
Abstract
Bone remodeling is tightly regulated by the RANK-RANKL-OPG axis to maintain skeletal integrity. We previously identified a RANK splicing variant, vRANK, which encodes a truncated protein that inhibits osteoclastogenesis and promotes apoptosis. This study examined the regulatory mechanisms of vRANK expression in vitro and its physiological role in vivo using transgenic mice with forced vRANK expression in the monocyte-macrophage lineage and in a systemic manner. In vitro, PMA and TGF-β1 specifically induced vRANK expression, which was completely suppressed by U0126, a MEK1/2 inhibitor, and significantly reduced by Sam68 knockdown, indicating its involvement in RNA splicing regulation. In vivo, RANK-Cre-driven vRANK expression had no skeletal impact, whereas LysM-Cre-driven expression resulted in increased bone mass and suppressed osteoclastogenesis. However, systemic overexpression of vRANK (CAG-CreER) caused perinatal lethality, severe cardiac fibrosis, and immune dysfunction. Notably, myocardial fibrosis in vRANK-overexpressing mice correlated with TGF-β1 upregulation in fibrotic foci, suggesting a pathological feedback loop exacerbating fibrosis. These findings suggest that vRANK functions similarly to OPG in inhibiting osteoclastogenesis while also potentially playing a role in cardiac remodeling and immune regulation.
Similar content being viewed by others


Introduction
Bone remodeling is a tightly regulated process essential for maintaining skeletal integrity, achieved through a dynamic balance between bone formation and resorption1,2,3. The latter process is primarily governed by the receptor activator of nuclear factor-κB ligand (RANKL)4,5, its receptor RANK6, and the decoy receptor osteoprotegerin (OPG)7. RANKL, predominantly secreted by osteoblasts and osteocytes, binds to RANK on osteoclast precursors, promoting their differentiation, activation, and survival8. In contrast, OPG, secreted by osteoblasts and other cell types, functions as a competitive inhibitor by binding to RANKL, thereby preventing its interaction with RANK and suppressing osteoclast-mediated bone resorption. By modulating the RANK-RANKL-OPG axis, bone metabolism is influenced by various systemic factors, including estrogen9, vitamin D10, and parathyroid hormone (PTH)11. Dysregulation of this system can lead to pathological bone loss1,12,13; for instance, excessive RANKL expression or OPG deficiency enhances osteoclast activity, contributing to diseases such as osteoporosis14, rheumatoid arthritis15,16, and osteolytic bone metastases1,13,17,18. Notably, the RANK-RANKL-OPG system plays a critical role not only in bone metabolism but also in broader physiological processes, including immune regulation19, cardiovascular function20, and thermoregulation21.
In our previous research22, we identified a splicing variant of RANK (vRANK), which introduced a premature stop codon in RANK mRNA, leading to the production of a truncated protein. In vitro studies demonstrated that vRANK inhibits osteoclastogenesis and promotes osteoclast apoptosis, thereby shortening osteoclast lifespan22. However, its functional significance in vivo remains unclear.
In this study, we first conducted in vitro analyses to investigate the factors regulating vRANK expression. Subsequently, we generated transgenic mouse models in which vRANK was forcibly expressed in the monocyte-macrophage lineage and systemically to elucidate its pathophysiological role at the organismal level in mice.
Materials and methods
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, USA), unless otherwise stated. Cell culture media, fetal bovine serum (FBS) and antibiotics-antimycotics were purchased from Life Technologies (Carlsbad, USA).
Genomic structure and organization of mouse Rank gene (Fig. 1)

Genomic organization of mouse RANK gene, splicing pattern, predicted 3D structure, and putative sam68 binding sites around exan2a. (A) In mice, the RANK gene harbors a splicing variant (vRANK) in addition to the known ten-exon structure. This variant includes an additional exon, designated as exon 2a, which is inserted between exon 1 and exon 2 (highlighted in blue, with the corresponding nucleotide sequence also marked in blue below). Within exon 2a, a stop codon is present (indicated in yellow in the nucleotide sequence). (B) Consequently, while the full-length RANK (fRANK) encodes a protein consisting of 625 amino acids, the vRANK transcript results in a truncated RANK protein comprising only 45 amino acids. The predicted three-dimensional structures of these protein products and a schematic diagram of the RANK protein is included, highlighting the signal peptide, cysteine-rich domain, transmembrane region, and intracellular domain. The vRANK variant corresponds primarily to the region encompassing the signal peptide and adjacent N-terminal sequences. (C) Furthermore, a motif search for Sam68 (U/AAA(N)15–20U/AAA,), a key factor involved in regulating splicing variations, identified four putative binding motifs within the nucleotide sequence, which are underlined in the lower panel.
We reported the identification of exon 2a within the Rank gene of mice, located between exon 1 and exon 2, in addition to the previously known ten exons (Fig. 1A)22. The presence of canonical ag and gu splice site sequences, which typically flank intronic regions, further supports the inclusion of exon 2a within the Rank gene structure. Additionally, a stop codon is located within exon 2a, resulting in the production of a truncated form of the RANK protein, composed of the first 45 amino acids from the N-terminus. The predicted three-dimensional structures of the full-length RANK (fRANK) protein and its truncated variant (vRANK), derived from their respective mRNA transcripts, are shown in Fig. 1B. The structural predictions were generated based on the amino acid sequences using a modeling tool available through the Protein Data Bank (PDB) website. A schematic diagram of the RANK protein is also included, highlighting the signal peptide, cysteine-rich domain, transmembrane region, and intracellular domain. The vRANK variant corresponds primarily to the region encompassing the signal peptide and adjacent N-terminal sequences. Furthermore, four putative binding sites for Sam68, a key regulator of mRNA splicing characterized by the consensus sequence U/AAA(N)₁₅₋₂₀U/AAA23, were identified in the regions surrounding exon 2a (Fig. 1C).
Expression analysis of fRANK and vRANK by reverse transcription polymerase chain reaction (RT-PCR)
RAW264.7 cells (RIKEN BioResource Center, Japan) were cultured and passaged according to previously established methods24. Subconfluent RAW264.7 cells were treated with 10 µM of phorbol 12-myristate 13-acetate (PMA), 10 µM of forskolin, and 20 ng/ml lipopolysaccharide (LPS). RNA was extracted 12 h post-treatment and subjected to RT-PCR. Bone marrow- and spleen-derived macrophages were isolated from 8-week-old male mice using a previously described method9,25. These primary cultured macrophages were then stimulated with PMA under the same conditions as those applied to RAW264.7 cells and subjected to RT-PCR analysis. For RT-PCR, primer set A was used to amplify both fRANK and vRANK, while primer set B was specifically designed to detect vRANK (Fig. 1) as previously described22. The amplification protocol consisted of an initial denaturation at 95 °C for 2 min, followed by 30-cycle of 92 °C for 30 s, 65 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 2 min. The RT-PCR products were electrophoresed on a 3% agarose gel. To investigate the temporal regulation of vRANK expression, time-course experiments were conducted following 10 µM of PMA stimulation. Additionally, cells were treated with 2 ng/ml of recombinant mouse transforming growth factor-β1 (TGF-β1, R&D Systems, MN, USA, Catalog #: 7666-MB) and recombinant human/mouse/rat bone morphogenetic protein-2 (BMP-2, R&D Systems, MN, USA, Catalog #: 355-BM), and the effects of intracellular signaling inhibitors SB203580, PD98059, and U0126 were assessed. Furthermore, to examine the involvement of Sam68, a putative regulator of Rank mRNA splicing, small interfering RNA (siRNA) targeting Sam68 was used (Silencer® Select Pre-designed siRNA Product, siRNA ID s20951 and s20953, Ambion, Thermo Fisher Scientific, Tokyo, Japan), and the impact of Sam68 knockdown on vRANK expression was evaluated according to the manufacture’s procedure. Control RT-PCR was also conducted using 1 µg of total RNA and the following set of GAPDH primers: sense: 5′-TGCACCACCAACTGCTTAG-3′, antisense: 5′-GGATGCAGGGATGATGTTC-3′ as described26.
Osteoclast differentiation process and generation of Cre-expressing mouse models (Fig. 2)

Schematic representation of osteoclastogenesis and the Cre driver construct used in vRANK overexpression mice generated in this study. Osteoclasts originate from monocyte/macrophage lineage cells and mature into pre-osteoclasts expressing RANK in the presence of M-CSF. The interaction between RANKL, expressed by osteoblast-lineage cells, and its receptor RANK on pre-osteoclasts induces the terminal differentiation of pre-osteoclasts into multinucleated mature osteoclasts. Osteoprotegerin (OPG) acts as a decoy receptor for RANKL, thereby inhibiting the RANK-RANKL signaling axis. To generate mice with systemic overexpression of vRANK, we utilized a combination of the CAG promoter (Chicken β-actin promoter + Cytomegalovirus immediate early enhancer) and tamoxifen-inducible CreER. Additionally, we established a system in which vRANK was broadly expressed in the monocyte/macrophage lineage using LysM-Cre. Furthermore, we employed RANK-Cre to enforce vRANK expression specifically at the pre-osteoclast stage and examined the phenotypic consequences in each mouse model.
As illustrated in the schematic diagram in Fig. 2, osteoclast differentiation occurs through the interaction between monocyte/macrophage lineage cells and the RANK-RANKL signaling pathway leading to the formation of mature osteoclasts. To investigate the role of vRANK in this process, three Cre-expressing mouse models were generated: CAG-CreER, which induces systemic overexpression of vRANK; LysM-Cre, which drives expression in early monocyte/macrophage lineage cells; and RANK-Cre, which specifically expresses Cre in pre-osteoclasts, where the RANK receptor is present. The generation of these models was carried out using the following methodology. Cre-Lox-regulated vRANK-overexpressing transgenic mice (vRANK conditional transgenic mice) were generated by Transgenic Inc. (Kobe, Japan) as a custom order, using the pCALNL-vRANK transgenic construct. The vRANK DNA fragment was inserted into the cloning site of the pCALNL vector (Addgene plasmid 13769). The vRANK DNA fragment was flanked by loxP sites on both sides of a stop sequence, positioned downstream of a CAG enhancer within the pCALNL vector.
Cre-mediated deletion of the stop sequence promoted the expression of the vRANK transgene. Prior to pronuclear injection for transgenic mouse production, the pCALNL-vRANK transgenic construct vector was linearized by Sfi I/Ssp I digestion. Overexpression of vRANK was achieved by breeding vRANK conditional Tg mice with Cre driver mice that express Cre recombinase, thereby deleting the stop sequence.
CAG-CreER, RANK-Cre, and LysM-Cre mice were crossed with vRANK conditional Tg mice as Cre driver lines. The CAG-CreER mice were obtained from The Jackson Laboratory (Stock No. 004682)27. The RANK-Cre mice have been previously described28. LysM-Cre mice were obtained from The Jackson Laboratory (Stock No. 004781)29. All strains were maintained on a C57BL/6J background (CLEA Japan, Tokyo). Tamoxifen-inducible CreER-mediated gene recombination around embryonic day 10 was induced by oral gavage administration of tamoxifen (100 mg/kg body weight) to pregnant mice on embryonic days 8, 9, and 10. Stock solutions of Tamoxifen (Sigma) were prepared at a concentration of 20 mg/mL in corn oil. All mice were housed in a specific pathogen-free (SPF) facility under climate-controlled conditions, following a 12 h light/dark cycle, with ad libitum access to water and a standard diet (Oriental Yeast). In the RANK-Cre and LysM-Cre mouse lines, five randomly selected 8-week-old male mice derived from three litters were used for analysis. In the CAG-CreER line, three 5-week-old male mice that survived the perinatal period were obtained from four litters and included in the study. To assess the specificity of vRANK expression in each mouse line, we utilized a fluorescent marker gene, DsRed, which was inserted downstream of the vRANK gene via an internal ribosome entry site (IRES). Tissue-specific expression of vRANK was verified based on the localization of DsRed fluorescence in the respective transgenic mice. Furthermore, RNAScope™ in situ hybridization was performed using a probe targeting DsRed (AF506025.1), according to the manufacturer’s protocol (Advanced Cell Diagnostics, Newark, CA, USA) (data not shown). All animal procedures were performed in accordance with the Basic Animal Care and Experimental Guidelines of the Ministry of Education, Culture, Sports, Science and Technology of Japan. All animal experimental procedures and protocols were approved by the Committee on Animal Research at Ehime University (Permit No. 05-KU-23-16). All procedures involving live animals were conducted in accordance with the ARRIVE guidelines (PLoS Biol 8(6), e1000412, 2010), and were approved by the Institutional Animal Care and Use Committee (Permit No. 05-KU-23-16). Anesthesia and euthanasia were performed using isoflurane and pentobarbital sodium, respectively, in accordance with commonly accepted veterinary best practices.
Histological analysis of mouse tissues
Histological examination of genetically modified mouse tissues was performed following standard fixation and embedding protocols. Harvested tissues were fixed overnight at 4 °C in 4% paraformaldehyde (PFA), dehydrated through an ethanol series, and subsequently embedded in paraffin. Tissue sections were then prepared at a thickness of 4 μm using a microtome. For morphological analysis, hematoxylin and eosin (H&E) staining was performed, while tartrate-resistant acid phosphatase (TRAcP) staining with a commercial kit was conducted to visualize osteoclasts. As parameters for bone morphometric analysis, bone volume/tissue volume (BV/TV, %) was measured as a structural parameter, and osteoclast surface/bone surface (Oc.S/BS, %) was evaluated as a bone resorption parameter. Measurement terminology and units were defined according to the guidelines of the American Society for Bone and Mineral Research30,31. For CAG-CreER/vRANK mice, in addition to the above parameters, the proportion of adipose tissue within the bone marrow space (Fat/BM, %) were quantified in a 1.0-mm segment extending proximally from a point 0.5 mm above the distal femoral growth plate. These values were compared with those of 5-week-old control mice.
Microcomputed tomography (μCT)
μCT analysis of the distal femurs was performed using the Scanco Medical μCT 35 System (SCANCO Medical). The region of interest was defined as a 1.0-mm segment extending proximally from a point 0.5 mm above the distal femoral growth plate with a resolution of 6 μm, and BV/TV (%) was three-dimensionally calculated as previously described32.
Evaluation of osteoclastogenesis in ex vivo culture
To assess osteoclast differentiation in vitro, splenic tissues were harvested from mice and mechanically dissociated using a 20 μm mesh filter. Following erythrocyte lysis with ammonium chloride, cells were cultured in the presence of macrophage colony-stimulating factor (M-CSF, SRP3221, Sigma-Aldrich, Japan). Osteoclast differentiation was induced by the addition of 100 ng/ml recombinant human soluble RANKL (sRANKL, Oriental Yeast Co., LTD., Tokyo, Japan). Osteoclast formation was evaluated by TRAcP staining, and the number of multinucleated giant cells with three or more nuclei per mm2 in each round cover slip inserted in culture dish was quantified under microscope.
Immunohistochemistry and in situ hybridization
For the analysis of mRNA expression, in situ hybridization was conducted using the RNAscope™ technique (Mm-Tnfrsf11b, NM_008764.3) to assess OPG mRNA expression levels. To verify mRNA preservation in tissue samples, the expression of ubiquitin C (Mm-Ubc, NM_019639.4) was used as a positive control. To investigate the localization of TGF-β1 protein expression, immunohistochemical analysis was performed on excised myocardial tissues from CAG-CreER/vRANK mice, with control mice used for comparison. Anti-TGF-β1 antibody (Abcam, ab215715, at 1:500 dilution) were utilized for detection. At embryonic day 10 (E10.0), hearts were harvested from control mouse embryos during the critical period of aortic valve development. RANK expression (NM_009399.3) was analyzed by RNAscope in situ hybridization. Cathepsin K expression was evaluated by immunohistochemistry using a monoclonal anti-cathepsin K antibody [3F9] (ab37259, Abcam, Cambridge, UK).
Statistical analyses
Statistical analyses were performed using GraphPad Prism 10, GraphPad Software, Boston, MA, USA). Data are expressed as mean ± standard deviation (SD). Differences between two groups were analyzed using Student’s t-test. Asterisks indicate significant upregulation or downregulation (*P < 0.05, **P < 0.01). NS indicates that a result was not significant.
Results
Analysis of vRANK expression regulation
Stimulation of RAW264.7 cells with PMA, forskolin, or LPS resulted in no significant changes in fRANK expression. However, vRANK expression was specifically induced following PMA treatment (Fig. 3A, upper panel). A time-course analysis of PMA-induced vRANK expression revealed that expression was detectable approximately 9 h after stimulation and gradually declined but remained detectable up to 48 h post-treatment (Fig. 3A, lower panel). To assess the responsiveness to PMA, primary cultured macrophages were examined. Induction of vRANK expression upon PMA stimulation was also observed in both spleen- and bone marrow-derived macrophages (Fig. 3B). In experiments with TGF-β1 and BMP-2, neither factor significantly altered fRANK expression. However, similar to PMA, TGF-β1 treatment induced vRANK expression, whereas BMP-2 had almost no effect (Fig. 3C, left). To further investigate intracellular signaling pathways regulating vRANK expression, pharmacological inhibitors were used. Notably, U0126, a MEK1/2 inhibitor, completely abolished PMA-induced vRANK expression (Fig. 3C, right). As shown in the lower panel of Fig. 1, since four Sam68 response elements were clustered around exon 2a of the RANK gene, Sam68 knockdown was performed using siRNA to assess Sam68’s functional relevance. As shown in Fig. 3D, Sam68 depletion by siRNA, alone or in combination, led to a marked suppression of vRANK expression in the presence of PMA.

Analysis of vRANK and fRANK expression in RAW264.7 cells and primary cultured macrophages by reverse transcriptase-PCR. (A) Upon treating RAW264.7 cells with PMA, Forskolin, and LPS to assess the inducibility of vRANK expression, PMA was found to induce vRANK expression. Time-course analysis of PMA treatment revealed that vRANK expression was observed at 9 h post-treatment, persisted for 24 h, and subsequently diminished while the expression level of GAPDH remained unchanged. (B) Induction of vRANK expression upon PMA stimulation was also observed in both spleen- and bone marrow-derived macrophages. (C) Treatment with TGF-β1 induced vRANK expression similarly to PMA, whereas BMP-2 treatment did not exhibit this effect. Investigation of the intracellular signaling pathways involved in PMA-induced vRANK expression using various inhibitors revealed that U0126, an MEK1/2 inhibitor, effectively blocked vRANK induction. (D) Knockdown of Sma68 was examined by treating cells with siRNA. The results showed that #1 (siRNA ID s20951) and #3 (siRNA ID s20953) each suppressed vRANK expression individually, as well as in combination while expression level of GAPDH unchanged.
Forced expression of vRANK under the control of RANK-Cre (Fig. 4)

Bone phenotype of mice in which vRANK is forcibly expressed at the pre-osteoclast level under the control of the RANK promoter using RANK-Cre system. Using the RANK-Cre system, we forcibly expressed vRANK in pre-osteoclast-level cells expressing RANK. Compared to control wild type (WT) mice, no macroscopic changes were observed in external appearance or internal organs. At the tissue level, no morphological differences were observed between the control WT group (A) and RANK-Cre/vRANK mice (B) in bone tissue. We examined osteoclast status using TRAcP staining, but there were no differences in the distribution or morphology of TRAcP-positive multinucleated cells between control WT mice (C) and RANK-Cre/vRANK mice (D). An enlarged image is inserted in the lower right corner (black arrows indicate TRAcP-positive osteoclasts). In histomorphometric analysis, there were no significant differences in either BV/TV or Oc.S/BS between control mice and RANK-Cre/vRANK mice (n = 5, all male, 8 weeks old).
Forced expression of vRANK under the control of RANK-Cre did not result in any overt phenotypic changes in mice at the macroscopic level. Histological analysis of bone tissues revealed no significant differences between vRANK-expressing mice and control mice in terms of growth plate width, trabecular structure and distribution, or cortical bone thickness (Fig. 4A, B). Similarly, TRAcP staining demonstrated that the morphology and distribution of osteoclasts remained unchanged between the two groups (Fig. 4C, D). Histomorphometric analysis revealed no significant differences between control and RANK-Cre/vRANK mice in either BV/TV (Fig. 4E) or Oc.S/BS (Fig. 4F). No apparent abnormalities were observed in the major organs—heart, lungs, liver, kidneys, intestines (small intestine), pancreas, thymus, skeletal muscle, femur, and vertebrae—either macroscopically or under microscopic examination (data not shown).
Forced expression of vRANK under the control of LysM-Cre (Fig. 5)

Phenotype of mice in which vRANK is forcibly expressed in monocyte/ macrophage-lineage cells using the LysM-Cre system. In LysM-Cre/vRANK mice, no macroscopic changes were observed in external appearance or internal organs. However, as shown by µCT (A), an increase in bone volume was observed. The BV/TV analysis (A, right) demonstrated a significant increase compared to control WT mice (*, P < 0.05). On the other hand, in control WT mice, thicker trabeculae were predominantly found in the metaphysis, gradually narrowing toward the diaphysis. In contrast, LysM-Cre/vRANK mice exhibited an irregular distribution of thick and thin trabeculae, suggesting a potential abnormality in bone remodeling. (B) Macrophages were cultured ex vivo from the spleens of control WT and LysM-Cre/vRANK mice, and osteoclast differentiation was induced by treatment with sRANKL. The upper panels show circular coverslips inserted into culture plates stained with TRAcP, and the lower panels present magnified views of these coverslips, where cultured multinucleated osteoclast-like cells are visible as red-stained cells. TRAcP-positive multinucleated cell formation was reduced in LysM-Cre/vRANK mice compared to WT. Quantification of TRAcP-positive multinucleated cells per unit area (B, right graph) revealed a significant decrease in osteoclast formation in LysM-Cre/vRANK mice (**, P < 0.02) (n = 5, all male, 8 weeks old).
In LysM-Cre-driven vRANK-overexpressing mice, no apparent macroscopic abnormalities were observed. However, µCT analysis (Fig. 5A, left) revealed a significant increase (P < 0.05) in bone volume fraction BV/TV in vRANK-expressing mice compared to controls (Fig. 5A, right). To evaluate osteoclastogenic potential, ex vivo cultures were performed (Fig. 5B, left). Osteoclast differentiation in response to sRANKL stimulation was significantly reduced (P < 0.01) in LysM-Cre/vRANK mice, in an inverse correlation with the observed increase in bone mass (Fig. 5B, right). Neither macroscopic inspection nor histological analysis revealed any marked changes in the heart, lungs, liver, kidneys, intestines (small intestine), pancreas, thymus, skeletal muscle, femur, and vertebrae (data not shown).
Forced expression of vRANK under the control of CAG-CreER (Bone, Fig. 6)

Phenotype of mice with systemic forced expression of vRANK using the CAG-CreER system (Bone). In H&E-stained bone sections, compared to control WT mice (A), CAG-CreER/vRANK mice (B) exhibited thinning of trabeculae and cortical bone, along with a reduction in bone marrow adipocytes. In TRAcP staining (C and D), numerous TRAcP-positive multinucleated cells were observed on the trabecular surface in controls (C), whereas their number was markedly reduced in CAG-CreER/vRANK mice (D). Histomorphometric analysis also revealed significantly decreased values in CAG-CreER/vRANK mice for (E) BV/TV, (F) Oc.S/BS, and (G) Fat/BM (the proportion of adipocytes per 1 mm2 bone marrow area) (*P < 0.05, P < 0.02; n = 3, all male, 5 weeks old).
Tamoxifen administration at embryonic days 8 and 9 resulted in complete perinatal lethality, with all pups dying shortly after birth. When tamoxifen was administered at embryonic day 10, CAG-CreER-driven systemic overexpression of vRANK also led to high rate of perinatal lethality; among 17 pups born from four pregnant dams, 14 died shortly after birth. The three surviving mice, all male, also exhibited progressive deterioration in general condition, and all died by approximately 5 weeks of age. At this time point, major organs—including the heart, lungs, kidneys, liver, and bone—were harvested from these mice for further analysis. Compared to 5-week-old control mice (Fig. 6A), femurs from CAG-CreER/vRANK mice (Fig. 6B) exhibited cortical bone thinning, reduced trabecular bone volume, and decreased bone marrow adiposity. TRAcP staining revealed a tendency toward reduced numbers of TRAcP-positive osteoclasts in CAG-CreER/vRANK mice (Fig. 6D), compared with controls (Fig. 6C). Quantitative histomorphometric analysis demonstrated a significant decrease in BV/TV in CAG-CreER/vRANK mice (P < 0.05; Fig. 6E). In parallel, a significant reduction in Oc.S/BS was observed in CAG-CreER/vRANK mice (P < 0.01; Fig. 6F). Additionally, the area occupied by adipocytes in the bone marrow was significantly lower in CAG-CreER/vRANK mice (P < 0.01; Fig. 6G).
Forced expression of vRANK under the control of CAG-CreER (Heart, Fig. 7)

Phenotype of mice with systemic forced expression of vRANK using the CAG-CreER system (Heart). In CAG-CreER/vRANK mice, systemic induction of vRANK resulted in significant cardiac enlargement and thinning of the left ventricular myocardial wall (yellow double-headed arrows) compared to control WT mice (A, B). At 200× magnification, myocardial tissue from WT mice displayed an orderly arrangement of cardiomyocytes (C), whereas in CAG-CreER/vRANK mice, myocardial cell loss and compensatory fibrosis were observed (D). At higher magnification (400×), cardiomyocyte swelling and myofiber disruption (black arrows), absent in the control (E), were sporadically observed in non-fibrotic regions of the myocardium in CAG-CreER/vRANK mice. Immunohistochemical analysis of TGF-β1 protein localization showed that WT mice had almost no positive cells (G, 400×), whereas in CAG-CreER/vRANK mice, TGF-β1 was localized in spindle-shaped fibroblasts within the fibrotic myocardial regions (H, 400×). In situ hybridization using the RNAscope method was performed to evaluate Opg mRNA expression in myocardial tissue. In both WT (I) and CAG-CreER/vRANK mice (J), Opg mRNA expression was observed as indicated by black arrows. Compared to WT mice, CAG-CreER/vRANK mice showed reduced expression of Opg mRNA (J, 400×). The inset in the lower right shows the expression of ubiquitin C as a positive control for each specimen, with strong signals detected in both groups, indicating that mRNA integrity was well preserved in all samples.
Histological examination of the heart revealed severe cardiac enlargement, thinning of the left ventricular wall, and focal fibrosis within the myocardium (Fig. 7A–D). Under high-magnification observation, while no cellular swelling or myofibrillar loss was observed in control mice (Fig. 7E), scattered cardiomyocytes exhibiting these pathological changes were present in CAG-CreER/vRANK mice (Fig. 7F). Notably, no inflammatory cell infiltration was observed in or around the fibrotic areas or the swollen cardiomyocytes. Immunohistochemical analyses revealed that, compared to the control (Fig. 7G), fibrotic regions in the CAG-CreER/vRANK heart tissue demonstrated strong TGF-β1 positivity (Fig. 7H). Although OPG mRNA expression levels were generally low in both control and CAG-CreER mice, a trend toward further reduction was observed in CAG-CreER mice (Fig. 7I, J). Additionally, systemic health deterioration was evident in CAG-CreER /vRANK mice the presence of pneumonia in lung tissues (Fig. 8). Microscopic examination revealed vascular congestion in the liver and kidney. No significant histological abnormalities were observed in other major organs, including the small intestine, pancreas, thymus, and skeletal muscle (data not shown).

Lung phenotypes of CAG-CreER/vRANK mice. At 5 weeks of age, CAG-CreER/vRANK mice exhibited severe systemic debilitation, and histological examination of the lung revealed features of pneumonia. In control mice (A), no cellular infiltration was observed in the alveolar spaces. In contrast, CAG-CreER/vRANK mice showed prominent infiltration of neutrophils within the alveolar spaces, consistent with acute bronchopneumonia. No evidence of vasculitis or specific inflammation was detected, and the pneumonia was pathologically interpreted as a terminal event secondary to systemic debilitation.
RANK and Cathepsin K Expression in the Embryonic Day 10 (E10.0) Mouse Heart (Fig. 9)

Analysis of Rank mRNA and Cathepsin K Protein Expression in Wild-Type Mouse Embryos at Embryonic Day 10 (E10.0) During Aortic Valve Development. Because myocardial fibrosis in CAG-CreER/vRANK mice was specifically observed when tamoxifen was administered at embryonic day 10 (E10.0), we focused on aortic valve formation, a critical event in cardiac development occurring at this stage. To investigate tissue-level molecular events, Rank mRNA expression and Cathepsin K protein expression were examined by RNAscope and immunohistochemistry, respectively. The upper panel shows a low-magnification view of a wild-type embryo, with the developing heart outlined in red, where aortic valve formation is actively occurring. In this region, Rank mRNA expression (lower left) was detected in the valve primordium (arrows), and Cathepsin K protein (lower right) was observed along the endocardium (arrows). These findings suggest that E10.0 corresponds to a critical window for aortic valve development, and that the timing of tamoxifen-induced vRANK expression may contribute to the cardiac phenotype observed in CAG-CreER/vRANK mice.
Given the presence of a critical period around embryonic day 10 (E10.0) during which CAG-CreER/vRANK mice appeared to experience significant cardiac impact, we investigated the expression of RANK and cathepsin K in the hearts of control embryos at E10.0. As previously reported33, RANK is expressed in the cells of the developing aortic valve primordium during valvulogenesis, while cathepsin K is induced on the endocardial side. We confirmed these expression patterns in our independent analysis (Fig. 9).
Osteoprotegerin expression in bone tissue of RANK-Cre, LyM-Cre, and CAG-CreER/vRANK mice (Fig. 10)

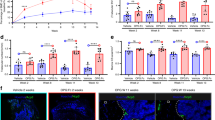
RNAscope-Based Analysis of Opg mRNA Expression in Bone Tissue of Control, RANK-Cre/vRANK, LyM-Cre/vRANK, and CAG-CreER/vRANK Mice. Opg mRNA expression in bone tissue of vRANK-overexpressing mice was examined using RNAscope. In control mice (A), Opg mRNA was detected in osteoblastic lineage cells and endothelial cells, as indicated by black arrows. In contrast, its expression was markedly reduced in (B) RANK-Cre/vRANK, (C) LyM-Cre/vRANK, and (D) CAG-CreER/vRANK mice. Ubiquitin C mRNA expression was also evaluated (E–H), and the number of grains per mm2 was quantified. When compared with the number of Opg grains, as shown in (I), a significant decrease in Opg mRNA expression was observed in all vRANK-overexpressing mice compared to controls. CAG-CreER/vRANK and control mice were analyzed at 5 weeks of age; all other groups were examined at 8 weeks.
Compared to control mice (Fig. 10A), bone tissues from RANK-Cre (Fig. 10B), LysM-Cre (Fig. 10C), and CAG-CreER (Fig. 10D) mice showed a marked suppression of OPG mRNA expression (indicated by arrows). Quantitative analysis comparing Opg mRNA expression with that observed in tissues expressing ubiquitin C (Fig. 10E–H), demonstrated that Opg mRNA expression was consistently reduced in all mice with enforced vRANK expression as summarized in Fig. 10I.
Discussion
In this study, we first studied the regulatory mechanism of vRANK expression in vitro. As shown in Fig. 3A, PMA-induced activation of PKC led to the upregulation of vRANK expression. Furthermore, the upregulation of vRANK expression was suppressed by U0126, a MEK1/2 inhibitor. Although the response to PMA differs from our previous observations22, this may be attributable to differences in culture conditions—particularly cell density—at the time of PMA treatment. These findings suggest that vRANK induction is primarily mediated through the MEK1/2 signaling pathway. Further sequence analysis of the RANK gene revealed the presence of four putative binding sites for Sam68 in the region surrounding exon 2a, suggesting its potential involvement in RNA splicing regulation. Sam68 is an RNA-binding protein (RBP) that plays a crucial role in post-transcriptional regulation, including alternative splicing, RNA transport, and stability control34,35. It is particularly important in cancer, immune responses, and cell division-related signaling pathways23,34,35. The activity of Sam68 is regulated by phosphorylation, with tyrosine phosphorylation mediated by the Src family of tyrosine kinases and serine/threonine phosphorylation induced by the MAPK signaling pathway35. Serine/threonine phosphorylation of Sam68 has been reported to modulate its function as a splicing regulatory factor, influencing exon selection in mRNAs such as Bcl-x36, CD4437, and Tau38, thereby affecting cell survival and differentiation. Although direct binding assays between Sam68 and RANK mRNA have not been conducted in this study, in vitro experiments using siRNA-mediated Sam68 knockdown (Fig. 3D) indirectly demonstrated that suppression of Sam68 inhibits the formation of splicing variants, supporting the proposed mechanism shown in Fig. 11. Based on these findings, we propose the following model: In the absence of phosphorylated Sam68, precursor RANK mRNA undergoes splicing where exon 2a is excluded, as Sam68 is unable to bind efficiently to the surrounding sequences. In contrast, when Sam68 is phosphorylated, it binds to the exon 2a region of precursor RANK mRNA, promoting the inclusion of exon 2a into the final transcript through the gu-ag splicing consensus sequences, leading to the formation of a splicing variant.

TGF-β1-Induced vRANK Expression via MEK1/2-ERK1/2 Signaling and Sam68-Mediated Alternative Splicing. TGF-β1 triggers a phosphorylation cascade through MEK1/2 and ERK1/2, leading to serine/threonine phosphorylation of Sam68 protein. As a result, as shown in the right figure, phosphorylated Sam68 binds to four Sam68-binding motifs located around RANK exon 2a, promoting the inclusion of exon 2a in a splicing variant of RANK. In fibrotic myocardial lesions, TGF-β1 production induces vRANK expression in infiltrating macrophages. This, in turn, promotes fibrosis progression, forming a vicious cycle that may contribute to disease exacerbation.
To investigate the pathophysiological function of the truncated RANK protein at the organismal level in mice, we generated a series of mouse models expressing vRANK. Since our current and previous in vitro studies demonstrate that vRANK negatively regulates osteoclastogenesis, a function similar to OPG, we expected the vRANK-expressing mice to have phenotypic similarity to OPG-genetically modified mice. When vRANK was forcibly expressed in RANK-expressing pre-osteoclast stage, as shown in Fig. 4A–F, no significant changes were observed in bone structure or systemic phenotype, suggesting that the effects of vRANK at this stage may be buffered by local regulatory factors such as OPG and M-CSF. Indeed, when examining Opg mRNA expression in bone tissues across all series of mouse models expressing vRANK, a consistent reduction in expression was observed (Fig. 8A–H), suggesting that Opg mRNA expression is suppressed via a feedback mechanism within the RANK-RANKL-OPG axis. Notably, it is presumed that cellular differentiation up to the stage at which RANK expression is initially induced in the monocyte/macrophage lineage by M-CSF39 is maintained. Moreover, the structure of vRANK contains a substantial portion of the signal peptide region40 of fRANK (Fig. 1B), suggesting that at the expression level under the control of the RANK promoter, vRANK may not completely suppress the endoplasmic reticulum transport and proper folding of fRANK. Therefore, terminal differentiation into osteoclasts via the positive feedback loop mediated by RANK-RANKL binding39 appears to be marginally preserved through the suppression of OPG expression. In contrast, when vRANK was overexpressed at an earlier stage of monocyte/macrophage lineage differentiation, a significant increase in bone mineral density (BMD) was observed (Fig. 5A). Furthermore, ex vivo cultures of macrophages isolated from the spleens of LysM-Cre/vRANK mice demonstrated a marked suppression of osteoclastogenesis (Fig. 5B), confirming that vRANK inhibits osteoclast differentiation and enhances bone mass at the organismal level. Moreover, as shown in Fig. 5A, µCT analysis of LysM-Cre/vRANK mice revealed irregular trabecular bone structure. This phenotype is reminiscent of previously reported OPG-overexpressing rat models41, in which apparent increases in bone mass were accompanied by an imbalance in bone remodeling, leading to increased bone fragility. The similarities between the vRANK- and OPG-overexpressing mice suggest that excess vRANK expression may disrupt normal bone remodeling dynamics, despite increasing overall bone mass.
When vRANK was globally induced at embryonic day 10 by tamoxifen administration under the control of CAG-CreER, the mice exhibited high perinatal lethality, and none survived beyond 5 weeks of age. As shown in Fig. 6, histological analysis of bone tissue from CAG-CreER/vRANK mice that survived to 5 weeks of age revealed narrowing of both cortical and trabecular bone. Quantitative bone morphometric analysis demonstrated a significant reduction in BV/TV (P < 0.05). Despite the marked reduction in osteoclast formation (Fig. 6D and F), a decrease in BV/TV was observed, suggesting that the bone phenotype is primarily attributable to a more severe impairment of bone formation. This finding further implies that the bone loss may be secondary to systemic deterioration in the overall condition of the mice. As shown in Fig. 6A, B, and G, the number of adipocytes in the bone marrow of CAG-CreER/vRANK mice was reduced compared to wild-type controls. Previous studies have reported that the number of adipocytes is increased in the bone marrow of Opg knockout mice42, suggesting that OPG functions to suppress adipogenesis within the bone marrow. The reduced number of adipocytes observed in the CAG-CreER/vRANK mice thus represents another point of similarity between the effects of vRANK expression and OPG activity.
Besides its inhibitory effect on osteoclasts, OPG is also recognized as a biomarker of vascular calcification43, and its overexpression has been reported to promote arterial wall calcification and contribute to the progression of atherosclerosis44. Additionally, elevated circulating OPG levels have been correlated with the severity of heart failure45, and excessive OPG expression has been implicated in cardiac hypertrophy46, and contractile dysfunction47. Conversely, OPG has also been suggested to exert a cardioprotective effect following myocardial infarction48, indicating a complex role in cardiac homeostasis and remodeling. Similar to its function in bone remodeling, both excessive expression and depletion of OPG may lead to pathological myocardial remodeling, potentially resulting in cardiac dysfunction49. Furthermore, it is known that the RANK–RANKL–OPG axis plays a role in cardiac development33. During the period of aortic valve formation, RANK is expressed in cells of the aortic valve primordium (bulbus cordis), while Cathepsin K is expressed on the endocardial side, contributing to valve morphogenesis33. We confirmed these findings in our own experiments using E10.0 control mouse embryos (Fig. 9). Therefore, induction of vRANK expression at E10.0 via tamoxifen, as in the present study, is presumed to interfere with endogenous RANK function during valve formation—at a level that allows survival up to approximately 5 weeks of age—potentially leading to aortic valve stenosis. The myocardial injury observed in CAG-CreER/vRANK mice appears to originate at the level of individual cardiomyocytes. Given the paucity of inflammatory infiltrates, this change is likely a non-inflammatory process induced by vRANK, which may exacerbate impaired cardiac remodeling in response to left ventricular pressure overload, ultimately resulting in fibrosis. These findings suggest the presence of a vicious cycle in which TGF-β released from fibrotic lesions induces vRANK expression, further exacerbating tissue injury and fibrosis. This extensive myocardial fibrosis likely contributed to a progressive decline in cardiac contractility. This, in turn, led to heart failure, cardiac enlargement, and myocardial wall thinning, resulting in the death of all affected mice by 5 weeks of age. The pathological features observed in CAG-CreER/vRANK mice seem to reflect clinical findings in patients with aortic stenosis, where myocardial fibrosis has been shown to be associated with increased OPG expression, as supported by studies using late gadolinium-enhanced cardiovascular magnetic resonance imaging, which demonstrated significantly higher circulating OPG levels in patients with chronic myocardial infarction fibrosis compared to those without fibrosis50. These extraskeletal phenotypic resemblance between vRANK and OPG-overexpressing mouse models suggest a potential functional overlap between vRANK and OPG in cardiac remodeling. Although time-course progression of lesions from birth could not be assessed, the cardiac lesions exhibited fibrosis consistent with scar formation over 2–4 weeks. In contrast, the pulmonary findings of neutrophilic infiltration suggest acute bronchopneumonia, likely representing a secondary event due to systemic deterioration, occurring within the past 2–3 days (Fig. 8).
Our findings indicate that vRANK exhibits functional similarity to OPG both in vitro and at the organismal level. OPG is not only a key regulator of bone metabolism but also circulates in the bloodstream, exerting systemic effects on the cardiovascular system and other tissues. In contrast, high expression of vRANK is presumed to exert its effects through intracellular competition involving signal peptides22, suggesting that its physiological and pathological effects may be more localized and operate at a more fine-tuned regulatory level than those of OPG. The influence of vRANK on bone metabolism and systemic homeostasis may be relatively limited, with its effects largely confined to vRANK-expressing cells, particularly macrophage-lineage cells, including osteoclasts.
Conclusions
vRANK may play a crucial role in the fine-tuning of local tissue remodeling. In irreversible pathological conditions, such as progressive myocardial fibrosis, vRANK-mediated regulation of local remodeling processes may contribute significantly to disease progression, highlighting its potential role in pathological tissue remodeling in both skeletal and cardiovascular tissues.
Future plan
In future studies, we plan to administer tamoxifen to CAG-CreER/vRANK mice at approximately 8 weeks of age to examine the resulting bone and cardiac phenotype. Additionally, we intend to establish an isoproterenol-induced heart failure model to investigate fibrotic remodeling in response to cardiac stress.
Limitations
In vitro analyses did not assess the direct interaction between Sam68 and RANK mRNA, nor did they comprehensively examine factors other than Sam68 involved in mRNA splicing.
At the time of this study’s publication, selectively analyzing vRANK mRNA expression in cardiac tissue using in situ hybridization was technically challenging, and thus, was not conducted.
Moreover, due to the absence of a specific antibody capable of detecting the truncated vRANK protein, immunohistochemical analysis of vRANK tissue expression could not be performed.
In the CAG-ERCre model, tamoxifen administration at embryonic day 10.0 induced myocardial fibrosis; however, only a limited number of mice survived after birth. Because the attrition of mice occurred in a non-random manner over time, the assumption of unbiased time-dependent survival could not be met, making it difficult to present a Kaplan–Meier survival curve.
Furthermore, the plasma concentration of OPG has not been measured in genetically modified animal models, and the impact of forced vRANK expression on circulating OPG levels remains unassessed.
Data availability
All data generated or analyzed during this study are included in this manuscript.
References
Kitazawa, S., Haraguchi, R. & Kitazawa, R. Roles of osteoclasts in pathological conditions. Pathol. Int. 75(2), 55–68 (2025).
Teitelbaum, S. L. & Ross, F. P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 4(8), 638–649 (2003).
Delaisse, J. M. et al. Re-thinking the bone remodeling cycle mechanism and the origin of bone loss. Bone 141, 115628 (2020).
Yasuda, H. et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. U S A 95(7), 3597–35602 (1998).
Lacey, D. L. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93(2), 165–176 (1998).
Nakagawa, N. et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 253(2), 395–400 (1998).
Simonet, W. S. et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 89(2), 309–319 (1997).
Yasuda, H. et al. A novel molecular mechanism modulating osteoclast differentiation and function. Bone 25(1), 109–113 (1999).
Kitazawa, R. et al. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J. Clin. Invest. 94(6), 2397–2406 (1994).
Lee, S. K. et al. 1,25(OH)2 vitamin D3-stimulated osteoclast formation in spleen-osteoblast cocultures is mediated in part by enhanced IL-1 alpha and receptor activator of NF-κB ligand production in osteoblasts. J. Immunol. 169(5), 2374–2380 (2002).
Kanatani, M. et al. Estrogen via the estrogen receptor blocks cAMP-mediated parathyroid hormone (PTH)-stimulated osteoclast formation. J. Bone Miner. Res. 13(5), 854–862 (1998).
Teitelbaum, S. L. Therapeutic implications of suppressing osteoclast formation versus function. Rheumatology 55(suppl 2), ii61–ii63 (2016).
Kitazawa, R. et al. Pathologic conditions of hard tissue: role of osteoclasts in osteolytic lesion. Histochem. Cell Biol. 149(4), 405–415 (2018).
Jabbar, S. et al. Osteoprotegerin, RANKL and bone turnover in postmenopausal osteoporosis. J. Clin. Pathol. 64(4), 354–357 (2011).
Mori, H. et al. RANK ligand, RANK, and OPG expression in type II collagen-induced arthritis mouse. Histochem. Cell Biol. 117(3), 283–292 (2002).
Yan, M. et al. ETS1 governs pathological tissue-remodeling programs in disease-associated fibroblasts. Nat. Immunol. 23(9), 1330–1341 (2022).
Kitazawa, S. & Kitazawa, R. RANK ligand is a prerequisite for cancer-associated osteolytic lesions. J. Pathol. 198(2), 228–236 (2002).
Kitazawa, S. et al. Modulation of αvβ3 integrin via transactivation of β3 integrin gene on murine bone marrow macrophages by 1,25(OH)2D3, retinoic acid and interleukin-4. Acta Histochem. Cytochem. 52(4), 77–83 (2019).
Okamoto, K. Crosstalk between bone and the immune system. J. Bone Miner. Metab. 42(4), 470–480 (2024).
Tamtaji, O. R. et al. The effects of fatty acids consumption on OPG/RANKL/RANK system in cardiovascular diseases: Current status and future perspectives for the impact of diet-gene interaction. J. Cell Biochem. 120(3), 2774–2781 (2019).
Hanada, R. et al. Central control of fever and female body temperature by RANKL/RANK. Nature 462(7272), 505–509 (2009).
Mukai, S. et al. Identification and analysis of function of a novel splicing variant of mouse receptor activator of NF-κB. Mol. Cell Biochem. 350(1–2), 29–38 (2011).
Naro, C. et al. Functional interaction between U1snRNP and Sam68 insures Proper 3’ end Pre-mRNA processing during germ cell differentiation. Cell Rep. 26(11), 2929-2941 e5 (2019).
Kitazawa, R. et al. CpG methylation of receptor activator NF-κB (RANK) gene promoter region delineates senescence-related decrease of RANK gene expression. Acta Histochem. Cytochem. 57(4), 137–147 (2024).
Takahashi, N. et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology 123(5), 2600–2602 (1988).
Kiyoshi, M. et al. Modulation of mouse RANKL gene expression by Runx2 and PKA pathway. J. Cell. Biochem. 98(6), 1629–1644 (2006).
Hayashi, S. & McMahon, A. P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244(2), 305–318 (2002).
Maeda, K. et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 18(3), 405–412 (2012).
Clausen, B. E. et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8(4), 265–277 (1999).
Dempster, D. W. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR histomorphometry nomenclature-committee. J. Bone Miner. Res. 28, 2–17 (2013).
Parfitt, A. M. et al. Bone histomorphometry: Standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committe. J. Bone Miner. Res. 2, 595–610 (1987).
Ito, M. et al. Multi-detector row CT imaging of vertebral microstructure for evaluation of fracture risk. J. Bone Miner. Res. 20, 1828–1836 (2005).
Alexander, W. L. et al. NFATc1 expression in the developing heart valves is responsible to the RANKL pathway and is required for endocardial expression of catepsin K. Dev. Biol. 292(2), 407–417 (2006).
Lukong, K. E. & Richard, S. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 1653(2), 73–86 (2003).
Matter, N., Herrlich, P. & Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420(6916), 691–695 (2002).
Paronetto, M. P. et al. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 176(7), 929–939 (2007).
Batsche, E., Yaniv, M. & Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 13(1), 22–29 (2006).
Stoss, O. et al. The STAR/GSG family protein rSLM-2 regulates the selection of alternative splice sites. J. Biol. Chem. 276(12), 8665–8673 (2001).
Kitazawa, R. et al. RANK-NFATc1 signaling forms positive feedback loop on rank gene expression via functional NFATc1 responsive element in rank gene promoter. Biochem. Biophys. Res. Commun. 572, 86–91 (2021).
Crockett, J. C. et al. Signal peptide mutations in RANK prevent downstream activation of NF-κB. J. Bone Miner. Res. 26(8), 1926–1938 (2011).
Ominsky, M. S. et al. One year of transgenic overexpression of osteoprotegerin in rats suppressed bone resorption and increased vertebral bone volume, density, and strength. J. Bone Miner. Res. 24(7), 1234–1246 (2009).
Zhang, L. et al. Role of osteoprotegerin (OPG) in bone marrow adipogenesis. Cell Physiol. Biochem. 40(3–4), 681–692 (2016).
Hofbauer, L. C. & Schoppet, M. Osteoprotegerin: A link between osteoporosis and arterial calcification?. Lancet 358(9278), 257–259 (2001).
Kiechl, S. et al. Osteoprotegerin is a risk factor for progressive atherosclerosis and cardiovascular disease. Circulation 109(18), 2175–2180 (2004).
Ueland, T. et al. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation 111(19), 2461–2468 (2005).
Tsuruda, T. et al. Angiotensin II stimulation of cardiac hypertrophy and functional decompensation in osteoprotegerin-deficient mice. Hypertension 67(5), 848–856 (2016).
Omland, T. et al. Plasma osteoprotegerin levels in the general population: Relation to indices of left ventricular structure and function. Hypertension 49(6), 1392–1398 (2007).
Lee, J. et al. Adipose-derived stem cell-released osteoprotegerin protects cardiomyocytes from reactive oxygen species-induced cell death. Stem Cell Res. Ther. 8(1), 195 (2017).
Shetelig, C. et al. Osteoprotegerin levels in ST-elevation myocardial infarction: Temporal profile and association with myocardial injury and left ventricular function. PLoS ONE 12(3), e0173034 (2017).
Loudon, B. L. et al. Osteoprotegerin and myocardial fibrosis in patients with aortic stenosis. Sci. Rep. 8(1), 14550 (2018).
Acknowledgements
We would like to thank Dr. Yuuki Imai (Division of Integrated Pathophysiology, Proteo-Science Center, Department of Pathophysiology, Graduate School of Medicine, Ehime University) for his help with µCT analyses. We also thank Ms. Chie Shiraishi and Ms. Yuki Takaoka for their excellent technical assistance.
Funding
The funders had no role in the design of the study, in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
Conceptualization: Sohei Kitazawa. Investigation and analyses: Riko Kitazawa and Ryuma Haraguchi. Writing-review and editing: All. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kitazawa, R., Haraguchi, R. & Kitazawa, S. A variant form of receptor activator of nuclear factor-κB functions as an osteoprotegerin mimic in bone and cardiac development. Sci Rep 15, 27919 (2025). https://doi.org/10.1038/s41598-025-13314-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13314-7
