
Abstract
Gestational diabetes mellitus (GDM) is a pregnancy-complicated disease that poses risks to maternal and infant health. However, its etiology has not yet been elucidated. This study investigated the associations between functional genetic variants of the GC vitamin D-binding protein (GC) gene and the risk of GDM. Subsequently, a nomogram predictive model was constructed for early risk identification in GDM. After adjusting for age and pre-pregnancy BMI, rs4752 A > G (AG vs AA: adjusted OR = 1.58, 95% CI: 1.19–2.10, P = 0.001; AG/GG vs AA: adjusted OR = 1.34, 95% CI: 1.04 – 1.71, P = 0.021), rs3733359 G > A (AA vs GG: adjusted OR = 0.70, 95% CI: 0.49–0.98, P = 0.039; AA vs GG/GA: adjusted OR = 0.71, 95% CI: 0.52 – 0.97, P = 0.031), and rs7041 A > C (AC vs AA: adjusted OR = 0.73, 95% CI: 0.57 – 0.94, P = 0.015; AC/CC vs AA: adjusted OR = 0.74, 95% CI: 0.58 – 0.94, P = 0.014) were significantly associated with GDM risk. In the MDR analysis, rs7041 was identified as the best single-locus model, while the two-loci model of rs4752 and rs7041 was the best multiple-factor interaction model for GDM risk prediction. It appears that rs4752 and rs7041 may alter post-transcriptional splicing, while rs3733359 alters transcription factor binding, thereby affecting individual susceptibility to GDM. A predictive nomogram model constructed with rs4752 and clinical indicators (Age, FPG, OGTT1h, OGTT2h and HbA1c) has ideal discriminant ability with a diagnostic AUC of 0.943. These findings still need to be confirmed through larger scale studies and molecular experiments in the future.
Similar content being viewed by others

Introduction
Gestational diabetes mellitus (GDM) is widely recognized as one of the most common metabolic complications of pregnant women, and its incidence has been rising in the past decades1. Currently, the global morbidity of GDM is around 2–20% and that is 14.8% in China2. GDM can lead to serious short-term complications, such as pre-eclampsia, preterm birth, and fetal macrosomia3,4, as well as long-term risks, including obesity and type 2 diabetes mellitus (T2DM) for both mothers and their offspring5,6. However, the specific etiological mechanism of GDM has not been fully revealed yet.
Clarifying the risk factors of GDM is essential for early intervention and improved prognosis7. Known risk factors include advanced maternal age, obesity, and family history of diabetes8,9. Epidemiological studies further indicate that GDM exhibits familial genetic characteristics, with at least 30% of affected women experiencing recurrence10. Family history of diabetes is also an independent risk factor, with closer familial relationships correlating with higher GDM risk11. These findings highlight the significant role of genetic factors in GDM etiology.
Single nucleotide polymorphisms (SNPs) are the most common genetic variants frequently occurring in the human genome12. SNPs located in different functional regions of genes may alter an individual’s susceptibility to complex diseases by affecting alternative splicing, mRNA levels, protein translation, or metabolic pathways13,14,15. Genome-wide association studies (GWASs) and candidate gene studies have identified a series of GDM susceptible SNPs. And our previous studies also have found numerous GDM genetic polymorphisms in the Guilin population, such as the OR2D2 gene rs1965211, RXR-g rs2134095, TSNARE1 rs7814359, XAB2 rs3760675, ERBB4 rs1595066, MTNR1B rs10830963, CDKAL (rs7756992 and rs7754840), and ACE2 (rs6632677 and rs2074192)16,17,18,19,20. Genetic polymorphisms were observed significant interaction with environmental factors in the GDM occurrence, such as age, pre-pregnancy BMI, blood glucose, or lipid levels21.
Disease prediction models are valuable tools for estimating an individual’s probability of developing specific conditions in the future22. Numerous nomogram models for GDM based on conventional clinical parameters have been developed to facilitate early prevention and treatment23,24,25. The nomogram calculates individual risk scores by weighted key risk factors and evaluates the risk or survival probability of an event based on these scores26. This approach provides clinicians with a highly accurate, personalized risk assessment tool to support decision-making27. However, current nomogram models primarily rely on clinical and demographic factors, with limited inclusion of genetic susceptibility markers. Given the significant role of genetic variants in GDM pathogenesis, there is a critical need to integrate genetic risk factors with traditional clinical indicators2,18,28. Such models would significantly enhance the precision of GDM risk prediction and inform targeted prevention strategies20,29.
Currently, a case–control study involving 567 GDM cases and 710 controls from the Guilin region of China has been conducted to clarify the association between candidate functional genetic variants of the GC gene and GDM risk. Besides, a nomogram model with identified significantly associated SNPs and key clinical indicators was constructed for early GDM prediction.
Methods
Study subjects
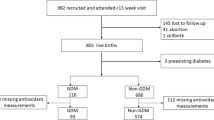
1277 participants were recruited from the Affiliated Hospital of Guilin Medical University, including 567 GDM cases and 710 normal pregnancies for case–control studies. According to the criteria of the International Association of Diabetes and Pregnancy Study Group (IADPSG)30, GDM can be diagnosed based on at least one threshold value reached or exceeded in the 75 g oral glucose tolerance test (OGTT) : fasting plasma glucose (FPG) (≥ 5.1mmol/L); 1 h (1h) blood glucose (≥ 10.0mmol/L); 2 h blood glucose (≥ 8.5mmol/L). Inclusion criteria required that participants have no familial relationships and be experiencing singleton pregnancy. Exclusion criteria included: (1) presence of endocrine diseases; (2) serious systemic diseases; (3) history of pre-pregnancy type 1 or type 2 diabetes; (4) long term use of glucose metabolism-affecting drugs; (5) other pregnancy complications before pregnancy. The Ethics Committee of Guilin Medical University has approved this study, and all the participants had signed the informed consent. The research was conducted in accordance with the Declaration of Helsinki.
Clinical and biochemical data collection
Clinical indicators were collected from the same questionnaires and hospital records, including age, height, weight, systolic blood pressure (SBP), and diastolic blood pressure (DBP), fasting plasma glucose (FPG), a-hour postprandial glucose (1hPG), 2-h postprandial glucose (2hPG), glycosylated hemoglobin (HbA1c), triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-c), low density lipoprotein cholesterol (LDL-c). Pre-pregnancy body mass index (pre-BMI) was calculated using formula:weight (kg)/height (m)2.
DNA extraction, variants selection and genotyping
Genomic DNA was extracted from EDTA-treated whole blood using a DNA extraction kit (Aidlab Biotechnologies Co., Ltd, China) and stored at −80 °C prior to PCR.
Potential functional variants in the GC gene region were identified using the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) and the SNPinfo Web Server (http://snpinfo.niehs.nih.gov/). Variants were screened based on a minimum allele frequency (MAF) > 0.05 in the Chinese Han population in Beijing (CHB). Four functional variants with a linkage disequilibrium (LD) threshold of less than 0.8 were selected, including splicing site variants (rs4752 A > G, rs4588 G > T and rs7041 A > C) and a transcription factor binding site (TFBS) variant (rs3733359 G > A).
Genotyping of candidate variants was performed using the Sequenom MassARRAY platform. The PCR reaction mixture consisted of the following components: 1 μl template DNA (20 ~ 100 ng/μl), 1.850 μl ddH2O, 0.625 μl of 1.25 × PCR buffer (15 mmol/L MgCl2), 0.325 μl of 25 mmol/L MgCl2, 0.1 μl of 25 mmol/L dNTP mix, 1 μl of 0.5 μmol/L primer mix, and 0.1 μl of 5 U/μl HotStar Taq polymerase. PCR cycling was conducted at 94 ℃ for 15 min, 45 cycles at 94 ℃ for 20 s, 56 ℃ for 30 s, and 72 ℃ for 1 min, with a final incubation at 72 ℃ for 3 min. The original data and genotyping plots were obtained using TYPER 4.0 software.
Functional analysis by bioinformatic tools
For SNPs located in transcription factor binding site, the Alibaba 2.1 tool (http://generegulation.com/pub/programs/alibaba2/index.html) was used to explore potential biological functions. The Alternative Splice Site Predictor (ASSP) (http://wangcomputing.com/assp/overview.html) was used to analyze their post-transcriptional splicing regulation. DNA sequence data obtained from public databases (NCBI), ranged from 100nt. DNA sequence containing wild-type and mutant alleles in FASTA format was inputted. Alibaba 2.1 compares these sequences with known transcription factor motif databases to predict SNP induced changes in binding sites. In the ASSP analysis, codon usage and stop codons for all three possible reading frames (F1-frame 1, F2-frame 2, F3-frame 3) and scores of the preprocessing models reflecting splice site strength are calculated.
Statistical analysis
Data analysis was performed using the software IBM SPSS Statistics 28 for Windows (IBM Corp., Armonk, NY, USA). The case and control groups were compared for continuous variables, which were expressed as mean ± standard deviation(‾x ± sd), using the independent samples t-test. Categorical variables were analyzed using the chi-square test, and the Hardy–Weinberg equilibrium (HWE) was assessed using a χ2 goodness-of-fit test. The odds ratios (ORs) and their corresponding 95% confidence intervals (CIs) were utilized to evaluate the association between genetic variants and GDM risk. A two-sided test was applied, and a P < 0.05 was considered as statistically significant.
Stratified analysis was conducted to assess the association between positive SNPs and GDM risk among different subgroups, which were categorized by the mean values of variables. Additionally, false-positive reporting probability (FPRP) analysis was employed to validate the robustness of the statistically significant associations between variants and GDM risk, using a preset FPRP cutoff value of 0.2 and a prior probability level of 0.1. Results with FPRP values < 0.2 were considered genuine associations31.
Multi-factor dimension reduction (MDR) software (version 3.0.2) was applied to investigate the interaction effects between variants. The best multifactorial model was identified with values of cross validation consistency (CVC) and testing balanced accuracy (TBA) variables with a preset 100-fold cross-validation.
A predictive nomogram composed of clinical risk factors and positive SNPs was eventually constructed using the R package ‘rms’. Variables for constructing the predictive nomogram were selected using the stepwise logistic regression method. The training set and validation set were randomly divided in a 7:3 ratio. The area under the receiver operating characteristic curve (AUC) was used to evaluate the model’s performance. The calibration curve was generated using internal validation with a bootstrap method involving 1000 resamples to evaluate the consistency between the predicted and observed values. Besides, decision curve analysis (DCA) was also conducted to evaluate the clinical effectiveness and benefit of the nomogram.
DeLong’s test was used to compare performance differences between ROC curves. A two-sided test was adopted, and P values < 0.05 were considered to indicate statistical significance.
Results
Characteristics of study subjects
The characteristics of recruited research subjects are shown inTable 1. Multiple clinical indicators in GDM cases were significantly higher than those in controls, including age, pre-BMI, SBP, DBP, TG, Hb, FPG, 1hPG and 2hPG (P < 0.001).
Association between studied variants and GDM risk
After testing, the distribution frequencies of the three genotypes of the studied variants in the control group were consistent with HWE, except for rs4752 and rs4588. Logistic regression analyses showed that rs4752 A > G was significantly associated with an increased risk of GDM after adjusting of age and pre-BMI. Compared with the AA genotype, the AG genotype significantly increased the risk of GDM (AG vs AA, adjusted OR = 1.58, 95% CI = 1.19 − 2.10, P = 0.001). Meanwhile, under the dominant model (AG/GG vs AA), the AG/GG genotype showed a significantly increased GDM risk compared to the AA genotype (adjusted OR = 1.34, 95% CI = 1.04 − 1.71, P = 0.021). After adjusting for age and pre-BMI, logistic regression analysis also demonstrated that rs3733359 G > A and GC rs7041 A > C were significantly associated with the decreased risk of GDM. Compared to GG genotype, rs3733359 AA significantly reduced the GDM risk by 30% (adjusted OR = 0.70, 95% CI = 0.49—0.98, P = 0.039). Moreover, under the recessive model (AA vs GG/GA), AA genotype carriers had a lower GDM risk compared with GG/GA (adjusted OR = 0.71, 95% CI = 0.52—0.97, P = 0.031). Additionally, rs7041 AC significantly decreased the GDM risk by 27% compared to the AA genotype (adjusted OR = 0.73, 95% CI = 0.57—0.94, P = 0.015). Under the dominant model (rs7041 AC/CC vs AA), the AC/CC genotype also showed a protective effect against individual susceptibility to GDM (adjusted OR = 0.74, 95% CI = 0.58—0.94, P = 0.014). However, no significant association between rs4588 and GDM risk was found, as shown in Table 2. Further stratified analysis showed that, similar to rs3733359, significant effects of rs4752 and rs7041 on GDM risk were detected in most subgroups (supplement Table 1–3).
False-positive reporting probability (FPRP) analysis
The FPRP was adopted to evaluate relationship between the studied variants (rs4752, rs3733359, rs7041) and GDM risk, using a prior probability of 0.1 and a relatively stringent FPRP cutoff value of 0.2. The association between rs4752 and GDM risk was found to be genuine in subjects under the heterozygous model (AG vs AA) (PFPRP = 0.028), with SBP ≤ 110.19mmHg (PFPRP = 0.123), 1hPG ≤ 8.23mmol/L (PFPRP = 0.068) and 2hPG ≤ 7.08mmol/L (PFPRP = 0.181). Meanwhile, for rs7041, the FPRP values were 0.135 under the dominant model, 0.151 under the heterozygous model, 0.192 for SBP > 110.19mmHg and 0.181 for DBP > 69.37mmHg subgroup, suggesting that the significant associations observed may be genuine. FPRP results indicated that all significant findings related to rs3733359 might be chance occurrences and should be cautiously accepted. See Supplement Table 4 and Table 5.
MDR analysis for variants interactions in GC gene with GDM risk
The MDR analysis results showed that rs7041 was the optimal single-point model for GDM risk (TBA: 0.5417, CVC: 81/100, P < 0.003). Additionally, the best multi-loci model was the two-locus model involving rs4752 and rs7041, suggesting that their interaction may significantly increase susceptibility to GDM (TBA: 0.5624, CVC: 100/100, P < 0.0001), as shown in Table 3.
Potential regulatory function analysis
According to the SNP function prediction results from SNPinfo Web Server, we found that the mutation sites rs4752 and rs7041 in the GC gene splicing region are significantly associated with GDM susceptibility. We then evaluated the effects of these variants on the post-transcriptional splicing and the structure or function of the GC coding protein, using the Alternative Splice Site Predictor (ASSP) tool. Figure. 1 suggests that rs4752 A > G may create a new splicing site at the 102bp position of the examined DNA sequence, but only with the rs4752 G but not the A allele. This mutation also caused a change at the putative splice site at the 58bp position, which transitioned from being defined as Alt. isoform/cryptic 5′ splice sites to an unclassified 5′ splice sites. This suggests that rs4752 may have different post-transcriptional splicing regulation effects under different alleles. Besides, rs4752 may lead to changes in the activity of putative splice sites, which are characterized by score activation, intron GC activation, alt. isoform/cryptic activation, constitutive activation, and confidence activation near the polymorphism. For instance, from the analysis results, we observed that the activity of the putative splice site at the 105bp position had a score activation of 5.706, intron GC activation of 0.486, alt. isoform/cryptic activation of 0.870, constitutive activation of 0.129, and confidence activation of 0.851 with the A allele of rs4752. In contrast, the rs4752 G allele had a score activation of 5.602, intron GC activation of 0.500, alt. isoform/cryptic activation of 0.887, constitutive activation of 0.113, and confidence activation of 0.873. Additionally, rs7041 A > C may also create a new splicing site at the 106bp position of the examined DNA sequence, but only with the rs4752 C allele, not the A allele. This suggests that rs7041 may similarly alter the activity of putative splice sites, like rs4752. We found that the score activation at the putative splice sites at the 93bp position with the rs7041 A allele was 7.002, while the score activation changed to 7.048 with rs7041 C allele. See Fig. 2.

GC rs4752 (A > G) potential regulatory function analyzed by the Alternative Splice Site Predictor (ASSP) tool (A) Schematic representation of splicing positions reflecting the GC rs4752; (B) Score plot for preprocessing model reflecting sequences of putative splice sites.

GC rs7041 (A > G) potential regulatory function analyzed by the Alternative Splice Site Predictor (ASSP) tool (A) Schematic representation of splicing positions reflecting the GC rs7041; (B) Score plot for preprocessing model reflecting sequences of putative splice sites.
Furthermore, AliBaba 2.1 tool was used to predict transcription factor binding sites by constructing matrices from TRANSFAC 4.0 sites for rs3733359 G > A. The results suggested that, compared with the rs3733359 G allele, the mutation caused a decrease in two segments (RAR-alph and T3R-alpha) in this sequence, which were identified as potential binding sites in the rs3733359 A allele, as shown in Fig. 3.

Prediction transcription factors binding by the AliBaba 2.1 tool for rs3733359 G>A.
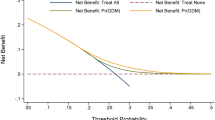
of variables in the stepwise regression model is shown in supplementary Table 6. Finally, six risk factors were selected to establish a nomogram based on the stepwise regression model, including Age, FPG, HAb1c, OGTT1h, OGTT2h and the dominant model of rs4752, see Fig. 4A. It can be observed that subjects with Age > 30.08 years, FPG > 4.77mmol/L, OGTT1h > 8.23mmol/L, OGTT 2 h > 7.08mmol/L, HbA1c > 5.16%, and carrying the AG/GG genotypes will obtain a total score of 312. The area under the ROC curve (AUC) was 0.943 in the training cohort and 0.959 in the validation cohort, demonstrating well-accepted predictive and discriminant performance (Fig. 4a-b). As shown in Fig. 4c-d, the calibration curve almost coincided with the ideal line, which also supports the good prediction ability of our nomogram. As shown in DCA (Fig. 4e-f), predictive nomogram model provides a higher clinical net benefit compared with the “treat all” or “treat none”strategies, indicating a good clinical value for the nomogram.

The establishment and validation of a nomogram model in predicting GDM risk. (A) A nomogram model constructed by Age, FPG, OGTT1h, OGTT2h, Hb, rs4752 based on stepwise logistic regression; Validation of the nomogram. (a) Receiver operating characteristic (ROC) curves in training set with an area under the curve (AUC) of 0.943.(b) ROC curve in validation set with a AUC of 0.959. (c) Calibration plot in training set (d) Calibration plot in validation (e) A decision curve analysis (DCA) in training set. (f) DCA curve in validation set.
To further evaluate the additional predictive value contributed by rs4752, we compared the model’s performance with and without genetic variant. The model incorporating only clinical variables (Age, FPG, HbA1c, OGTT1h, and OGTT2h) achieved an AUC of 0.923, while inclusion of the rs4752 variant increased the AUC to 0.943. However, DeLong’s test revealed this 0.020 improvement did not reach statistical significance (P = 0.19). See supplementary Fig. 1.
Discussion
Gestational diabetes mellitus (GDM) has become a critical public health challenge due to its increasing prevalence and its potential to cause health risks for both mothers and their offspring32,33. Despite extensive research, the underlying genetic mechanisms contributing to GDM remain incompletely characterized. This study investigated the associations between key functional variants in the GC gene, encoding vitamin D-binding protein, and GDM susceptibility in a Chinese cohort.
In this study, we observed that GC gene variants rs4752, rs3733359 and rs7041 are significantly associated with the risk of GDM. These associations may arise through changes in an individual’s genetic susceptibility and/or by regulating metabolism of blood glucose, blood lipids, and other clinical traits to participate in the pathogenesis of GDM. Interestingly, in the MDR analysis, a two-variant interaction model of rs4752 and rs7041 made the best multi-loci model for predicting GDM risk. Based on this, we conclude that there is a synergistic effect of genes and gene-environment in the onset of GDM. Furthermore, FPRP analysis suggests the associations between rs4752 and rs7041 and GDM risk in specific subjects are likely to be genuine. To some extent, the individualized differences of people suffering from GDM are likely due to GC gene variants and their interactions with key environmental factors.
We aimed to identify the potential biological functions underlying the significant association between variant and GDM pathogenesis. Specifically, the GC gene splicing site variants rs4752 and rs7041 are likely to regulate post-transcriptional splicing of GC mRNA by modifying the activity of existing splicing sites or by creating new splicing sites under different variant alleles. Additionally, rs3733359 A can affect the transcription factors (RAR-alpha and T3R-alpha) binding to the specific binding motifs in the GC gene promoter, potentially alter GC gene transcription. Therefore, under the same environmental risk exposure, GC variants may modify the genetic background of individuals with GDM. The above findings provide some new clues for revealing the biological mechanisms of susceptibility to GDM.
The continuous development of medical data science has made the construction of risk prediction models, such as logistic regression models, decision tree models, random forest models, support vector machine models, neural network models, etc., an important tool for predicting individual disease risk34. Given this, constructing an early risk prediction model based on key pathogenic factors and their effects may minimize the harm of the GDM35. An R et al.′s study found that the nomogram including clinical indicators such as AST/ALT in early pregnancy, maternal age, pre-BMI, waist circumference, and glucose and lipid metabolism, shows good predictive ability for the occurrence of GDM with an area under the ROC curve of 0.77836. However, current prediction models are primarily based on clinical indicators or environmental variables rather than predictive models that incorporate genetic effects.
In the present study, a nomogram model established with clinical parameters of Age, FPG, OGTT1h, OGTT2h, HbA1c and genetic variant rs4752, suggesting a higher accuracy (AUC = 0.943) and reliability in predicting GDM risk. This model quantifies the risk scores of various high-risk factors for GDM and visualizes the probability of individuals developing GDM in the future, providing guidance for personalized prevention of GDM. However, compared to the prediction model that excluded rs4752, a non-significant improvement in AUC was observed in the inclusion of rs4752 model. We speculate that this may be due to the fact that the impact of a single SNP on complex diseases is usually“minor”and does not play a dominant role in all factors related to GDM, such as environmental and genetic factors. However, there are a vast number of SNP variants in the human genome, and we have reason to believe that the overall effect of these potential’GDM related variants’in the genome plays an undeniable role in the etiology of GDM.
Our study further explored the contribution of genetic variants to GDM, building upon existing literature. We extended the findings of Mo et al37 on the GC gene by providing the association study of genetic variants (rs4752, rs3733359) and conducting a comprehensive functional analysis of SNPs within the GC gene. By elucidating how these genetic variants influence specific molecular mechanisms, our findings provide novel insights into the etiology of GDM. Furthermore, we innovatively integrated genetic variants with clinical indicators to construct a highly accurate nomogram model, significantly enhancing predictive accuracy.
However, several limitations should be acknowledged. First, as a hospital-based case–control study, selection bias is inevitable. Second, potential confounding factors, such as smoking status, obstetrics history, malnutrition, and socioeconomic factors were not assessed, which may influence the observed associations between genetic variants and GDM risk. Third, while the sample size of this study is relatively large, it may still be insufficient to detect extremely low-frequency genotypes. Fourthly, the deviation from Hardy–Weinberg equilibrium observed in the genotype frequencies of rs4752 and rs4588 could be due to factors such as the low frequency of these variant genotypes, the limited sample size, and potential interference effects from the multiplex PCR reactions in the Sequenom MassArray platform. Therefore, the association between these variants and GDM risk should be further validated in future studies with larger sample sizes. Additionally, this study explored the potential biological functions of positively associated SNPs using bioinformatics tools but did not confirm through cellular and molecular biology experiments. Finally, the validation cohort was derived from a random split of the original cohort, which may compromise the independence and reliability of the validation results. Independent external validation should be incorporated in future studies to better confirm the model and enhance its practical value.
In conclusion, our findings confirm a significant genetic association between functional variants in the GC gene and GDM susceptibility. The predictive nomogram model based on GC gene variants and key clinical parameters can effectively distinguish individual GDM risk. However, the findings of this study need to be further validated in larger and more diverse samples, and the biological functions of GC gene variants should also be elucidated through cellular and molecular biology experiments.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
References
Juan, J. & Yang, H. Prevalence, prevention, and lifestyle intervention of gestational diabetes mellitus in China. Int. J. Environ. Res. Public Health 17, 9517 (2020).
Liang, Q. et al. Genetic susceptibility, mendelian randomization and nomogram model construction of gestational diabetes mellitus. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgae200 (2024).
Bodmer-Roy, S., Morin, L., Cousineau, J. & Rey, E. Pregnancy outcomes in women with and without gestational diabetes mellitus according to the International association of the diabetes and pregnancy study groups criteria. Obstet. Gynecol. 120, 746–752 (2012).
Poomalar, G. K. Changing trends in management of gestational diabetes mellitus. World J. Diabetes 6, 284–295 (2015).
Bellamy, L., Casas, J. P., Hingorani, A. D. & Williams, D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet 373, 1773–1779 (2009).
Nar, G. et al. The relationship between epicardial fat thickness and gestational diabetes mellitus. Diabetol. Metab. Syndr. 6, 120 (2014).
Leonco, L. et al. Does universal screening for gestational diabetes mellitus improve neonatal outcomes in a socially vulnerable population: A prospective study in french guiana. Front. Endocrinol. 12, 644770 (2021).
Zhang, C. & Ning, Y. Effect of dietary and lifestyle factors on the risk of gestational diabetes: Review of epidemiologic evidence. Am. J. Clin. Nutr. 94, 1975s–1979s (2011).
Schwartz, N., Nachum, Z. & Green, M. S. The prevalence of gestational diabetes mellitus recurrence–effect of ethnicity and parity: A metaanalysis. Am. J. Obstet. Gynecol. 213, 310–317 (2015).
Forouhi, N. G., Luan, J., Cooper, A., Boucher, B. J. & Wareham, N. J. Baseline serum 25-hydroxy vitamin d is predictive of future glycemic status and insulin resistance: The medical research council ely prospective study 1990–2000. Diabetes 57, 2619–2625 (2008).
Wei, W. et al. Gestational diabetes mellitus: The genetic susceptibility behind the disease. Horm. Metab. Res. 53, 489–498 (2021).
Kim, S. & Misra, A. SNP genotyping: Technologies and biomedical applications. Annu. Rev. Biomed. Eng. 9, 289–320 (2007).
Prasad, R. B., Kristensen, K., Katsarou, A. & Shaat, N. Association of single nucleotide polymorphisms with insulin secretion, insulin sensitivity, and diabetes in women with a history of gestational diabetes mellitus. BMC Med. Genom. 14, 274 (2021).
Li, G. Expression quantitative trait loci analysis in multiple tissues. Methods Mol. Biol. (Clifton, N.J.) 2082(231), 237 (2020).
Azizzadeh-Roodpish, S., Garzon, M. H. & Mainali, S. Classifying single nucleotide polymorphisms in humans. Mol. Genet. Genom. 296, 1161–1173 (2021).
Liang, Q. et al. Genetic susceptibility, mendelian randomization, and nomogram model construction of gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 109, 2802–2814 (2024).
Yu, X. Y. et al. Association between functional genetic variants in retinoid X receptor-α/γ and the risk of gestational diabetes mellitus in a southern Chinese population. Biosci. Rep. 41, BSR20211338 (2021).
Li, R. et al. Genetic variants of ERBB4 gene and risk of gestational diabetes mellitus: A susceptibility and diagnostic nomogram study. Front. Endocrino.l 14, 1283539 (2023).
Huang, G. et al. Association of ACE2 gene functional variants with gestational diabetes mellitus risk in a southern Chinese population. Front. Endocrinol. 13, 1052906 (2022).
Huang, G. et al. Functional genetic variants and susceptibility and prediction of gestational diabetes mellitus. Sci. Rep. 14, 18123 (2024).
Behavioural interventions for type 2 diabetes: an evidence-based analysis. Ont Health Technol Assess Ser 9 1-45. (2009)
Sammani, A., Baas, A. F., Asselbergs, F. W. & Te Riele, A. Diagnosis and risk prediction of dilated cardiomyopathy in the era of big data and genomics. J. Clin. Med. 10, 921 (2021).
Tong, J. N. et al. Fasting plasma glucose in the first trimester is related to gestational diabetes mellitus and adverse pregnancy outcomes. Endocrine 75, 70–81 (2022).
Zhang, D. et al. A clinical model and nomogram for early prediction of gestational diabetes based on common maternal demographics and routine clinical parameters. J. Obstet. Gynaecol. Res. 48, 2738–2747 (2022).
Zhang, X. et al. Risk prediction model of gestational diabetes mellitus based on nomogram in a Chinese population cohort study. Sci. Rep. 10, 21223 (2020).
Li, L. et al. Establishment and validation of a predictive nomogram for gestational diabetes mellitus during early pregnancy term: A retrospective study. Front. Endocrinol 14, 1087994 (2023).
Li, S., Li, H., Li, C., He, X. & Wang, Y. Development and validation of a nomogram for predicting the risk of pregnancy-induced hypertension: A retrospective cohort study. J. Women’s Health 30, 1182–1191 (2021).
Tian, Y. & Li, P. Genetic risk score to improve prediction and treatment in gestational diabetes mellitus. Front. Endocrinol. 13, 955821 (2022).
Cheng, J., Meng, C., Li, J., Kong, Z. & Zhou, A. Integrating polygenic risk scores in the prediction of gestational diabetes risk in China. Front. Endocrinol. 15, 1391296 (2024).
Metzger, B. E. et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 33, 676–682 (2010).
Wacholder, S., Chanock, S., Garcia-Closas, M., El Ghormli, L. & Rothman, N. Assessing the probability that a positive report is false: An approach for molecular epidemiology studies. J. Natl. Cancer Inst. 96, 434–442 (2004).
Senat, M. V. & Deruelle, P. Gestational diabetes mellitus. Gynecol. Obstet. Fertil. 44, 244–247 (2016).
Lindsay, R. S., Mackin, S. T. & Nelson, S. M. Gestational diabetes mellitus-right person, right treatment, right time?. BMC Med. 15, 163 (2017).
Hossain, M. E., Khan, A., Moni, M. A. & Uddin, S. Use of electronic health data for disease prediction: A comprehensive literature review. IEEE/ACM Trans. Comput. Biol. Bioinform. 18, 745–758 (2021).
Lamain-de Ruiter, M. et al. Prediction models for the risk of gestational diabetes: A systematic review. Diagn. Progn. Res. 1, 3 (2017).
An, R. et al. AST-to-ALT ratio in the first trimester and the risk of gestational diabetes mellitus. Front. Endocrinol. 13, 1017448 (2022).
Mo, M. et al. The association of gene variants in the vitamin D metabolic pathway and its interaction with vitamin D on gestational diabetes mellitus: A prospective cohort study. Nutrients 13, 4220 (2021).
Funding
This study was supported by the Guangxi Natural Science Foundation (2025GXNSFHA069270), Guilin Science Research and Technology Development Plan Project (20230135–2-1), self-funded research project of the Health Committee of Guangxi (Z-C20241580), the Open Project Program of Guangxi Key Laboratory of Environmental Exposomics and Entire Lifecycle Health (2024-GKLEH-02) and Maternal and Child Health Research Project of Guangxi Bagui Scholars (J.Z.).
Author information
Authors and Affiliations
Contributions
Conceptualization: X.Y.Y., G.Y.Q.; Data curation: M.Y.X., S.Y.Q., L.J.N.; Formal analysis: R.Q.L., Z.H.W.; Investigation: Q.L.L.; Methodology: X.Y.Y., R.Q.L.; Software: R.Q.L., Q.L.L.; Visualization: L.J.N., Z.H.W.; Writing-original draft: R.Q.L., Z.H.W., X.Y.Y.; Writing-review & editing: X.Y.Y., G.Y.Q.; All authors reviewed the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, R., Wang, Z., Xiang, M. et al. GC vitamin D-binding protein gene functional genetic variants and gestational diabetes mellitus risk and prediction. Sci Rep 15, 27807 (2025). https://doi.org/10.1038/s41598-025-13483-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13483-5

{kind=link}