022-0180en (2024).
Gotoh, M., Iguchi, A., Kakumu, S., Hirooka, Y. & Smythe, G. A. Central suppressive effect of octreotide on the hyperglycemic response to 2-deoxy-d-glucose injection or cold-swim stress in awake rats: possible mediation role of hypothalamic noradrenergic drive. Brain Res. 895, 146–152 (2001).
Pardridge, W. M. Drug delivery to the brain. J. Cereb. Blood Flow. Metab. 17, 713–731. https://doi.org/10.1097/00004647-199707000-00001 (1997).
Lerner, E. N., van Zanten, E. H. & Stewart, G. R. Enhanced delivery of octreotide to the brain via transnasal iontophoretic administration. J. Drug Target. 12, 273–280. https://doi.org/10.1080/10611860400000938 (2004).
Hassanshahi, A. et al. Preventive putative effect of Agmatine on cognitive and molecular outcomes in ventral tegmental area of male offspring following physical and psychological prenatal stress. Dev. Psychobiol. 65, 22410 (2023).
Saffarpour, S. et al. The alteration of hippocampal BDNF expression is associated with anxiety-like behavior following the injury to the sciatic nerve. Arch. Neurosci. 6 (2019).
Razavinasab, M., Moazzami, K. & Shabani, M. Maternal mobile phone exposure alters intrinsic electrophysiological properties of CA1 pyramidal neurons in rat offspring. Toxicol. Ind. Health. 32, 968–979. https://doi.org/10.1177/0748233714525497 (2016).
Hassanshahi, A. et al. Administration of Agmatine prior to physical or psychological stress in pregnant mice ameliorates behavioural and cognitive deficits in female offspring. Int. J. Dev. Neurosci. (2023).
Khaleghi, M. et al. Estrogen attenuates physical and psychological stress-induced cognitive impairments in ovariectomized rats. Brain Behav. 11, e02139. https://doi.org/10.1002/brb3.2139 (2021).
Shabani, M., Ilaghi, M., Naderi, R. & Razavinasab, M. The hyperexcitability of laterodorsal tegmentum cholinergic neurons accompanies adverse behavioral and cognitive outcomes of prenatal stress. Sci. Rep. 13, 6011. https://doi.org/10.1038/s41598-023-33016-2 (2023).
Razavinasab, M. et al. Early environmental enrichment prevents cognitive impairments and developing addictive behaviours in a mouse model of prenatal psychological and physical stress. Int. J. Dev. Neurosci. 82, 72–84. https://doi.org/10.1002/jdn.10161 (2022).
Giacchino, J. L. & Henriksen, S. J. Systemic morphine and local opioid effects on neuronal activity in the medial prefrontal cortex. Neuroscience. 70, 941–949. https://doi.org/10.1016/0306-4522(95)00409-2 (1996).
Ayieng’a, E. O. et al. Morphine aggravates inflammatory, behavioral, and hippocampal structural deficits in septic rats. Sci. Rep. 13, 21460. https://doi.org/10.1038/s41598-023-46427-y (2023).
Pirmoradi, Z. et al. Resveratrol and 1,25-dihydroxyvitamin D decrease Lingo-1 levels, and improve behavior in harmaline-induced essential tremor, suggesting potential therapeutic benefits. Sci. Rep. 14, 9864. https://doi.org/10.1038/s41598-024-60518-4 (2024).
Release, S. (2021).
Greenwood, J. R., Calkins, D., Sullivan, A. P. & Shelley, J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric States of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604 (2010).
Lu, C. et al. Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Jacobson, M. P., Friesner, R. A., Xiang, Z. & Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608 (2002).
Jacobson, M. P. et al. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 55, 351–367 (2004).
Halgren, T. A. et al. Glide: a new approach for rapid, accurate Docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate Docking and scoring. 1. Method and assessment of Docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein – ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Acknowledgements
We would like to thank Kerman Neuroscience Research Center and Pharmaceutical Sciences and Cosmetic Products Research Center for their help and support.
Funding
Kerman University of Medical Sciences (Grant No. 400000157) provided funding for this study.
Author information
Authors and Affiliations
Contributions
M.S.H. created Figs. 2, 3, 4, 5, 6 and 7 and helped with the discussion, Z.P. created Fig. 8 that is the pathology part and helped with animal handling, M.R. helped with animal handling and cannula insertion, A.H. helped with animal handling, R.N. helped with the cannula insertion, Z.E. helped with animal handling, M.N.E. helped with writing the initial draft and creating Fig. 1, M.I. helped with writing the initial draft, M.A.L. helped with the docking sections and creating Figs. 9 and 10 and A.H.M. helped with the docking sections and creating Figs. 9 and 10 and L.R. helped with writing the initial draft, M.A.Z. helped with the PCR section, M.A. helped with the PCR section, K.A.K. created the final draft, S.A. helped with animal handling, H.F. is the second supervisor, M.B. is the first supervisor.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All experiments were done in accordance with the ARRIVE guidelines and National Institutes of Health Guide for the Care and Use of Laboratory Animals) NIH Publication No. 80-23, revised 1996). All experiments were performed in accordance with the protocols approved by Kerman University of Medical Sciences Laboratory Animal Care and was accepted by the Institutional Review Board (IRB) of Kerman University of Medical Sciences (IRB code: IR.KMU.REC.1400.242).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shabani, M., Pirmoradi, Z., Razavinasab, M. et al. Unveiling the therapeutic potential of octreotide in treating morphine dependence. Sci Rep 15, 29853 (2025). https://doi.org/10.1038/s41598-025-12761-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-12761-6
